
Antibody-Based Immunotherapies as a Tool for Tackling Multidrug-Resistant Bacterial Infections




Abstract
:1. Introduction
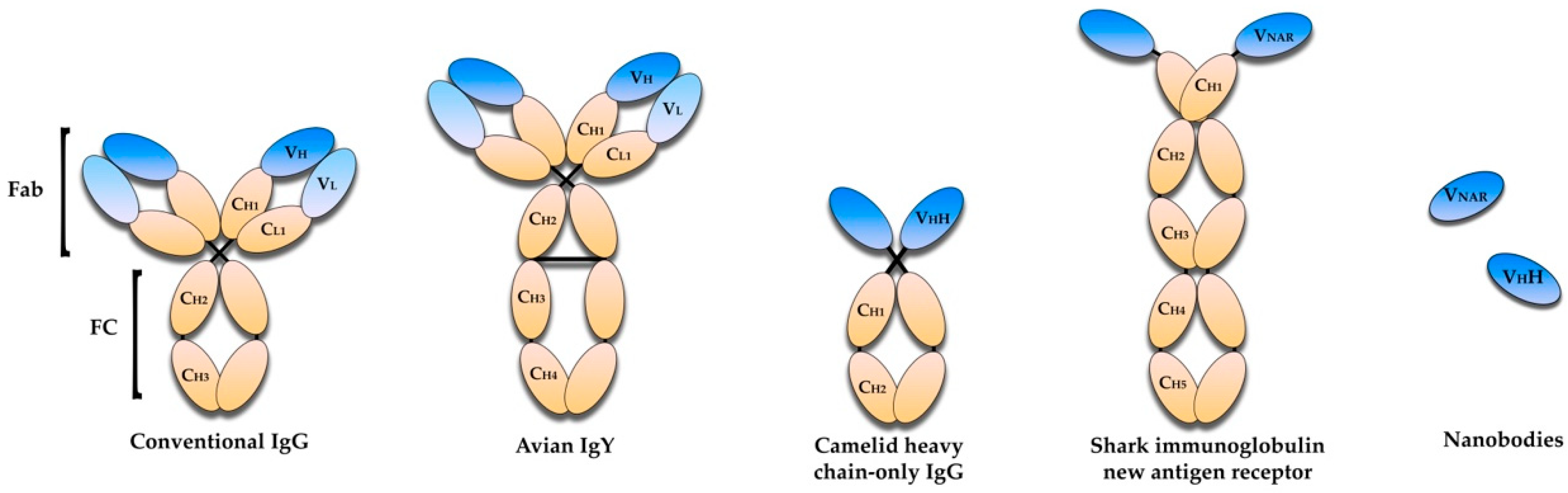
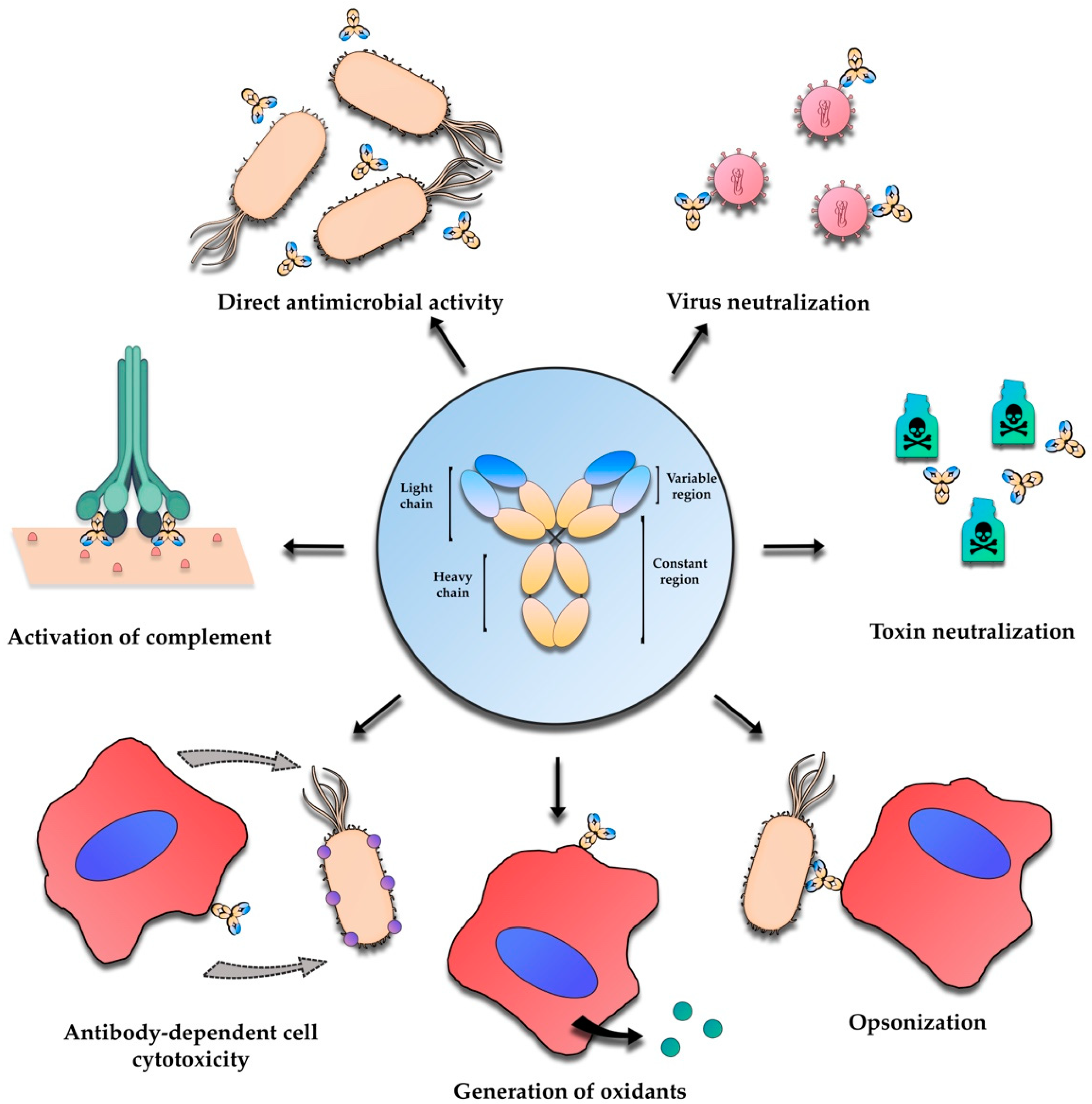
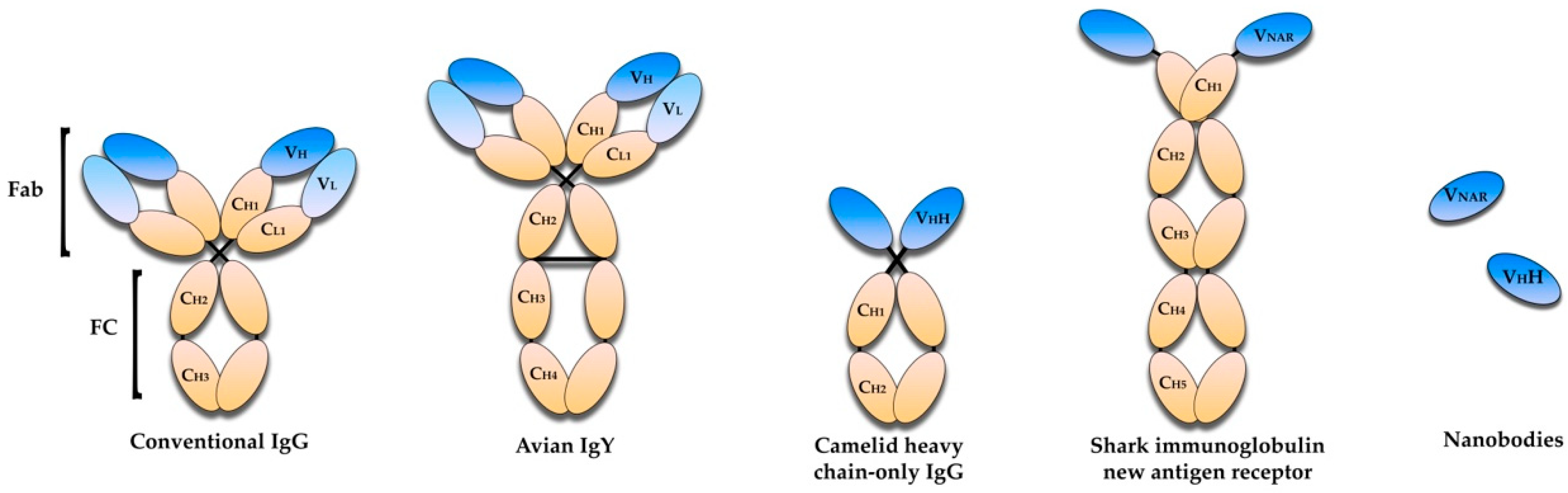
2. Antibody-Based Immunotherapies—An Overview
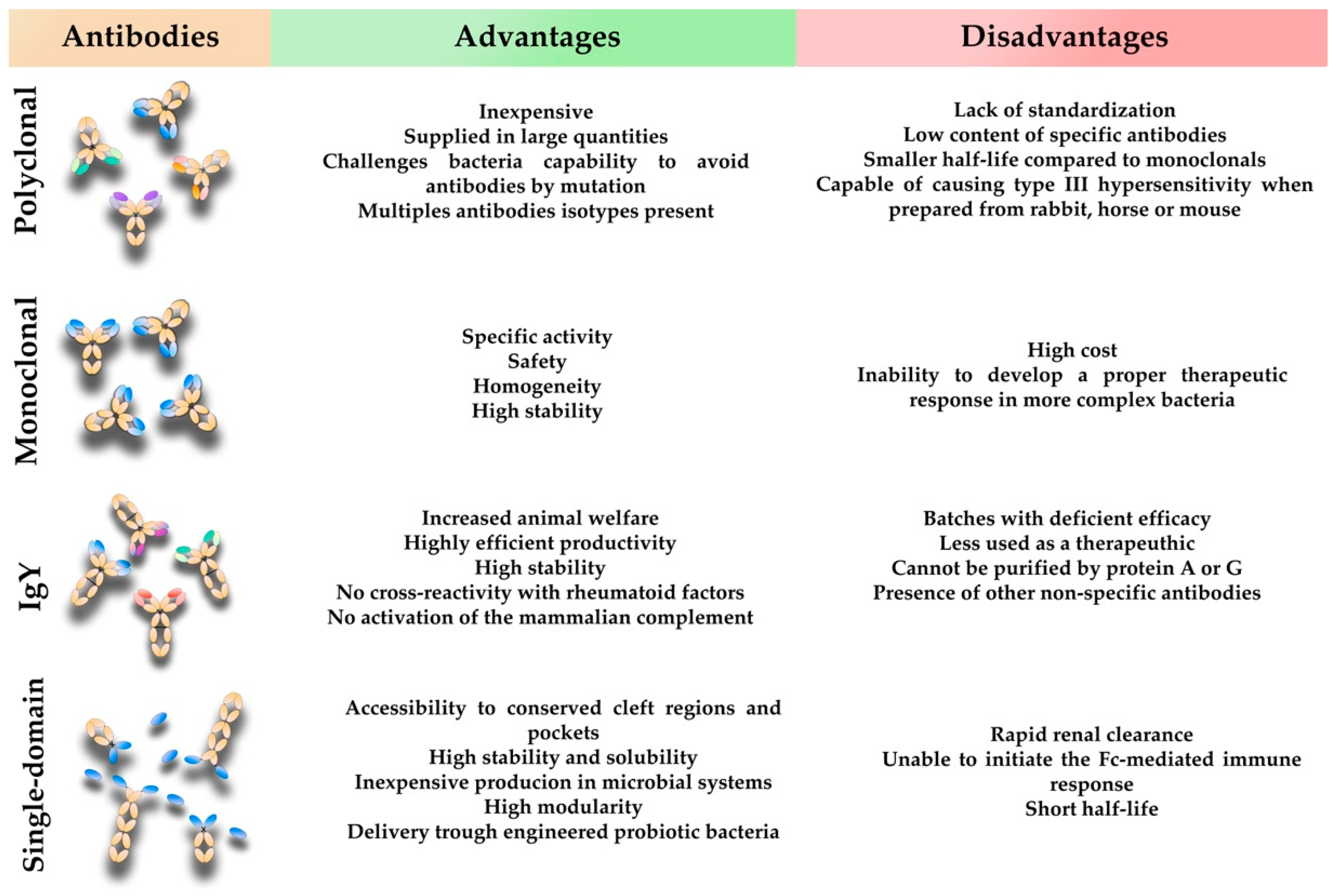
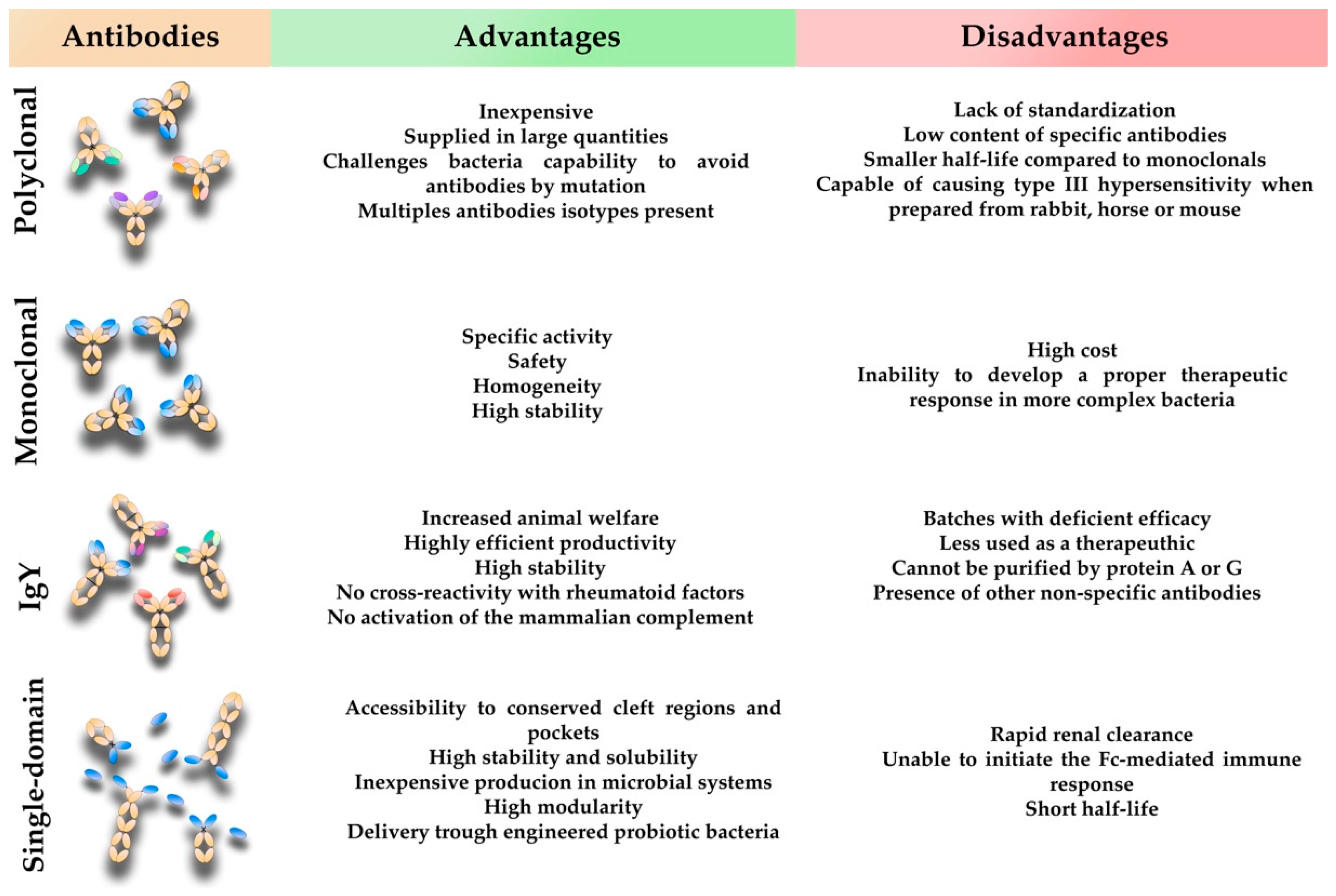
3. Conventional Mammalian Polyclonal Antibodies
4. Conventional Mammalian Monoclonal Antibodies
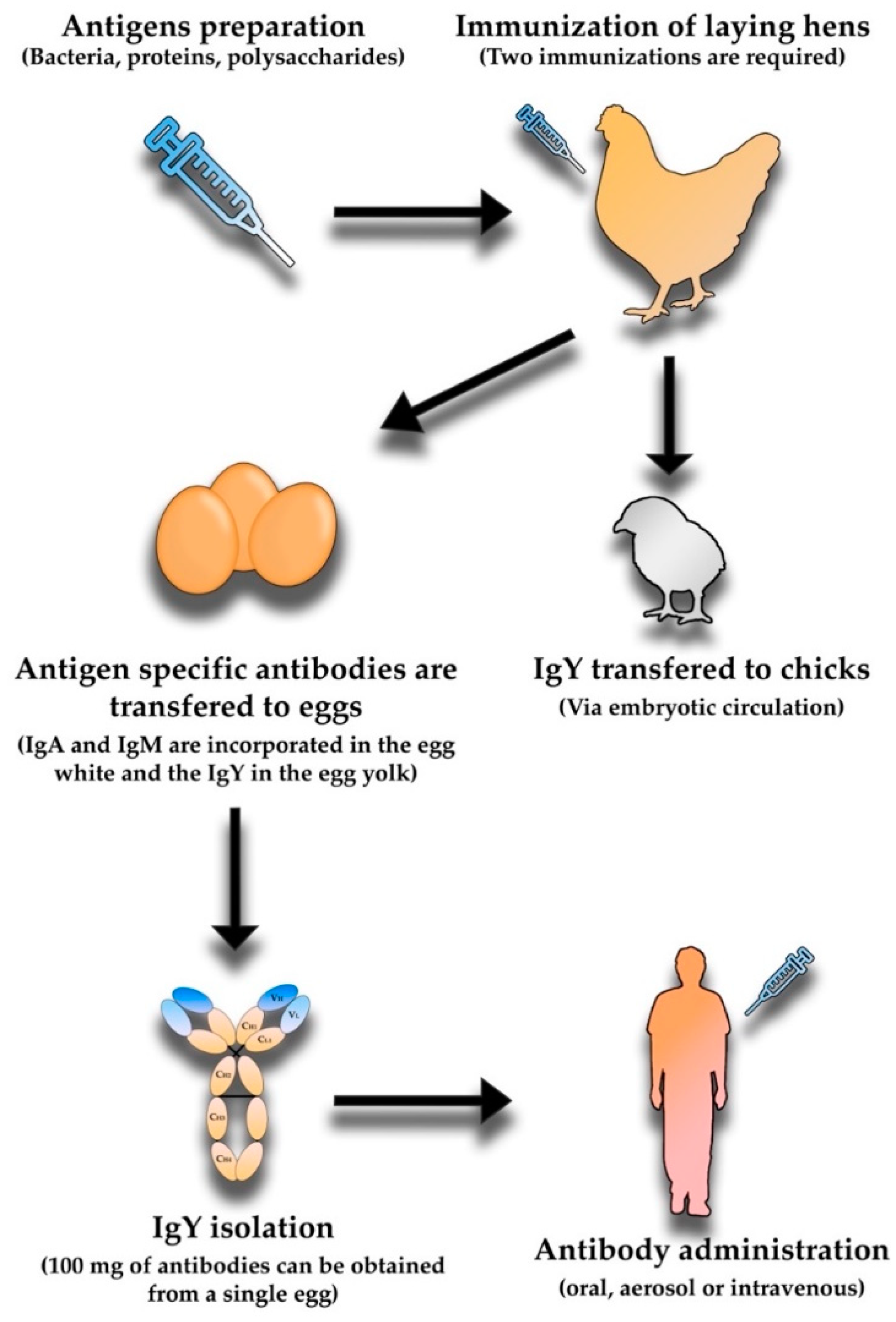
5. Avian Immunoglobulin Y Antibodies
6. Single-Domain Antibodies
7. Conclusions and Future Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Casadevall, A.; Scharff, M.D. Serum therapy revisited: Animal models of infection and development of passive antibody therapy. Antimicrob. Agents Chemother. 1994, 38, 1695–1702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Casadevall, A.; Dadachova, E.; Pirofski, L.A. Passive antibody therapy for infectious diseases. Nat. Rev. Microbiol. 2004, 2, 695–703. [Google Scholar] [CrossRef] [PubMed]
- Lewis, K. Platforms for antibiotic discovery. Nat. Rev. Drug Discov. 2013, 12, 371–387. [Google Scholar] [CrossRef] [PubMed]
