



Lysophosphatidylcholine Impairs the Mitochondria Homeostasis Leading to Trophoblast Dysfunction in Gestational Diabetes Mellitus
 , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Design and Sample Collection
2.2. Histology Analysis
2.3. Transmission Electron Microscopy (TEM)
2.4. Human LDL Isolation
2.5. Lipid Extraction
2.6. LC/MSE Analysis
2.7. Cell Culture
2.8. Confocal Microscopy
2.9. Mitochondria Isolation
2.10. Western Blot Analysis
2.11. Statistic Analysis
3. Results
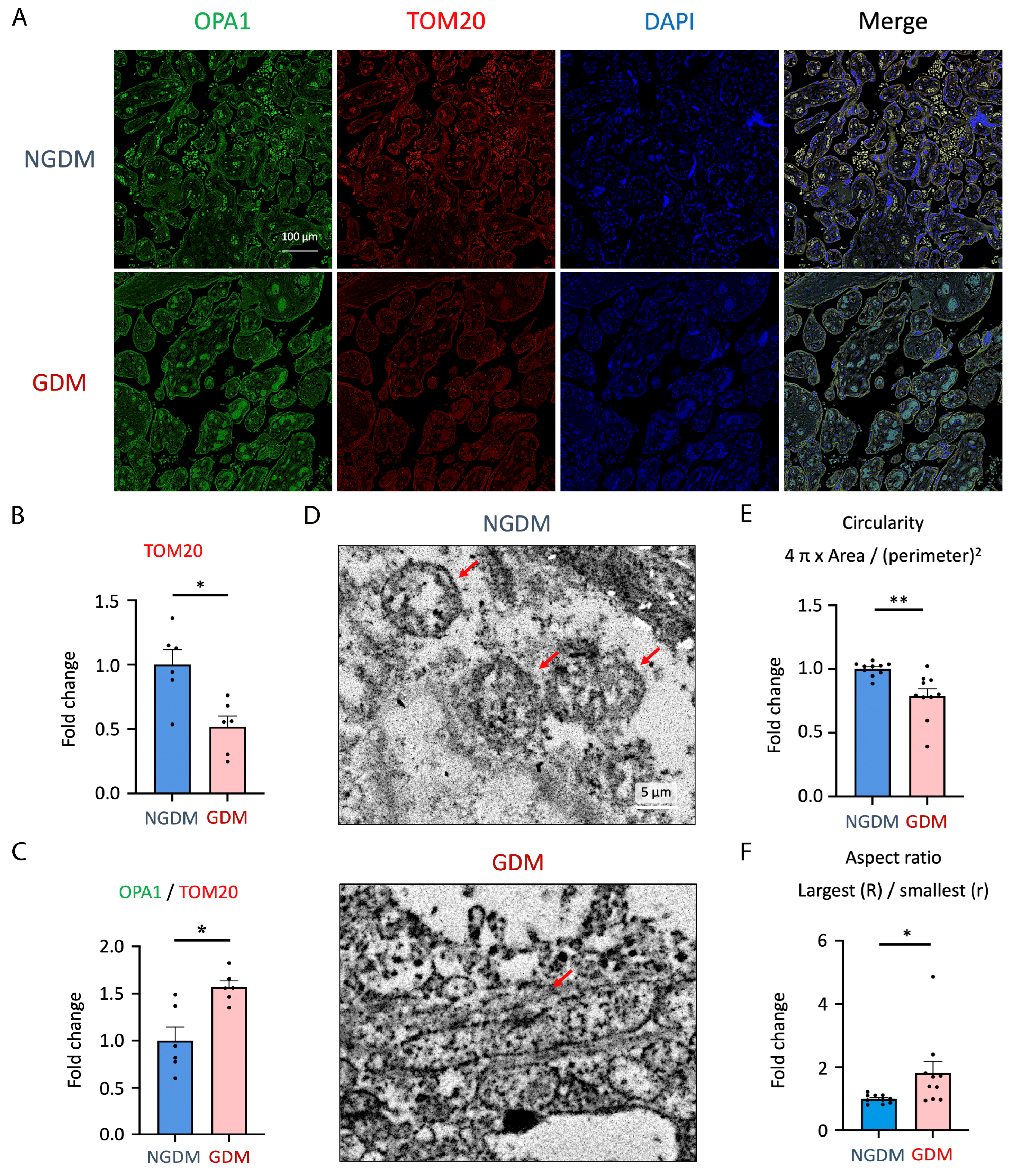
3.1. Modification of GDM Placenta with Abnormal Structure and Increased LOX-1 Expression
3.2. Elevated Mitochondrial Fusion and Decreased Mitochondria Mass in GDM Placenta
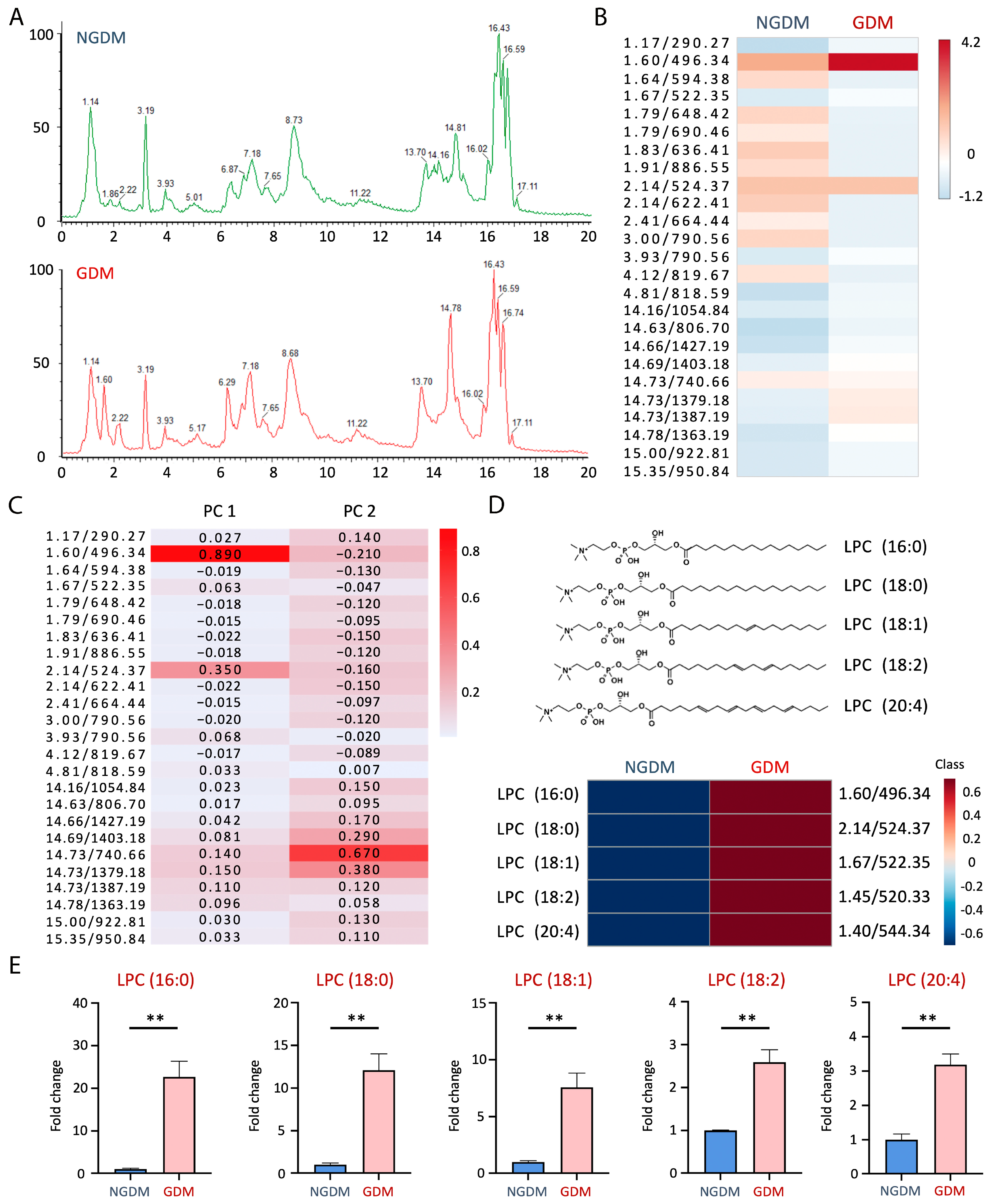
3.3. Lysophosphatidylcholine Increased in GDM Cord Blood
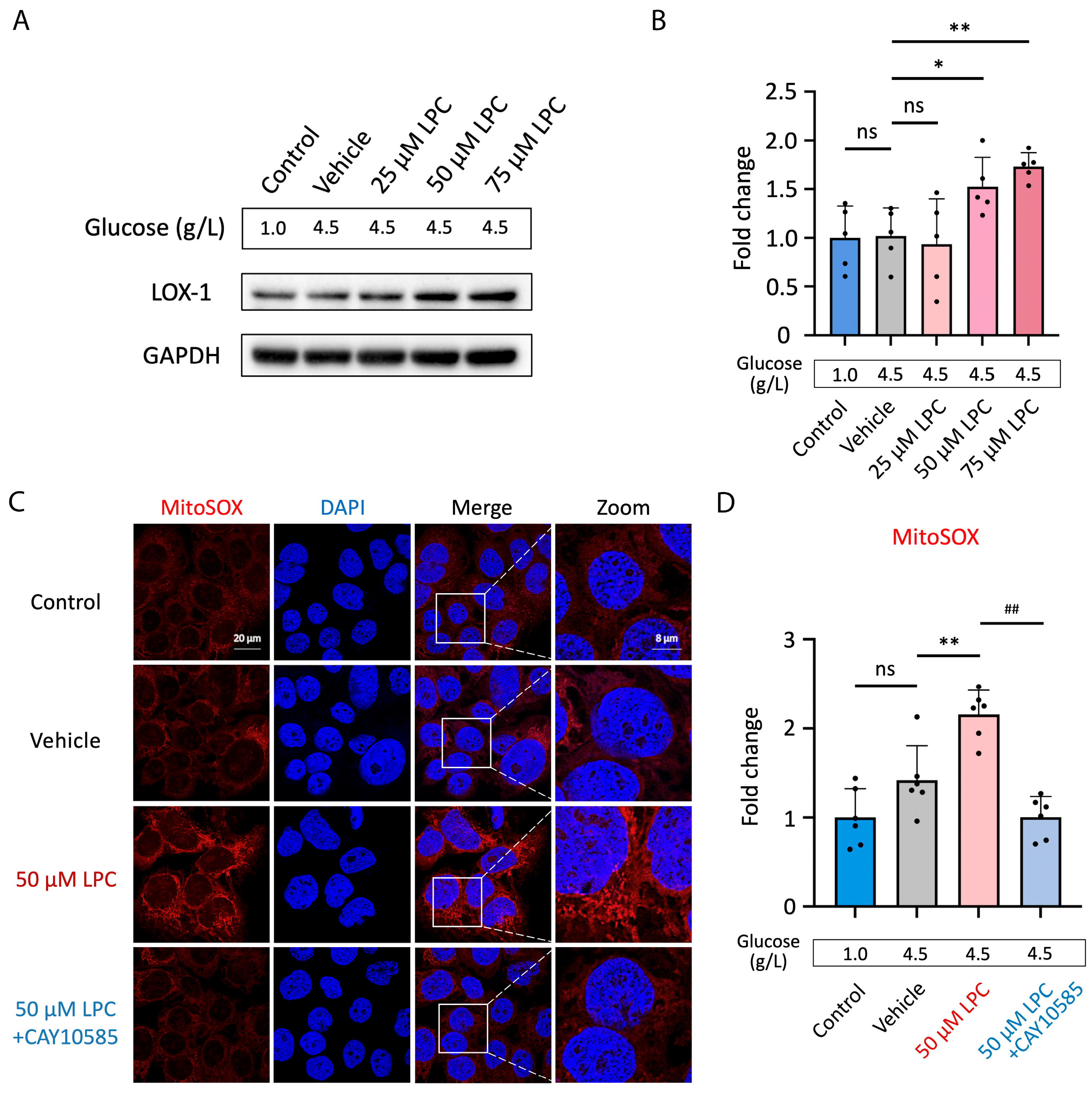
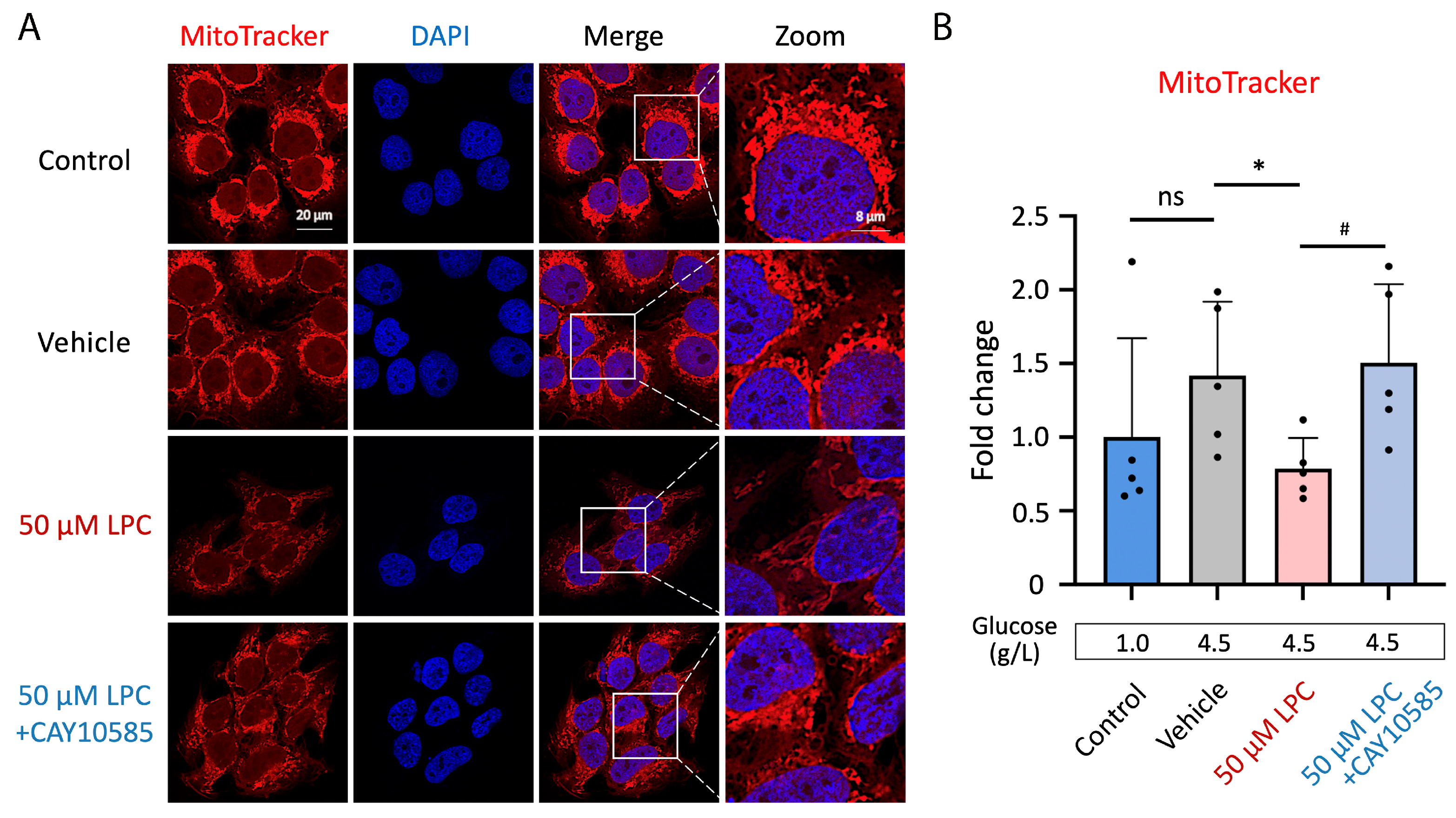
3.4. LPC Induces Excessive Mitochondrial ROS Production
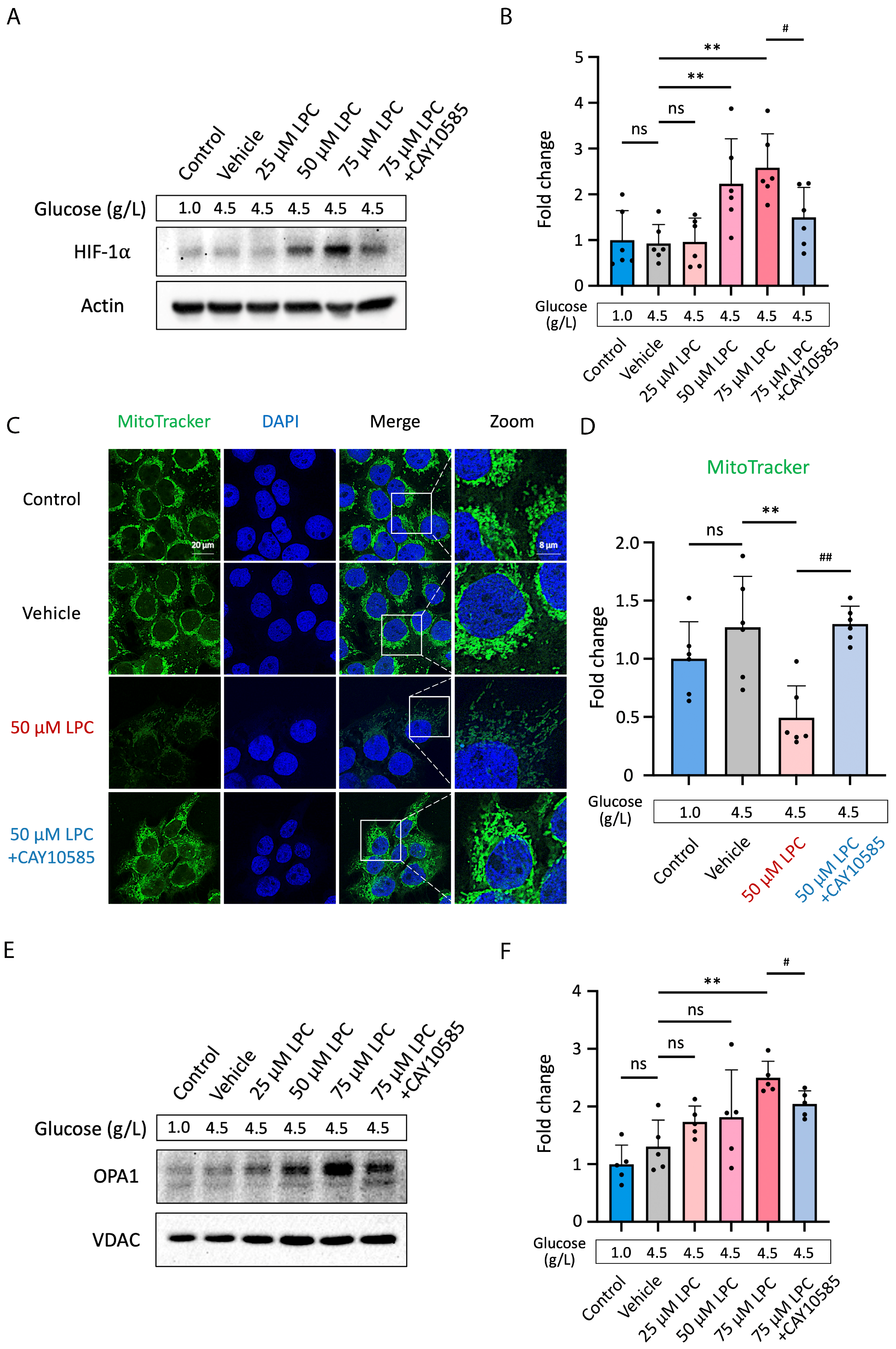
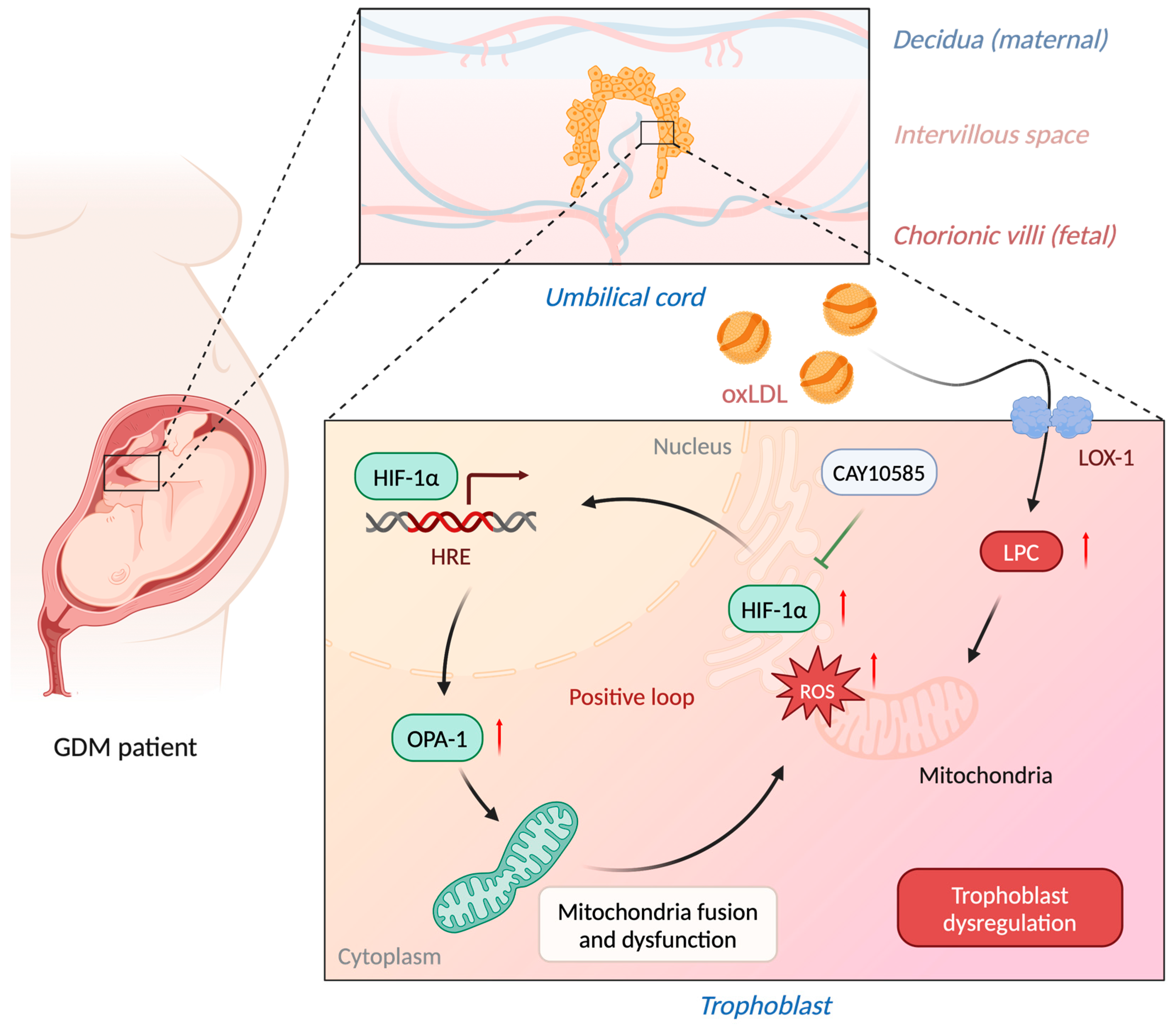
3.5. LPC Impairs Mitochondrial Homeostasis through HIF-1α Pathway
3.6. LPC Impaired Mitochondria Electron Transport Chain of GDM Trophoblast
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ACN | acetonitrile |
| AKT | protein kinase B |
| ApoE | apolipoprotein E |
| apoE−/− | apolipoprotein E-deficient |
| ASCVD | atherosclerotic cardiovascular disease |
| BCRC | Bioresource Collection and Research Center |
| cPLA2 | cytosolic phospholipase A2 |
| CVD | cardiovascular disease |
| CRP | C-reactive protein |
| CSH | charged surface hybrid |
| DAB | 3,3′-diaminobenzidine |
| DAPI | 4′,6-diamidino-2-phenylindole |
| DG | diacylglycerol |
| EDTA | ethylenediaminetetraacetic acid |
| ER | endoplasmic reticulum |
| ESI | electrospray ionization interface |
| EVT | extravillous trophoblast |
| FGF-2 | fibroblast-growth factor-2 |
| GDM | gestational diabetes mellitus |
| H&E | Hematoxylin and eosin |
| HDL-C | high-density lipoprotein cholesterol |
| HIF-1α | hypoxia-inducible factor-1α |
| HRP | horseradish peroxidase |
| IRB | institutional review board |
| IADPSG | International Association of Diabetes and Pregnancy Study Groups |
| ICAM-1 | intercellular adhesion molecule-1 |
| IgG | immunoglobulin G (IgG) |
| IPA | isopropanol |
| iPLA2 | Ca2+-independent phospholipase A2 |
| JEG3 | choriocarcinoma cells |
| KMUH | Kaohsiung Medical University Hospital |
| L5-LDL | the most electronegative subfraction of low-density lipoprotein |
| LDL(−) | electronegative low-density lipoprotein |
| LDL-C | low-density lipoprotein-cholesterol |
| LDLR | low-density lipoprotein receptor |
| LC/MS | liquid chromatography/mass spectrometry |
| LC/MSE | liquid chromatography/data-independent collection mode mass spectrometry |
| LOX-1 | lectin-like oxidized low-density lipoprotein receptor 1 |
| LPA | lysophosphatidic acid |
| LPC | lysophosphatidylcholine |
| Lp-PLA2 | lipoprotein-associated phospholipase A2 |
| MEM | minimum essential media |
