

Cystathionine Gamma-Lyase Regulates TNF-α-Mediated Injury Response in Human Colonic Epithelial Cells and Colonoids

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Cell Culture
2.3. Organoid Culture
2.4. Western Blot Analysis
2.5. Small-Interfering RNA (siRNA)-Mediated CSE Silencing
2.6. NF-ĸB Translocation Assay
2.7. Separation of Cytoplasmic and Nuclear Extracts
2.8. Measuring H2S Production in Live Cells
2.9. Assessing Transepithelial Electrical Resistance (TEER)
2.10. Wound Scratch Assay
2.11. Data and Statistical Analysis
3. Results
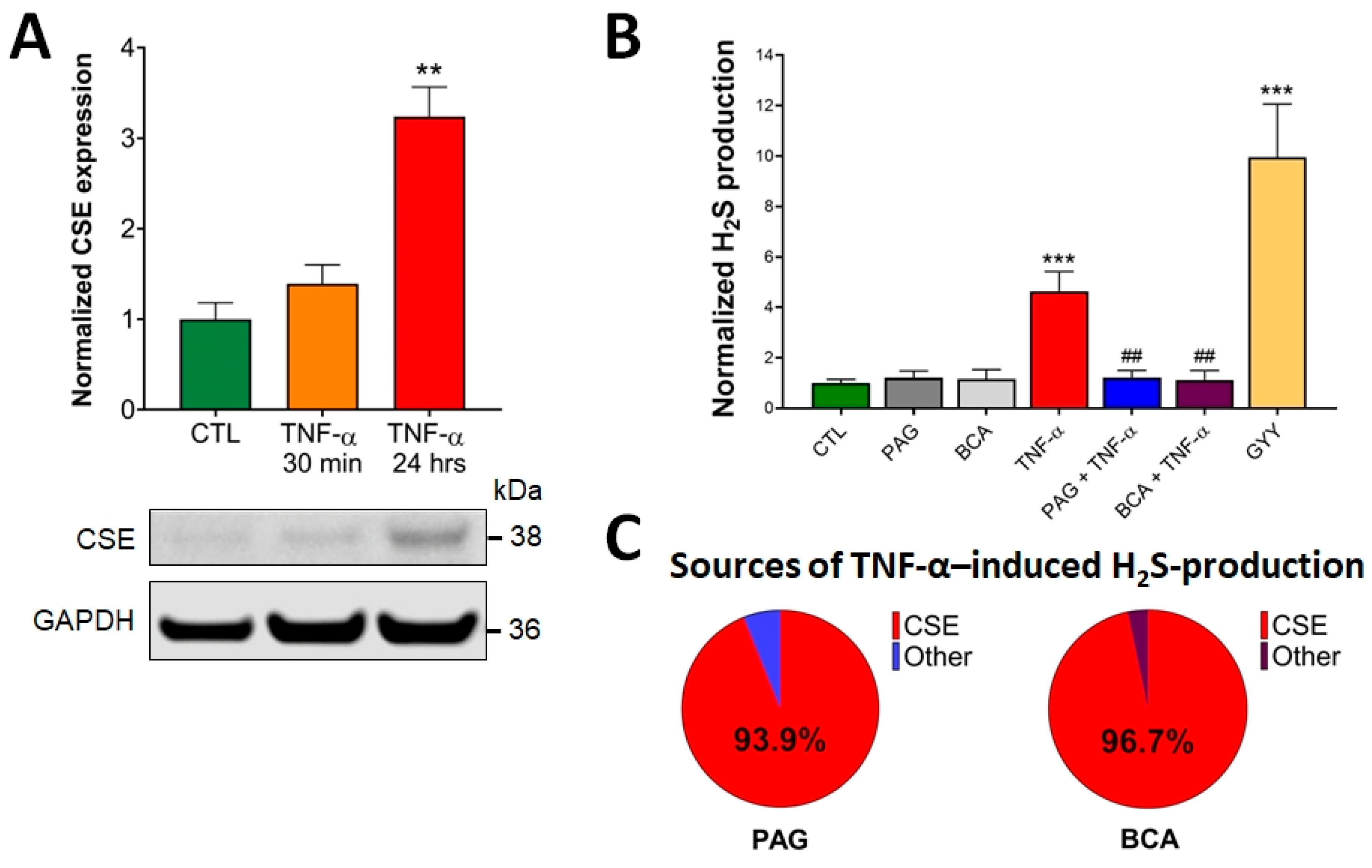
3.1. CSE Expression Is Elevated by TNF-α Stimulus in Colonocytes
3.2. CSE-Derived H2S Production Is Increased by TNF-α
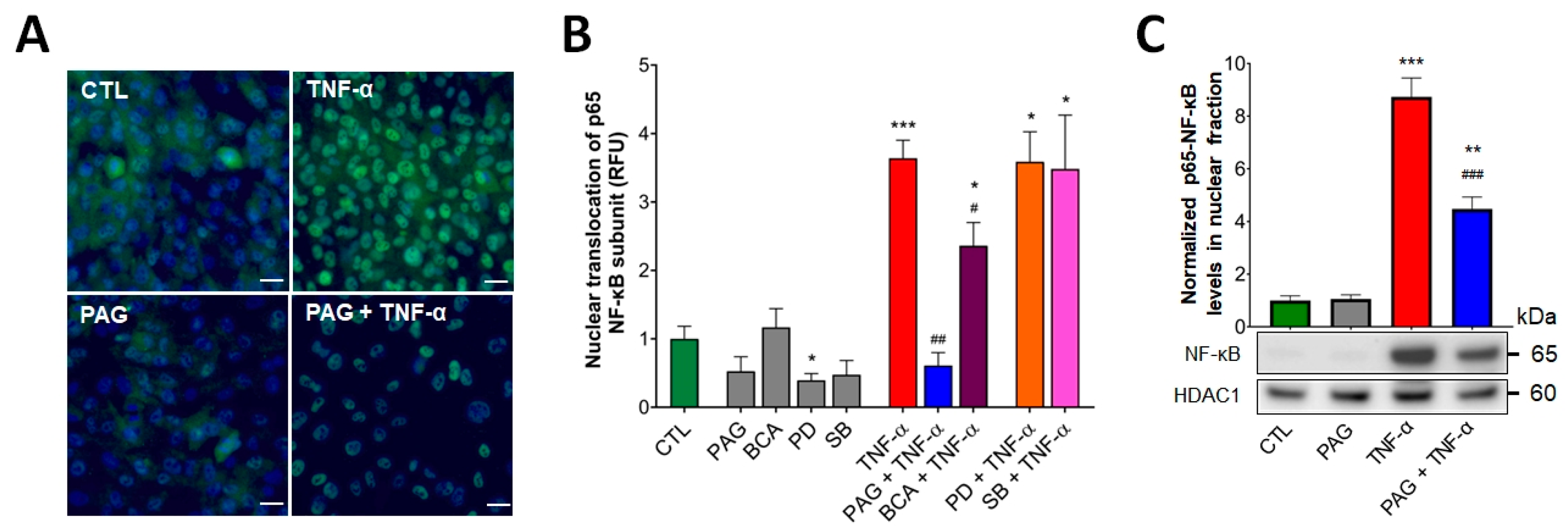
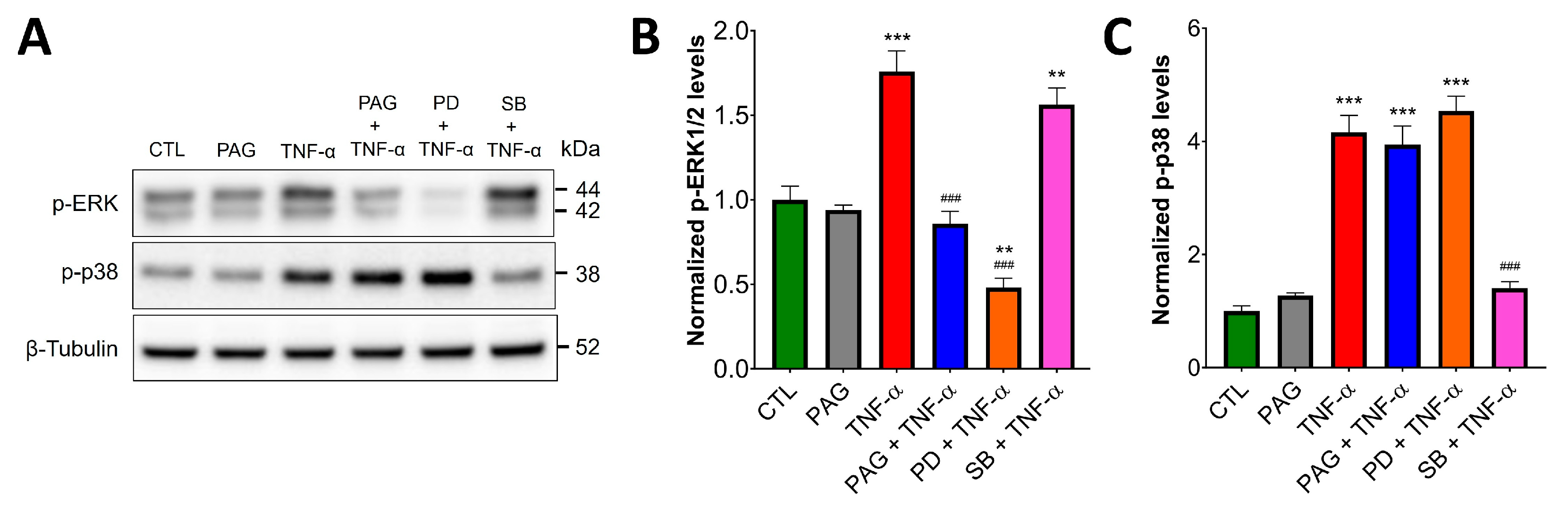
3.3. CSE/H2S Axis Regulates TNF-α-Induced MAPK and NF-ĸB Signaling
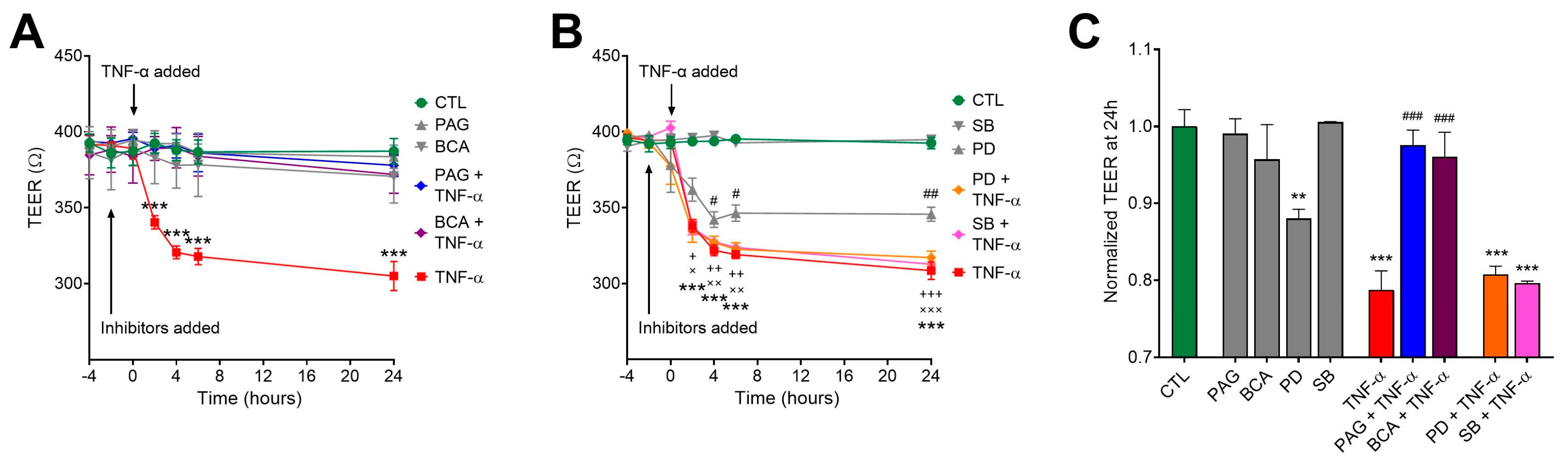
3.4. CSE Inhibition Blocks TNF-α-Mediated Transepithelial Permeability Increase Independently of MAPK Activity
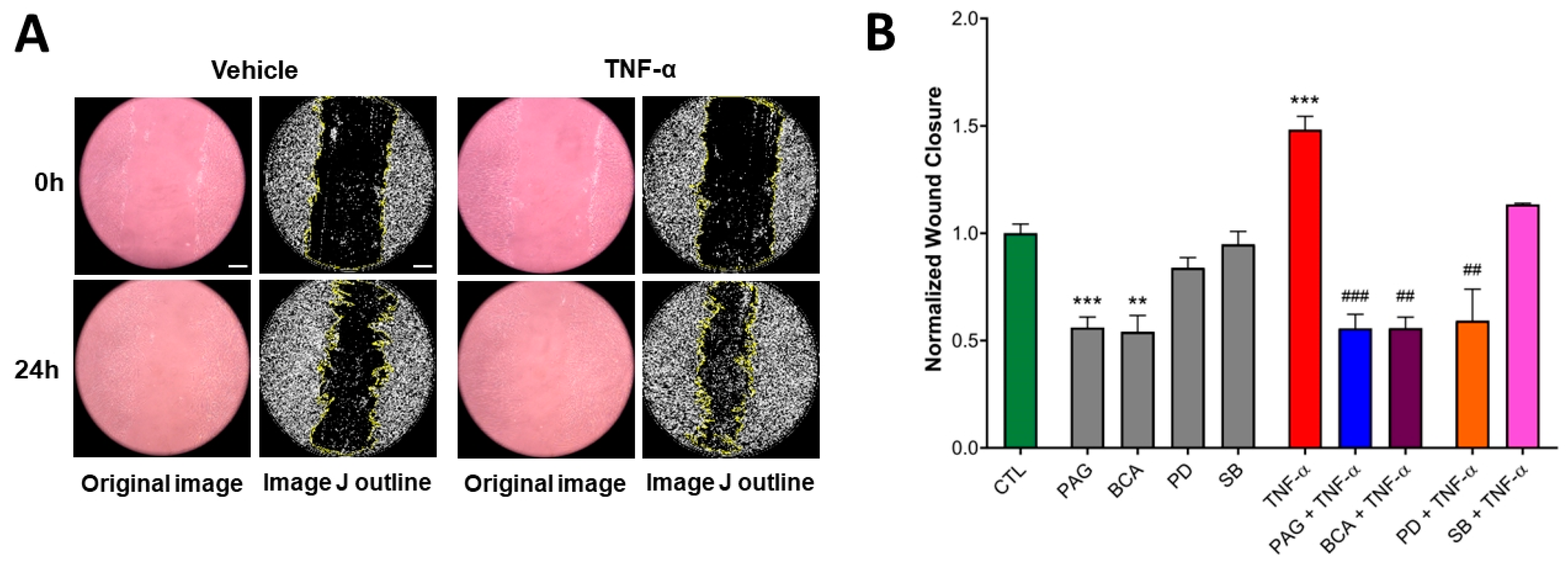
3.5. CSE Activity Stimulates TNF-α-Mediated Wound Healing
3.6. Colonoid Cultures Confirm CSE Regulation of TNF-α Signaling Identified in HCECs
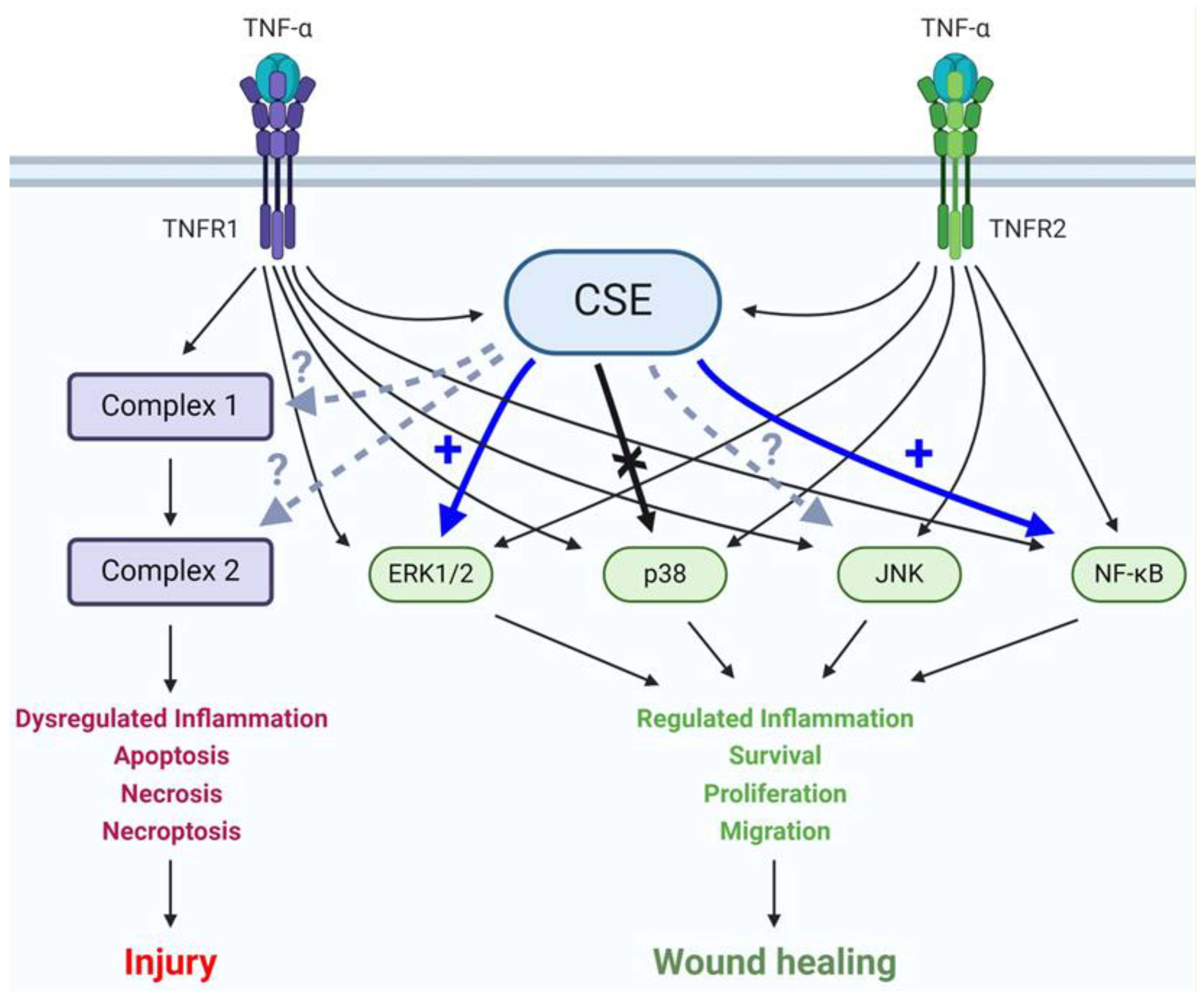
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Iizuka, M.; Konno, S. Wound healing of intestinal epithelial cells. World J. Gastroenterol. 2011, 17, 2161–2171. [Google Scholar] [CrossRef] [PubMed]
- Sommer, K.; Wiendl, M.; Muller, T.M.; Heidbreder, K.; Voskens, C.; Neurath, M.F.; Zundler, S. Intestinal Mucosal Wound Healing and Barrier Integrity in IBD-Crosstalk and Trafficking of Cellular Players. Front. Med. 2021, 8, 643973. [Google Scholar] [CrossRef] [PubMed]
- Vereecke, L.; Beyaert, R.; van Loo, G. Enterocyte death and intestinal barrier maintenance in homeostasis and disease. Trends Mol. Med. 2011, 17, 584–593. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, R.; Koide, T.; Tokoro, C.; Yamamoto, T.; Godai, T.; Morohashi, T.; Fujita, Y.; Takahashi, D.; Kawana, I.; Suzuki, S.; et al. Quantitive cytokine mRNA expression profiles in the colonic mucosa of patients with steroid naive ulcerative colitis during active and quiescent disease. Inflamm. Bowel Dis. 2009, 15, 328–334. [Google Scholar] [CrossRef]
- Ordás, I.; Eckmann, L.; Talamini, M.; Baumgart, D.C.; Sandborn, W.J. Ulcerative colitis. Lancet 2012, 380, 1606–1619. [Google Scholar] [CrossRef] [PubMed]