- Antonelli, G.; Cappelli, L.; Cinelli, P.; Cuffaro, R.; Manca, B.; Nicchi, S.; Tondi, S.; Vezzani, G.; Viviani, V.; Delany, I.; et al. Strategies to tackle antimicrobial resistance: The example of Escherichia coli and Pseudomonas aeruginosa. Int. J. Mol. Sci. 2021, 22, 4943. [Google Scholar] [CrossRef] [PubMed]
- European Centre for Disease Prevention and Control. Antimicrobial Resistance in the EU/EEA (EARS-Net) Annual Epidemiological Report 2019; ECDC: Stockholm, Sweden, 2020. [Google Scholar]
- Hotinger, J.A.; Morris, S.T.; May, A.E. The case against antibiotics and for anti-virulence therapeutics. Microorganisms 2021, 9, 2049. [Google Scholar] [CrossRef] [PubMed]
- Boucher, H.W.; Talbot, G.H.; Bradley, J.S.; Edwards, J.E.; Gilbert, D.; Rice, L.B.; Scheld, M.; Spellberg, B.; Bartlett, J. Bad bugs, no drugs: No ESKAPE! An update from the Infectious Diseases Society of America. Clin. Infect. Dis. 2009, 48, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Higgins, P.G.; Dammhayn, C.; Hackel, M.; Seifert, H. Global spread of carbapenem-resistant Acinetobacter baumannii. J. Antimicrob. Chemother. 2010, 65, 233–238. [Google Scholar] [CrossRef] [Green Version]
- López-Siles, M.; Corral-Lugo, A.; McConnell, M.J. Vaccines for multidrug resistant Gram negative bacteria: Lessons from the past for guiding future success. FEMS Microbiol. Rev. 2021, 45, fuaa054. [Google Scholar] [CrossRef]
- Baxter, D. Active and passive immunity, vaccine types, excipients and licensing. Occup. Med. 2007, 57, 552–556. [Google Scholar] [CrossRef] [Green Version]