| MFSD2A | major facilitator superfamily domain-containing protein 2 |
| MS | mass spectrometry |
| MSE | data-independent collection mode mass spectrometry |
| NGDM | non-gestational diabetes |
| NO | nitric oxide |
| NP-40 | 4-Nonylphenyl-polyethylene glycol, Nonidet P™-40 |
| OGTT | oral glucose tolerance test |
| OPA1 | mechanochemical GTPase optic atrophy 1 |
| OsO4 | osmium tetroxide |
| oxLDL | oxidized low-density lipoprotein |
| PAS | placenta accreta spectrum |
| PBS | phosphate-buffered saline |
| PI3K | phosphatidylinositol 3-kinase |
| PVDF | polyvinylidene difluoride membrane |
| RIPA | radioimmunoprecipitation assay buffer |
| ROS | reactive oxygen species |
| QTOF | quadrupole time of flight mass spectrometry |
| SDS | sodium dodecyl sulfate |
| SEM | standard error of the mean |
| SM | Sphingomyelin |
| T2D | type 2 diabetes mellitus |
| TC | total cholesterol |
| TEM | transmission electron microscopy (TEM) |
| TEMED | N, N, N′, N′-Tetramethyl ethylenediamine |
| TG | triglyceride |
| TOM20 | mitochondrial import receptor subunit |
| Tris-HCl | TRIS Hydrochloride |
| UPLC | ultra-performance liquid chromatography |
| VCAM | vascular cell adhesion molecule-1 |
| VDAC | voltage-dependent anion channel |
References
- Quintanilla Rodriguez, B.S.; Mahdy, H. Gestational Diabetes. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2022. [Google Scholar]
- International Association of Diabetes and Pregnancy Study Groups Consensus Panel; Metzger, B.E.; Gabbe, S.G.; Persson, B.; Buchanan, T.A.; Catalano, P.A.; Damm, P.; Dyer, A.R.; Leiva, A.; Hod, M.; et al. International association of diabetes and pregnancy study groups recommendations on the diagnosis and classification of hyperglycemia in pregnancy. Diabetes Care 2010, 33, 676–682. [Google Scholar] [CrossRef]
- Moon, J.H.; Jang, H.C. Gestational Diabetes Mellitus: Diagnostic Approaches and Maternal-Offspring Complications. Diabetes Metab. J. 2022, 46, 3–14. [Google Scholar] [CrossRef]
- HAPO Study Cooperative Research Group; Metzger, B.E.; Lowe, L.P.; Dyer, A.R.; Trimble, E.R.; Chaovarindr, U.; Coustan, D.R.; Hadden, D.R.; McCance, D.R.; Hod, M.; et al. Hyperglycemia and adverse pregnancy outcomes. N. Engl. J. Med. 2008, 358, 1991–2002. [Google Scholar] [PubMed]
- Fadl, H.E.; Ostlund, I.K.; Magnuson, A.F.; Hanson, U.S. Maternal and neonatal outcomes and time trends of gestational diabetes mellitus in Sweden from 1991 to 2003. Diabet. Med. 2010, 27, 436–441. [Google Scholar] [CrossRef]
- Kc, K.; Shakya, S.; Zhang, H. Gestational diabetes mellitus and macrosomia: A literature review. Ann. Nutr. Metab. 2015, 66 (Suppl. S2), 14–20. [Google Scholar] [CrossRef]
- DeSisto, C.L.; Kim, S.Y.; Sharma, A.J. Prevalence estimates of gestational diabetes mellitus in the United States, Pregnancy Risk Assessment Monitoring System (PRAMS), 2007–2010. Prev. Chronic Dis. 2014, 11, E104. [Google Scholar] [CrossRef]