- Porter, R.J.; Kalla, R.; Ho, G.T. Ulcerative colitis: Recent advances in the understanding of disease pathogenesis. F1000Research 2020, 9, 294. [Google Scholar] [CrossRef] [PubMed]
- Singh, U.P.; Singh, N.P.; Murphy, E.A.; Price, R.L.; Fayad, R.; Nagarkatti, M.; Nagarkatti, P.S. Chemokine and cytokine levels in inflammatory bowel disease patients. Cytokine 2016, 77, 44–49. [Google Scholar] [CrossRef] [PubMed]
- Kalliolias, G.D.; Ivashkiv, L.B. TNF biology, pathogenic mechanisms and emerging therapeutic strategies. Nat. Rev. Rheumatol. 2016, 12, 49–62. [Google Scholar] [CrossRef]
- Delgado, M.E.; Brunner, T. The many faces of tumor necrosis factor signaling in the intestinal epithelium. Genes. Immun. 2019, 20, 609–626. [Google Scholar] [CrossRef]
- Bradford, E.M.; Ryu, S.H.; Singh, A.P.; Lee, G.; Goretsky, T.; Sinh, P.; Williams, D.B.; Cloud, A.L.; Gounaris, E.; Patel, V.; et al. Epithelial TNF Receptor Signaling Promotes Mucosal Repair in Inflammatory Bowel Disease. J. Immunol. 2017, 199, 1886–1897. [Google Scholar] [CrossRef]
- Guan, Q. A Comprehensive Review and Update on the Pathogenesis of Inflammatory Bowel Disease. J. Immunol. Res. 2019, 2019, 7247238. [Google Scholar] [CrossRef]
- Dube, P.E.; Punit, S.; Polk, D.B. Redeeming an old foe: Protective as well as pathophysiological roles for tumor necrosis factor in inflammatory bowel disease. Am. J. Physiol. Gastrointest. Liver Physiol. 2015, 308, G161–G170. [Google Scholar] [CrossRef] [PubMed]
- Ruder, B.; Atreya, R.; Becker, C. Tumour Necrosis Factor Alpha in Intestinal Homeostasis and Gut Related Diseases. Int. J. Mol. Sci. 2019, 20, 1887. [Google Scholar] [CrossRef] [PubMed]
- Ma, T.Y.; Boivin, M.A.; Ye, D.; Pedram, A.; Said, H.M. Mechanism of TNF-{α} modulation of Caco-2 intestinal epithelial tight junction barrier: Role of myosin light-chain kinase protein expression. Am. J. Physiol. Gastrointest. Liver Physiol. 2005, 288, G422–G430. [Google Scholar] [CrossRef] [PubMed]
- Marchiando, A.M.; Shen, L.; Graham, W.V.; Edelblum, K.L.; Duckworth, C.A.; Guan, Y.; Montrose, M.H.; Turner, J.R.; Watson, A.J. The epithelial barrier is maintained by in vivo tight junction expansion during pathologic intestinal epithelial shedding. Gastroenterology 2011, 140, 1208–1218.e2. [Google Scholar] [CrossRef]
- Ngo, B.; Farrell, C.P.; Barr, M.; Wolov, K.; Bailey, R.; Mullin, J.M.; Thornton, J.J. Tumor necrosis factor blockade for treatment of inflammatory bowel disease: Efficacy and safety. Curr. Mol. Pharmacol. 2010, 3, 145–152. [Google Scholar] [CrossRef]