- Henrique, I.M.; Sacerdoti, F.; Ferreira, R.L.; Henrique, C.; Amaral, M.M.; Piazza, R.M.F.; Luz, D. Therapeutic Antibodies Against Shiga Toxins: Trends and Perspectives. Front. Cell. Infect. Microbiol. 2022, 12, 825856. [Google Scholar] [CrossRef]
- Vidarsson, G.; Dekkers, G.; Rispens, T. IgG subclasses and allotypes: From structure to effector functions. Front. Immunol. 2014, 5, 520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keyt, B.A.; Baliga, R.; Sinclair, A.M.; Carroll, S.F.; Peterson, M.S. Structure, Function, and Therapeutic Use of IgM Antibodies. Antibodies 2020, 9, 53. [Google Scholar] [CrossRef] [PubMed]
- Hand, T.W.; Reboldi, A. Production and Function of Immunoglobulin A. Annu. Rev. Immunol. 2021, 39, 695–718. [Google Scholar] [CrossRef]
- Sterlin, D.; Gorochov, G. When Therapeutic IgA Antibodies Might Come of Age. Pharmacology 2021, 106, 9–19. [Google Scholar] [CrossRef]
- Rajam, G.; Sampson, J.; Carlone, G.M.; Ades, E.W. An Augmented Passive Immune Therapy to Treat Fulminant Bacterial Infections. Recent Pat. Antiinfect. Drug Discov. 2010, 5, 157–167. [Google Scholar] [CrossRef] [PubMed]
- Leitner, W.W.; Haraway, M.; Pierson, T.; Bergmann-Leitner, E.S. Role of opsonophagocytosis in immune protection against malaria. Vaccines 2020, 8, 264. [Google Scholar] [CrossRef] [PubMed]