- Lavery, J.A.; Friedman, A.M.; Keyes, K.M.; Wright, J.D.; Ananth, C.V. Gestational diabetes in the United States: Temporal changes in prevalence rates between 1979 and 2010. BJOG 2017, 124, 804–813. [Google Scholar] [CrossRef] [PubMed]
- Su, F.L.; Lu, M.C.; Yu, S.C.; Yang, C.P.; Yang, C.C.; Tseng, S.T.; Yan, Y.H. Increasing trend in the prevalence of gestational diabetes mellitus in Taiwan. J. Diabetes Investig. 2021, 12, 2080–2088. [Google Scholar] [CrossRef]
- Zhou, T.; Du, S.; Sun, D.; Li, X.; Heianza, Y.; Hu, G.; Sun, L.; Pei, X.; Shang, X.; Qi, L. Prevalence and Trends in Gestational Diabetes Mellitus Among Women in the United States, 2006–2017: A Population-Based Study. Front. Endocrinol. 2022, 13, 868094. [Google Scholar] [CrossRef] [PubMed]
- Nguyen-Ngo, C.; Jayabalan, N.; Salomon, C.; Lappas, M. Molecular pathways disrupted by gestational diabetes mellitus. J. Mol. Endocrinol. 2019, 63, R51–R72. [Google Scholar] [CrossRef]
- Meakin, C.; Barrett, E.S.; Aleksunes, L.M. Extravillous trophoblast migration and invasion: Impact of environmental chemicals and pharmaceuticals. Reprod. Toxicol. 2022, 107, 60–68. [Google Scholar] [CrossRef]
- Zhao, H.; Wong, R.J.; Stevenson, D.K. The Impact of Hypoxia in Early Pregnancy on Placental Cells. Int. J. Mol. Sci. 2021, 22, 9675. [Google Scholar] [CrossRef]
- Ciampa, E.J.; Flahardy, P.; Srinivasan, H.; Jacobs, C.; Tsai, L.; Karumanchi, S.A.; Parikh, S.M. Hypoxia-inducible factor 1 signaling drives placental aging and can provoke preterm labor. eLife 2023, 12, RP85597. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Huang, C.; Wang, P.; Gao, J.; Liu, X.; Li, Y.; Yan, S.; Shi, Y. HIF-1alpha affects trophoblastic apoptosis involved in the onset of preeclampsia by regulating FOXO3a under hypoxic conditions. Mol. Med. Rep. 2020, 21, 2484–2492. [Google Scholar] [PubMed]
- Iriyama, T.; Wang, W.; Parchim, N.F.; Song, A.; Blackwell, S.C.; Sibai, B.M.; Kellems, R.E.; Xia, Y. Hypoxia-independent upregulation of placental hypoxia inducible factor-1alpha gene expression contributes to the pathogenesis of preeclampsia. Hypertension 2015, 65, 1307–1315. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Friera, L.; Fuster, V.; Lopez-Melgar, B.; Oliva, B.; Garcia-Ruiz, J.M.; Mendiguren, J.; Bueno, H.; Pocock, S.; Ibanez, B.; Fernandez-Ortiz, A.; et al. Normal LDL-Cholesterol Levels Are Associated With Subclinical Atherosclerosis in the Absence of Risk Factors. J. Am. Coll. Cardiol. 2017, 70, 2979–2991. [Google Scholar] [CrossRef]
- Ghaneei, A.; Yassini, S.; Ghanei, M.E.; Shojaoddiny-Ardekani, A. Increased serum oxidized low-density lipoprotein levels in pregnancies complicated by gestational diabetes mellitus. Iran. J. Reprod. Med. 2015, 13, 421–424. [Google Scholar]
- Stancel, N.; Chen, C.C.; Ke, L.Y.; Chu, C.S.; Lu, J.; Sawamura, T.; Chen, C.H. Interplay between CRP, Atherogenic LDL, and LOX-1 and Its Potential Role in the Pathogenesis of Atherosclerosis. Clin. Chem. 2016, 62, 320–327. [Google Scholar] [CrossRef]