- Genaro, L.M.; Gomes, L.E.M.; Franceschini, A.; Ceccato, H.D.; de Jesus, R.N.; Lima, A.P.; Nagasako, C.K.; Fagundes, J.J.; Ayrizono, M.L.S.; Leal, R.F. Anti-TNF therapy and immunogenicity in inflammatory bowel diseases: A translational approach. Am. J. Transl. Res. 2021, 13, 13916–13930. [Google Scholar]
- Kucharzik, T.; Koletzko, S.; Kannengiesser, K.; Dignass, A. Ulcerative Colitis-Diagnostic and Therapeutic Algorithms. Dtsch. Arztebl. Int. 2020, 117, 564–574. [Google Scholar] [CrossRef]
- Gisbert, J.P.; Chaparro, M. Primary Failure to an Anti-TNF Agent in Inflammatory Bowel Disease: Switch (to a Second Anti-TNF Agent) or Swap (for Another Mechanism of Action)? J. Clin. Med. 2021, 10, 5318. [Google Scholar] [CrossRef] [PubMed]
- Gisbert, J.P.; Marin, A.C.; McNicholl, A.G.; Chaparro, M. Systematic review with meta-analysis: The efficacy of a second anti-TNF in patients with inflammatory bowel disease whose previous anti-TNF treatment has failed. Aliment. Pharmacol. Ther. 2015, 41, 613–623. [Google Scholar] [CrossRef]
- Gough, P.; Myles, I.A. Tumor Necrosis Factor Receptors: Pleiotropic Signaling Complexes and Their Differential Effects. Front. Immunol. 2020, 11, 585880. [Google Scholar] [CrossRef] [PubMed]
- van Loo, G.; Bertrand, M.J.M. Death by TNF: A road to inflammation. Nat. Rev. Immunol. 2023, 23, 289–303. [Google Scholar] [CrossRef]
- Al-Sadi, R.; Guo, S.; Ye, D.; Ma, T.Y. TNF-alpha modulation of intestinal epithelial tight junction barrier is regulated by ERK1/2 activation of Elk-1. Am. J. Pathol. 2013, 183, 1871–1884. [Google Scholar] [CrossRef]
- Kim, J.A.; Kim, D.K.; Kang, O.H.; Choi, Y.A.; Park, H.J.; Choi, S.C.; Kim, T.H.; Yun, K.J.; Nah, Y.H.; Lee, Y.M. Inhibitory effect of luteolin on TNF-alpha-induced IL-8 production in human colon epithelial cells. Int. Immunopharmacol. 2005, 5, 209–217. [Google Scholar] [CrossRef]
- Waetzig, G.H.; Seegert, D.; Rosenstiel, P.; Nikolaus, S.; Schreiber, S. p38 mitogen-activated protein kinase is activated and linked to TNF-alpha signaling in inflammatory bowel disease. J. Immunol. 2002, 168, 5342–5351. [Google Scholar] [CrossRef] [PubMed]
- Wullaert, A.; Bonnet, M.C.; Pasparakis, M. NF-kappaB in the regulation of epithelial homeostasis and inflammation. Cell Res. 2011, 21, 146–158. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.C. NF-kappaB signaling in inflammation. Signal Transduct. Target Ther. 2017, 2, 17023. [Google Scholar] [CrossRef]