- Heesterbeek, D.A.C.; Angelier, M.L.; Harrison, R.A.; Rooijakkers, S.H.M. Complement and Bacterial Infections: From Molecular Mechanisms to Therapeutic Applications. J. Innate Immun. 2018, 10, 455–464. [Google Scholar] [CrossRef]
- Pai, J.; Sutherland, J.; Maynard, J. Progress Towards Recombinant Anti-Infective Antibodies. Recent Pat. Antiinfect. Drug Discov. 2009, 4, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Mullard, A. FDA approves 100th monoclonal antibody product. Nat. Rev. Drug Discov. 2021, 20, 491–495. [Google Scholar] [CrossRef]
- Casadevall, A. Antibody-mediated immunity against intracellular pathogens: Two-dimensional thinking comes full circle. Infect. Immun. 2003, 71, 4225–4228. [Google Scholar] [CrossRef]
- Zurawski, D.V.; McLendon, M.K. Monoclonal antibodies as an antibacterial approach against bacterial pathogens. Antibiotics 2020, 9, 155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- García-Quintanilla, M.; Pulido, M.R.; Carretero-Ledesma, M.; McConnell, M.J. Vaccines for Antibiotic-Resistant Bacteria: Possibility or Pipe Dream? Trends Pharmacol. Sci. 2016, 37, 143–152. [Google Scholar] [CrossRef] [PubMed]
- Beaber, J.W.; Hochhut, B.; Waldor, M.K. SOS response promotes horizontal dissemination of antibiotic resistance genes. Nature 2004, 427, 72–74. [Google Scholar] [CrossRef] [PubMed]