- Wang, Y.C.; Lee, A.S.; Lu, L.S.; Ke, L.Y.; Chen, W.Y.; Dong, J.W.; Lu, J.; Chen, Z.; Chu, C.S.; Chan, H.C.; et al. Human electronegative LDL induces mitochondrial dysfunction and premature senescence of vascular cells in vivo. Aging Cell 2018, 17, e12792. [Google Scholar] [CrossRef]
- Hofmann, A.; Brunssen, C.; Morawietz, H. Contribution of lectin-like oxidized low-density lipoprotein receptor-1 and LOX-1 modulating compounds to vascular diseases. Vascul Pharmacol. 2017, 107, 1–11. [Google Scholar] [CrossRef]
- Kobayashi, H.; Matsubara, S.; Yoshimoto, C.; Shigetomi, H.; Imanaka, S. Current understanding of the pathogenesis of placenta accreta spectrum disorder with focus on mitochondrial function. J. Obstet. Gynaecol. Res. 2024, 50, 929–940. [Google Scholar] [CrossRef] [PubMed]
- Andreescu, M. Correlation Between Maternal-Fetus Interface and Placenta-Mediated Complications. Cureus 2024, 16, e62457. [Google Scholar] [CrossRef] [PubMed]
- Burton, G.J.; Fowden, A.L. The placenta: A multifaceted, transient organ. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2015, 370, 20140066. [Google Scholar] [CrossRef]
- Gundacker, C.; Ellinger, I. The unique applicability of the human placenta to the Adverse Outcome Pathway (AOP) concept: The placenta provides fundamental insights into human organ functions at multiple levels of biological organization. Reprod. Toxicol. 2020, 96, 273–281. [Google Scholar] [CrossRef]
- Wang, Y.; Zhao, S. Vascular Biology of the Placenta; Morgan & Claypool Life Sciences: San Rafael, CA, USA, 2010. [Google Scholar]
- Valent, A.M.; Choi, H.; Kolahi, K.S.; Thornburg, K.L. Hyperglycemia and gestational diabetes suppress placental glycolysis and mitochondrial function and alter lipid processing. FASEB J. 2021, 35, e21423. [Google Scholar] [CrossRef] [PubMed]
- Gauster, M.; Moser, G.; Wernitznig, S.; Kupper, N.; Huppertz, B. Early human trophoblast development: From morphology to function. Cell. Mol. Life Sci. 2022, 79, 345. [Google Scholar] [CrossRef]
- Carrasco-Wong, I.; Moller, A.; Giachini, F.R.; Lima, V.V.; Toledo, F.; Stojanova, J.; Sobrevia, L.; San Martin, S. Placental structure in gestational diabetes mellitus. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165535. [Google Scholar] [CrossRef]
- Zeljkovic, A.; Ardalic, D.; Vekic, J.; Antonic, T.; Vladimirov, S.; Rizzo, M.; Gojkovic, T.; Ivanisevic, J.; Mihajlovic, M.; Vujcic, S.; et al. Effects of Gestational Diabetes Mellitus on Cholesterol Metabolism in Women with High-Risk Pregnancies: Possible Implications for Neonatal Outcome. Metabolites 2022, 12, 959. [Google Scholar] [CrossRef]
- Bao, W.; Dar, S.; Zhu, Y.; Wu, J.; Rawal, S.; Li, S.; Weir, N.L.; Tsai, M.Y.; Zhang, C. Plasma concentrations of lipids during pregnancy and the risk of gestational diabetes mellitus: A longitudinal study. J. Diabetes 2018, 10, 487–495. [Google Scholar] [CrossRef] [PubMed]
- Pazhohan, A.; Rezaee Moradali, M.; Pazhohan, N.; Sadeghyanifar, A. Indicators of Obesity and Serum Levels of Triglycerides in the First Trimester of Pregnancy as the Predictors of Gestational Diabetes Mellitus among Urmia Women. Mod. Care J. 2018, 15, e67186. [Google Scholar] [CrossRef]