- Pearson, G.; Robinson, F.; Beers Gibson, T.; Xu, B.E.; Karandikar, M.; Berman, K.; Cobb, M.H. Mitogen-activated protein (MAP) kinase pathways: Regulation and physiological functions. Endocr. Rev. 2001, 22, 153–183. [Google Scholar] [CrossRef]
- Dilek, N.; Papapetropoulos, A.; Toliver-Kinsky, T.; Szabo, C. Hydrogen sulfide: An endogenous regulator of the immune system. Pharmacol. Res. 2020, 161, 105119. [Google Scholar] [CrossRef]
- Linden, D.R. Hydrogen sulfide signaling in the gastrointestinal tract. Antioxid. Redox Signal 2014, 20, 818–830. [Google Scholar] [CrossRef]
- Wallace, J.L.; Blackler, R.W.; Chan, M.V.; Da Silva, G.J.; Elsheikh, W.; Flannigan, K.L.; Gamaniek, I.; Manko, A.; Wang, L.; Motta, J.P.; et al. Anti-inflammatory and cytoprotective actions of hydrogen sulfide: Translation to therapeutics. Antioxid. Redox Signal 2015, 22, 398–410. [Google Scholar] [CrossRef] [PubMed]
- Blachier, F.; Beaumont, M.; Kim, E. Cysteine-derived hydrogen sulfide and gut health: A matter of endogenous or bacterial origin. Curr. Opin. Clin. Nutr. Metab. Care 2019, 22, 68–75. [Google Scholar] [CrossRef] [PubMed]
- Cai, W.J.; Wang, M.J.; Ju, L.H.; Wang, C.; Zhu, Y.C. Hydrogen sulfide induces human colon cancer cell proliferation: Role of Akt, ERK and p21. Cell Biol. Int. 2010, 34, 565–572. [Google Scholar] [CrossRef] [PubMed]
- Flannigan, K.L.; Ferraz, J.G.; Wang, R.; Wallace, J.L. Enhanced synthesis and diminished degradation of hydrogen sulfide in experimental colitis: A site-specific, pro-resolution mechanism. PLoS ONE 2013, 8, e71962. [Google Scholar] [CrossRef]
- Stummer, N.; Weghuber, D.; Feichtinger, R.G.; Huber, S.; Mayr, J.A.; Kofler, B.; Neureiter, D.; Klieser, E.; Hochmann, S.; Lauth, W.; et al. Hydrogen Sulfide Metabolizing Enzymes in the Intestinal Mucosa in Pediatric and Adult Inflammatory Bowel Disease. Antioxidants 2022, 11, 2235. [Google Scholar] [CrossRef]
- Wallace, J.L.; Vong, L.; McKnight, W.; Dicay, M.; Martin, G.R. Endogenous and exogenous hydrogen sulfide promotes resolution of colitis in rats. Gastroenterology 2009, 137, 569–578.e1. [Google Scholar] [CrossRef]
- Scheller, A.S.; Philipp, T.M.; Klotz, L.O.; Steinbrenner, H. Altered Capacity for H(2)S Production during the Spontaneous Differentiation of Caco-2 Cells to Colonocytes Due to Reciprocal Regulation of CBS and SELENBP1. Antioxidants 2022, 11, 1957. [Google Scholar] [CrossRef] [PubMed]
- Hirata, I.; Naito, Y.; Takagi, T.; Mizushima, K.; Suzuki, T.; Omatsu, T.; Handa, O.; Ichikawa, H.; Ueda, H.; Yoshikawa, T. Endogenous hydrogen sulfide is an anti-inflammatory molecule in dextran sodium sulfate-induced colitis in mice. Dig. Dis. Sci. 2011, 56, 1379–1386. [Google Scholar] [CrossRef]