- De Vlieger, D.; Ballegeer, M.; Rossey, I.; Schepens, B.; Saelens, X. Single-Domain Antibodies and Their Formatting to Combat Viral Infections. Antibodies 2019, 8, 1. [Google Scholar] [CrossRef] [Green Version]
- McConnell, M.J. Where are we with monoclonal antibodies for multidrug-resistant infections? Drug Discov. Today 2019, 24, 1132–1138. [Google Scholar] [CrossRef]
- Casadevall, A. Antibody-Based Therapies for Emerging Infectious Diseases. Emerg. Infect. Dis. 1996, 2, 200–208. [Google Scholar] [CrossRef]
- Carlander, D.; Kollberg, H.; Wejåker, P.E.; Larsson, A. Peroral immunotheraphy with yolk antibodies for the prevention and treatment of enteric infections. Immunol. Res. 2000, 21, 1–6. [Google Scholar] [CrossRef]
- Mayor, A.; Chesnay, A.; Desoubeaux, G.; Ternant, D.; Heuzé-Vourc’h, N.; Sécher, T. Therapeutic antibodies for the treatment of respiratory tract infections—Current overview and perspectives. Vaccines 2021, 9, 151. [Google Scholar] [CrossRef]
- Döring, G.; Pier, G.B. Vaccines and immunotherapy against Pseudomonas aeruginosa. Vaccine 2008, 26, 1011–1024. [Google Scholar] [CrossRef]
- Yang, F.; Gu, J.; Yang, L.; Gao, C.; Jing, H.; Wang, Y.; Zeng, H.; Zou, Q.; Lv, F.; Zhang, J. Protective Efficacy of the Trivalent Pseudomonas aeruginosa Vaccine Candidate PcrV-OprI-Hcp1 in Murine Pneumonia and Burn Models. Sci. Rep. 2017, 7, 3957. [Google Scholar] [CrossRef]