- Enthoven, L.F.; Shi, Y.; Fay, E.; Kim, A.; Moreni, S.; Mao, J.; Isoherranen, N.; Totah, R.A.; Hebert, M.F. Effects of Pregnancy on Plasma Sphingolipids Using a Metabolomic and Quantitative Analysis Approach. Metabolites 2023, 13, 1026. [Google Scholar] [CrossRef] [PubMed]
- Hou, G.; Gao, Y.; Poon, L.C.; Ren, Y.; Zeng, C.; Wen, B.; Syngelaki, A.; Lin, L.; Zi, J.; Su, F.; et al. Maternal plasma diacylglycerols and triacylglycerols in the prediction of gestational diabetes mellitus. BJOG 2023, 130, 247–256. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Buckley, J.P.; Bartell, T.R.; Hong, X.; Pearson, C.; Wang, X. Gestational Diabetes Mellitus, Postpartum Lipidomic Signatures, and Subsequent Risk of Type 2 Diabetes: A Lipidome-Wide Association Study. Diabetes Care 2023, 46, 1223–1230. [Google Scholar] [CrossRef]
- Wang, Y.; Wu, P.; Huang, Y.; Ye, Y.; Yang, X.; Sun, F.; Ye, Y.X.; Lai, Y.; Ouyang, J.; Wu, L.; et al. BMI and lipidomic biomarkers with risk of gestational diabetes in pregnant women. Obesity 2022, 30, 2044–2054. [Google Scholar] [CrossRef] [PubMed]
- Villalobos-Labra, R.; Liu, R.; Spaans, F.; Saez, T.; Semeria Maitret, T.; Quon, A.; Sawamura, T.; Cooke, C.M.; Davidge, S.T. Placenta-Derived Extracellular Vesicles From Preeclamptic Pregnancies Impair Vascular Endothelial Function via Lectin-like Oxidized LDL Receptor-1. Hypertension 2023, 80, 2226–2238. [Google Scholar] [CrossRef]
- Akhmedov, A.; Sawamura, T.; Chen, C.H.; Kraler, S.; Vdovenko, D.; Luscher, T.F. Lectin-like oxidized low-density lipoprotein receptor-1 (LOX-1): A crucial driver of atherosclerotic cardiovascular disease. Eur. Heart J. 2021, 42, 1797–1807. [Google Scholar] [CrossRef]
- Munno, M.; Mallia, A.; Greco, A.; Modafferi, G.; Banfi, C.; Eligini, S. Radical Oxygen Species, Oxidized Low-Density Lipoproteins, and Lectin-like Oxidized Low-Density Lipoprotein Receptor 1: A Vicious Circle in Atherosclerotic Process. Antioxidants 2024, 13, 583. [Google Scholar] [CrossRef]
- Truthe, S.; Klassert, T.E.; Schmelz, S.; Jonigk, D.; Blankenfeldt, W.; Slevogt, H. Role of Lectin-like Oxidized Low-Density Lipoprotein Receptor-1 in Inflammation and Pathogen-Associated Interactions. J. Innate Immun. 2024, 16, 105–132. [Google Scholar] [CrossRef]
- Li, H.S.; Zhou, Y.N.; Li, L.; Li, S.F.; Long, D.; Chen, X.L.; Zhang, J.B.; Feng, L.; Li, Y.P. HIF-1alpha protects against oxidative stress by directly targeting mitochondria. Redox Biol. 2019, 25, 101109. [Google Scholar] [CrossRef]
- Monteiro, L.B.; Davanzo, G.G.; de Aguiar, C.F.; Moraes-Vieira, P.M.M. Using flow cytometry for mitochondrial assays. MethodsX 2020, 7, 100938. [Google Scholar] [CrossRef]
- von der Malsburg, A.; Sapp, G.M.; Zuccaro, K.E.; von Appen, A.; Moss, F.R., 3rd; Kalia, R.; Bennett, J.A.; Abriata, L.A.; Dal Peraro, M.; van der Laan, M.; et al. Structural mechanism of mitochondrial membrane remodelling by human OPA1. Nature 2023, 620, 1101–1108. [Google Scholar] [CrossRef]