- Motta, J.P.; Flannigan, K.L.; Agbor, T.A.; Beatty, J.K.; Blackler, R.W.; Workentine, M.L.; Da Silva, G.J.; Wang, R.; Buret, A.G.; Wallace, J.L. Hydrogen sulfide protects from colitis and restores intestinal microbiota biofilm and mucus production. Inflamm. Bowel Dis. 2015, 21, 1006–1017. [Google Scholar] [CrossRef]
- Wallace, J.L.; Dicay, M.; McKnight, W.; Martin, G.R. Hydrogen sulfide enhances ulcer healing in rats. FASEB J. 2007, 21, 4070–4076. [Google Scholar] [CrossRef] [PubMed]
- Thanki, K.K.; Johnson, P.; Higgins, E.J.; Maskey, M.; Phillips, C.N.; Dash, S.; Almenas, F.A.; Govar, A.A.; Tian, B.; Villéger, R.; et al. Deletion of cystathionine-γ-lyase in bone marrow-derived cells promotes colitis-associated carcinogenesis. Redox Biol. 2022, 55, 102417. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Zuo, S.; Zhu, J.; Yue, T.; Bu, D.; Wang, X.; Wang, P.; Pan, Y.; Liu, Y. Decreased Expression of Cystathionine beta-Synthase Exacerbates Intestinal Barrier Injury in Ulcerative Colitis. J. Crohns Colitis 2019, 13, 1067–1080. [Google Scholar] [CrossRef]
- Zanardo, R.C.; Brancaleone, V.; Distrutti, E.; Fiorucci, S.; Cirino, G.; Wallace, J.L. Hydrogen sulfide is an endogenous modulator of leukocyte-mediated inflammation. FASEB J. 2006, 20, 2118–2120. [Google Scholar] [CrossRef] [PubMed]
- Flannigan, K.L.; Agbor, T.A.; Blackler, R.W.; Kim, J.J.; Khan, W.I.; Verdu, E.F.; Ferraz, J.G.; Wallace, J.L. Impaired hydrogen sulfide synthesis and IL-10 signaling underlie hyperhomocysteinemia-associated exacerbation of colitis. Proc. Natl. Acad. Sci. USA 2014, 111, 13559–13564. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.; Liang, F.; Shah Masood, W.; Yan, X. Hydrogen sulfide protected gastric epithelial cell from ischemia/reperfusion injury by Keap1 s-sulfhydration, MAPK dependent anti-apoptosis and NF-kappaB dependent anti-inflammation pathway. Eur. J. Pharmacol. 2014, 725, 70–78. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Yan, R.; Zhou, X.; Ji, F.; Zhang, B. Hydrogen sulfide improves colonic barrier integrity in DSS-induced inflammation in Caco-2 cells and mice. Int. Immunopharmacol. 2016, 39, 121–127. [Google Scholar] [CrossRef] [PubMed]
- Roig, A.I.; Eskiocak, U.; Hight, S.K.; Kim, S.B.; Delgado, O.; Souza, R.F.; Spechler, S.J.; Wright, W.E.; Shay, J.W. Immortalized epithelial cells derived from human colon biopsies express stem cell markers and differentiate in vitro. Gastroenterology 2010, 138, 1012–1021.e5. [Google Scholar] [CrossRef]