- Buyel, J.F.; Twyman, R.M.; Fischer, R. Very-large-scale production of antibodies in plants: The biologization of manufacturing. Biotechnol. Adv. 2017, 35, 458–465. [Google Scholar] [CrossRef] [PubMed]
- Hassani, M.; Patel, M.C.; Pirofski, L.A. Vaccines for the prevention of diseases caused by potential bioweapons. Clin. Immunol. 2004, 111, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Cosson, P.; Hartley, O. Recombinant antibodies for academia: A practical approach. CHIMIA Int. J. Chem. 2016, 70, 893–897. [Google Scholar] [CrossRef] [PubMed]
- Casadevall, A.; Pirofski, L.A.; Joyner, M.J. The principles of antibody therapy for infectious diseases with relevance for covid-19. mBio 2021, 12, e03372-20. [Google Scholar] [CrossRef] [PubMed]
- Berry, C.M. Antibody immunoprophylaxis and immunotherapy for influenza virus infection: Utilization of monoclonal or polyclonal antibodies? Hum. Vaccines Immunother. 2018, 14, 796–799. [Google Scholar] [CrossRef] [PubMed]
- Ian Gust, A.O. Role of passive immunotherapies in managing infectious outbreaks. Biologicals 2012, 40, 196–199. [Google Scholar] [CrossRef]