- Neikirk, K.; Marshall, A.G.; Kula, B.; Smith, N.; LeBlanc, S.; Hinton, A., Jr. MitoTracker: A useful tool in need of better alternatives. Eur. J. Cell Biol. 2023, 102, 151371. [Google Scholar] [CrossRef]
- Law, S.H.; Chan, M.L.; Marathe, G.K.; Parveen, F.; Chen, C.H.; Ke, L.Y. An Updated Review of Lysophosphatidylcholine Metabolism in Human Diseases. Int. J. Mol. Sci. 2019, 20, 1149. [Google Scholar] [CrossRef] [PubMed]
- Goncalves, I.; Edsfeldt, A.; Ko, N.Y.; Grufman, H.; Berg, K.; Bjorkbacka, H.; Nitulescu, M.; Persson, A.; Nilsson, M.; Prehn, C.; et al. Evidence supporting a key role of Lp-PLA2-generated lysophosphatidylcholine in human atherosclerotic plaque inflammation. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 1505–1512. [Google Scholar] [CrossRef]
- Mahmut, A.; Boulanger, M.C.; El Husseini, D.; Fournier, D.; Bouchareb, R.; Despres, J.P.; Pibarot, P.; Bosse, Y.; Mathieu, P. Elevated expression of lipoprotein-associated phospholipase A2 in calcific aortic valve disease: Implications for valve mineralization. J. Am. Coll. Cardiol. 2014, 63, 460–469. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Zhang, W.; Liu, J.; Yang, H.; Hu, Y.; Zhang, M.; Bai, T.; Chang, F. Lysophosphatidylcholine promotes intercellular adhesion molecule-1 and vascular cell adhesion molecule-1 expression in human umbilical vein endothelial cells via an orphan G protein receptor 2-mediated signaling pathway. Bioengineered 2021, 12, 4520–4535. [Google Scholar] [CrossRef] [PubMed]
- Chan, H.C.; Chan, H.C.; Liang, C.J.; Lee, H.C.; Su, H.; Lee, A.S.; Shiea, J.; Tsai, W.C.; Ou, T.T.; Wu, C.C.; et al. Role of Low-Density Lipoprotein in Early Vascular Aging Associated With Systemic Lupus Erythematosus. Arthritis Rheumatol. 2020, 72, 972–984. [Google Scholar] [CrossRef] [PubMed]
- Chan, H.C.; Ke, L.Y.; Chu, C.S.; Lee, A.S.; Shen, M.Y.; Cruz, M.A.; Hsu, J.F.; Cheng, K.H.; Chan, H.C.; Lu, J.; et al. Highly electronegative LDL from patients with ST-elevation myocardial infarction triggers platelet activation and aggregation. Blood 2013, 122, 3632–3641. [Google Scholar] [CrossRef]
- Uhlen, M.; Fagerberg, L.; Hallstrom, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, A.; Kampf, C.; Sjostedt, E.; Asplund, A.; et al. Proteomics. Tissue-based map of the human proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef]
- Sawamura, T.; Kume, N.; Aoyama, T.; Moriwaki, H.; Hoshikawa, H.; Aiba, Y.; Tanaka, T.; Miwa, S.; Katsura, Y.; Kita, T.; et al. An endothelial receptor for oxidized low-density lipoprotein. Nature 1997, 386, 73–77. [Google Scholar] [CrossRef]
- Chen, C.H.; Jiang, T.; Yang, J.H.; Jiang, W.; Lu, J.; Marathe, G.K.; Pownall, H.J.; Ballantyne, C.M.; McIntyre, T.M.; Henry, P.D.; et al. Low-density lipoprotein in hypercholesterolemic human plasma induces vascular endothelial cell apoptosis by inhibiting fibroblast growth factor 2 transcription. Circulation 2003, 107, 2102–2108. [Google Scholar] [CrossRef] [PubMed]