- Modis, K.; Ramanujam, V.S.; Govar, A.A.; Lopez, E.; Anderson, K.E.; Wang, R.; Szabo, C. Cystathionine-gamma-lyase (CSE) deficiency increases erythropoiesis and promotes mitochondrial electron transport via the upregulation of coproporphyrinogen III oxidase and consequent stimulation of heme biosynthesis. Biochem. Pharmacol. 2019, 169, 113604. [Google Scholar] [CrossRef]
- Han, Y.; Brasier, A.R. Mechanism for biphasic rel A. NF-kappaB1 nuclear translocation in tumor necrosis factor alpha-stimulated hepatocytes. J. Biol. Chem. 1997, 272, 9825–9832. [Google Scholar] [CrossRef] [PubMed]
- Coletta, C.; Modis, K.; Szczesny, B.; Brunyanszki, A.; Olah, G.; Rios, E.C.; Yanagi, K.; Ahmad, A.; Papapetropoulos, A.; Szabo, C. Regulation of Vascular Tone, Angiogenesis and Cellular Bioenergetics by the 3-Mercaptopyruvate Sulfurtransferase/H2S Pathway: Functional Impairment by Hyperglycemia and Restoration by DL-alpha-Lipoic Acid. Mol. Med. 2015, 21, 1–14. [Google Scholar] [CrossRef]
- Srinivasan, B.; Kolli, A.R.; Esch, M.B.; Abaci, H.E.; Shuler, M.L.; Hickman, J.J. TEER measurement techniques for in vitro barrier model systems. J. Lab. Autom. 2015, 20, 107–126. [Google Scholar] [CrossRef] [PubMed]
- Phillips, C.M.; Zatarain, J.R.; Nicholls, M.E.; Porter, C.; Widen, S.G.; Thanki, K.; Johnson, P.; Jawad, M.U.; Moyer, M.P.; Randall, J.W.; et al. Upregulation of Cystathionine-beta-Synthase in Colonic Epithelia Reprograms Metabolism and Promotes Carcinogenesis. Cancer Res. 2017, 77, 5741–5754. [Google Scholar] [CrossRef]
- Wallace, J.L.; Ianaro, A.; de Nucci, G. Gaseous Mediators in Gastrointestinal Mucosal Defense and Injury. Dig. Dis. Sci. 2017, 62, 2223–2230. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Cen, L.; Zhang, X.; Tang, C.; Chen, Y.; Zhang, Y.; Yu, M.; Lu, C.; Li, M.; Li, S.; et al. MPST deficiency promotes intestinal epithelial cell apoptosis and aggravates inflammatory bowel disease via AKT. Redox Biol. 2022, 56, 102469. [Google Scholar] [CrossRef]
- Marsal, J.; Barreiro-de Acosta, M.; Blumenstein, I.; Cappello, M.; Bazin, T.; Sebastian, S. Management of Non-response and Loss of Response to Anti-tumor Necrosis Factor Therapy in Inflammatory Bowel Disease. Front. Med. 2022, 9, 897936. [Google Scholar] [CrossRef]
- Huang, C.Y.; Yao, W.F.; Wu, W.G.; Lu, Y.L.; Wan, H.; Wang, W. Endogenous CSE/H2 S system mediates TNF-alpha-induced insulin resistance in 3T3-L1 adipocytes. Cell Biochem. Funct. 2013, 31, 468–475. [Google Scholar] [CrossRef]