- Baum, A.; Fulton, B.O.; Wloga, E.; Copin, R.; Pascal, K.E.; Russo, V.; Giordano, S.; Lanza, K.; Negron, N.; Ni, M.; et al. Antibody cocktail to SARS-CoV-2 spike protein prevents rapid mutational escape seen with individual antibodies. Science 2020, 369, 1014–1018. [Google Scholar] [CrossRef]
- Berry, J.D.; Gaudet, R.G. Antibodies in infectious diseases: Polyclonals, monoclonals and niche biotechnology. N. Biotechnol. 2011, 28, 489–501. [Google Scholar] [CrossRef]
- Beigel, J.H.; Voell, J.; Kumar, P.; Raviprakash, K.; Wu, H.; Jiao, J.A.; Sullivan, E.; Luke, T.; Davey, R.T., Jr. Safety and tolerability of a novel, polyclonal human anti-MERS coronavirus antibody produced from transchromosomic cattle: A phase 1 randomised, double-blind, single-dose-escalation study. Lancet Infect. Dis. 2018, 18, 410–418. [Google Scholar] [CrossRef] [Green Version]
- Seixas, A.M.M.; Sousa, S.A.; Feliciano, J.R.; Gomes, S.C.; Ferreira, M.R.; Moreira, L.M.; Leitão, J.H. A Polyclonal Antibody Raised against the Burkholderia cenocepacia OmpA-like Protein BCAL2645 Impairs the Bacterium Adhesion and Invasion of Human Epithelial Cells In Vitro. Biomedicines 2021, 9, 1788. [Google Scholar] [CrossRef]
- Rupp, M.E.; Holley, H.P., Jr.; Lutz, J.; Dicpinigaitis, P.V.; Woods, C.W.; Levine, D.P.; Veney, N.; Fowler, V.G., Jr. Phase II, randomized, multicenter, double-blind, placebo-controlled trial of a polyclonal anti-Staphylococcus aureus capsular polysaccharide immune globulin in treatment of Staphylococcus aureus bacteremia. Antimicrob. Agents Chemother. 