- Zeya, B.; Chandra, N.C. LOX-1: Its cytotopographical variance and disease stress. J. Biochem. Mol. Toxicol. 2019, 33, e22375. [Google Scholar] [CrossRef] [PubMed]
- Murdocca, M.; De Masi, C.; Pucci, S.; Mango, R.; Novelli, G.; Di Natale, C.; Sangiuolo, F. LOX-1 and cancer: An indissoluble liaison. Cancer Gene Ther. 2021, 28, 1088–1098. [Google Scholar] [CrossRef] [PubMed]
- Ke, L.Y.; Chan, H.C.; Chan, H.C.; Kalu, F.C.U.; Lee, H.C.; Lin, I.L.; Jhuo, S.J.; Lai, W.T.; Tsao, C.R.; Sawamura, T.; et al. Electronegative Low-Density Lipoprotein L5 Induces Adipose Tissue Inflammation Associated With Metabolic Syndrome. J. Clin. Endocrinol. Metab. 2017, 102, 4615–4625. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Du, G.; Tong, L.; Guo, X.; Wei, Y. Overexpression of LOX-1 in hepatocytes protects vascular smooth muscle cells from phenotype transformation and wire injury induced carotid neoatherosclerosis through ALOX15. Biochim. Biophys. Acta Mol. Basis Dis. 2023, 1869, 166805. [Google Scholar] [CrossRef] [PubMed]
- Zuniga, F.A.; Ormazabal, V.; Gutierrez, N.; Aguilera, V.; Radojkovic, C.; Veas, C.; Escudero, C.; Lamperti, L.; Aguayo, C. Role of lectin-like oxidized low density lipoprotein-1 in fetoplacental vascular dysfunction in preeclampsia. Biomed. Res. Int. 2014, 2014, 353616. [Google Scholar] [CrossRef] [PubMed]
- Satoh, H.; Kiyota, E.; Terasaki, Y.; Sawamura, T.; Takagi, K.; Mizuta, H.; Takeya, M. Expression and localization of lectin-like oxidized low-density lipoprotein receptor-1 (LOX-1) in murine and human placentas. J. Histochem. Cytochem. 2008, 56, 773–784. [Google Scholar] [CrossRef]
- Ethier-Chiasson, M.; Forest, J.C.; Giguere, Y.; Masse, A.; Marseille-Tremblay, C.; Levy, E.; Lafond, J. Modulation of placental protein expression of OLR1: Implication in pregnancy-related disorders or pathologies. Reproduction 2008, 136, 491–502. [Google Scholar] [CrossRef] [PubMed]
- Sheikhi, A.; Razdar, S.; Rahmanpour, H.; Mousavinasab, N.; Ganji, H.B.; Jafarzadeh, A. Higher expression of HSP70 and LOX-1 in the placental tissues of pre-eclampsia pregnancies. Clin. Exp. Hypertens. 2015, 37, 128–135. [Google Scholar] [CrossRef]
- Leslie, C.C. Cytosolic phospholipase A(2): Physiological function and role in disease. J. Lipid Res. 2015, 56, 1386–1402. [Google Scholar] [CrossRef]
- Dennis, E.A.; Cao, J.; Hsu, Y.H.; Magrioti, V.; Kokotos, G. Phospholipase A2 enzymes: Physical structure, biological function, disease implication, chemical inhibition, and therapeutic intervention. Chem. Rev. 2011, 111, 6130–6185. [Google Scholar] [CrossRef]
- Schanstra, J.P.; Luong, T.T.; Makridakis, M.; Van Linthout, S.; Lygirou, V.; Latosinska, A.; Alesutan, I.; Boehme, B.; Schelski, N.; Von Lewinski, D.; et al. Systems biology identifies cytosolic PLA2 as a target in vascular calcification treatment. JCI Insight 2019, 4, e125638. [Google Scholar] [CrossRef] [PubMed]