- Fox, B.; Schantz, J.T.; Haigh, R.; Wood, M.E.; Moore, P.K.; Viner, N.; Spencer, J.P.; Winyard, P.G.; Whiteman, M. Inducible hydrogen sulfide synthesis in chondrocytes and mesenchymal progenitor cells: Is H2S a novel cytoprotective mediator in the inflamed joint? J. Cell Mol. Med. 2012, 16, 896–910. [Google Scholar] [CrossRef] [PubMed]
- Sen, N.; Paul, B.D.; Gadalla, M.M.; Mustafa, A.K.; Sen, T.; Xu, R.; Kim, S.; Snyder, S.H. Hydrogen sulfide-linked sulfhydration of NF-kappaB mediates its antiapoptotic actions. Mol. Cell 2012, 45, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Liao, R.; Qiang, Z.; Zhang, C. Pro-inflammatory cytokine-driven PI3K/Akt/Sp1 signalling and H(2)S production facilitates the pathogenesis of severe acute pancreatitis. Biosci. Rep. 2017, 37, BSR20160483. [Google Scholar] [CrossRef]
- Bibli, S.I.; Hu, J.; Looso, M.; Weigert, A.; Ratiu, C.; Wittig, J.; Drekolia, M.K.; Tombor, L.; Randriamboavonjy, V.; Leisegang, M.S.; et al. Mapping the Endothelial Cell S-Sulfhydrome Highlights the Crucial Role of Integrin Sulfhydration in Vascular Function. Circulation 2021, 143, 935–948. [Google Scholar] [CrossRef]
- Bibli, S.I.; Hu, J.; Sigala, F.; Wittig, I.; Heidler, J.; Zukunft, S.; Tsilimigras, D.I.; Randriamboavonjy, V.; Wittig, J.; Kojonazarov, B.; et al. Cystathionine gamma Lyase Sulfhydrates the RNA Binding Protein HuR to Preserve Endothelial Cell Function and Delay Atherogenesis. Circulation 2018, 139, 101–114. [Google Scholar] [CrossRef] [PubMed]
- Yang, G.; Wu, L.; Jiang, B.; Yang, W.; Qi, J.; Cao, K.; Meng, Q.; Mustafa, A.K.; Mu, W.; Zhang, S.; et al. H2S as a physiologic vasorelaxant: Hypertension in mice with deletion of cystathionine gamma-lyase. Science 2008, 322, 587–590. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.J.; Wang, H.X.; Yao, Y.S.; Guo, L.Y.; Liu, P. The role of Ca2+/calmodulin-dependent protein kinase II and calcineurin in TNF-alpha-induced myocardial hypertrophy. Braz. J. Med. Biol. Res. 2012, 45, 1045–1051. [Google Scholar] [CrossRef]
- Sun, Q.; Collins, R.; Huang, S.; Holmberg-Schiavone, L.; Anand, G.S.; Tan, C.H.; van-den-Berg, S.; Deng, L.W.; Moore, P.K.; Karlberg, T.; et al. Structural basis for the inhibition mechanism of human cystathionine gamma-lyase, an enzyme responsible for the production of H(2)S. J. Biol. Chem. 2009, 284, 3076–3085. [Google Scholar] [CrossRef]
- Kimura, H. Production and physiological effects of hydrogen sulfide. Antioxid. Redox Signal 2014, 20, 783–793. [Google Scholar] [CrossRef] [PubMed]