- Osada-Oka, M.; Takahashi, M.; Akiba, S.; Sato, T. Involvement of Ca2+-independent phospholipase A2 in the translocation of hypoxia-inducible factor-1alpha to the nucleus under hypoxic conditions. Eur. J. Pharmacol. 2006, 549, 58–62. [Google Scholar] [CrossRef] [PubMed]
- Zheng, L.; Li, M.; Li, H. High Glucose Promotes and Aggravates the Senescence and Dysfunction of Vascular Endothelial Cells in Women with Hyperglycemia in Pregnancy. Biomolecules 2024, 14, 329. [Google Scholar] [CrossRef] [PubMed]
- Zhu, A.; Qi, S.; Li, W.; Chen, D.; Zheng, X.; Xu, J.; Feng, Y. Hyperglycemia-induced endothelial exosomes trigger trophoblast dysregulation and abnormal placentation through PUM2-mediated repression of SOX2. Hum. Exp. Toxicol. 2023, 42, 9603271221149656. [Google Scholar] [CrossRef] [PubMed]
- Sweeting, A.; Hannah, W.; Backman, H.; Catalano, P.; Feghali, M.; Herman, W.H.; Hivert, M.F.; Immanuel, J.; Meek, C.; Oppermann, M.L.; et al. Epidemiology and management of gestational diabetes. Lancet 2024, 404, 175–192. [Google Scholar] [CrossRef] [PubMed]
- Diaz, P.; Powell, T.L.; Jansson, T. The role of placental nutrient sensing in maternal-fetal resource allocation. Biol. Reprod. 2014, 91, 82. [Google Scholar] [CrossRef] [PubMed]
- Bowman, C.E.; Arany, Z.; Wolfgang, M.J. Regulation of maternal-fetal metabolic communication. Cell. Mol. Life Sci. 2021, 78, 1455–1486. [Google Scholar] [CrossRef]
- Estruch, M.; Sanchez-Quesada, J.L.; Ordonez Llanos, J.; Benitez, S. Electronegative LDL: A circulating modified LDL with a role in inflammation. Mediat. Inflamm. 2013, 2013, 181324. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| NGDM (n = 6) | GDM (n = 6) | p-Value | |
|---|---|---|---|
| Age, y (mean ± SD) | 34.5 ± 2.88 | 33.5 ± 5.13 | 0.7316 |
| Fasting glucose | 83.3 ± 3.5 | 96.3 ± 6.7 | ** 0.0022 |
| 1-h OGTT | 124.0 ± 16.1 | 160.0 ± 39.4 | 0.1797 |
| 2-h OGTT | 120.0 ± 15.5 | 141.0 ± 32.4 | 0.4567 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hung, S.-C.; Chan, T.-F.; Chan, H.-C.; Wu, C.-Y.; Chan, M.-L.; Jhuang, J.-Y.; Tan, J.-Q.; Mei, J.-B.; Law, S.-H.; Ponnusamy, V.K.; et al. Lysophosphatidylcholine Impairs the Mitochondria Homeostasis Leading to Trophoblast Dysfunction in Gestational Diabetes Mellitus. Antioxidants 2024, 13, 1007. https://doi.org/10.3390/antiox13081007
Hung S-C, Chan T-F, Chan H-C, Wu C-Y, Chan M-L, Jhuang J-Y, Tan J-Q, Mei J-B, Law S-H, Ponnusamy VK, et al. Lysophosphatidylcholine Impairs the Mitochondria Homeostasis Leading to Trophoblast Dysfunction in Gestational Diabetes Mellitus. Antioxidants. 2024; 13(8):1007. https://doi.org/10.3390/antiox13081007
Chicago/Turabian StyleHung, Shao-Chi, Te-Fu Chan, Hsiu-Chuan Chan, Chia-Ying Wu, Mei-Lin Chan, Jie-Yang Jhuang, Ji-Qin Tan, Jia-Bin Mei, Shi-Hui Law, Vinoth Kumar Ponnusamy, and et al. 2024. "Lysophosphatidylcholine Impairs the Mitochondria Homeostasis Leading to Trophoblast Dysfunction in Gestational Diabetes Mellitus" Antioxidants 13, no. 8: 1007. https://doi.org/10.3390/antiox13081007