- Whiteman, M.; Le Trionnaire, S.; Chopra, M.; Fox, B.; Whatmore, J. Emerging role of hydrogen sulfide in health and disease: Critical appraisal of biomarkers and pharmacological tools. Clin. Sci. 2011, 121, 459–488. [Google Scholar] [CrossRef]
- Asimakopoulou, A.; Panopoulos, P.; Chasapis, C.T.; Coletta, C.; Zhou, Z.; Cirino, G.; Giannis, A.; Szabo, C.; Spyroulias, G.A.; Papapetropoulos, A. Selectivity of commonly used pharmacological inhibitors for cystathionine beta synthase (CBS) and cystathionine gamma lyase (CSE). Br. J. Pharmacol. 2013, 169, 922–932. [Google Scholar] [CrossRef]
- Zhao, K.; Ju, Y.; Li, S.; Altaany, Z.; Wang, R.; Yang, G. S-sulfhydration of MEK1 leads to PARP-1 activation and DNA damage repair. EMBO Rep. 2014, 15, 792–800. [Google Scholar] [CrossRef]
- Qin, M.; Long, F.; Wu, W.; Yang, D.; Huang, M.; Xiao, C.; Chen, X.; Liu, X.; Zhu, Y.Z. Hydrogen sulfide protects against DSS-induced colitis by inhibiting NLRP3 inflammasome. Free Radic. Biol. Med. 2019, 137, 99–109. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Arroyo Almenas, F.; Törő, G.; Szaniszlo, P.; Maskey, M.; Thanki, K.K.; Koltun, W.A.; Yochum, G.S.; Pinchuk, I.V.; Chao, C.; Hellmich, M.R.; et al. Cystathionine Gamma-Lyase Regulates TNF-α-Mediated Injury Response in Human Colonic Epithelial Cells and Colonoids. Antioxidants 2024, 13, 1067. https://doi.org/10.3390/antiox13091067
Arroyo Almenas F, Törő G, Szaniszlo P, Maskey M, Thanki KK, Koltun WA, Yochum GS, Pinchuk IV, Chao C, Hellmich MR, et al. Cystathionine Gamma-Lyase Regulates TNF-α-Mediated Injury Response in Human Colonic Epithelial Cells and Colonoids. Antioxidants. 2024; 13(9):1067. https://doi.org/10.3390/antiox13091067
Chicago/Turabian StyleArroyo Almenas, Francisco, Gábor Törő, Peter Szaniszlo, Manjit Maskey, Ketan K. Thanki, Walter A. Koltun, Gregory S. Yochum, Irina V. Pinchuk, Celia Chao, Mark R. Hellmich, and et al. 2024. "Cystathionine Gamma-Lyase Regulates TNF-α-Mediated Injury Response in Human Colonic Epithelial Cells and Colonoids" Antioxidants 13, no. 9: 1067. https://doi.org/10.3390/antiox13091067
APA StyleArroyo Almenas, F., Törő, G., Szaniszlo, P., Maskey, M., Thanki, K. K., Koltun, W. A., Yochum, G. S., Pinchuk, I. V., Chao, C., Hellmich, M. R., & Módis, K. (2024). Cystathionine Gamma-Lyase Regulates TNF-α-Mediated Injury Response in Human Colonic Epithelial Cells and Colonoids. Antioxidants, 13(9), 1067. https://doi.org/10.3390/antiox13091067