2007, 51, 4249–4254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeJonge, M.; Burchfield, D.; Bloom, B.; Duenas, M.; Walker, W.; Polak, M.; Jung, E.; Millard, D.; Schelonka, R.; Eyal, F.; et al. Clinical Trial of Safety and Efficacy of IHN-A21 for the Prevention of Nosocomial Staphylococcal Bloodstream Infection in Premature Infants. J. Pediatr. 2007, 151, 260–265. [Google Scholar] [CrossRef] [PubMed]
- Köhler, G.; Milstein, C. Continuous cultures of fused cells secreting antibody of predefined specificity. Nature 1975, 256, 495–497. [Google Scholar] [CrossRef] [PubMed]
- Chames, P.M.; Regenmortel, M.V.; Weiss, E.; Baty, D. Therapeutic antibodies: Successes, limitations and hopes for the future. Br. J. Pharmacol. 2009, 157, 220–233. [Google Scholar] [CrossRef] [PubMed]
- Weiner, L.M. Fully human therapeutic monoclonal antibodies. J. Immunother 2006, 29, 1–9. [Google Scholar] [CrossRef]
- Duvall, M.; Bradley, N.; Fiorini, R.N. A novel platform to produce human monoclonal antibodies: The next generation of therapeutic human monoclonal antibodies discovery. In MAbs; Taylor & Francis: Oxford, UK, 2011; Volume 3, pp. 203–208. [Google Scholar] [CrossRef] [Green Version]
- Hussack, G.; Tanha, J. An update on antibody-based immunotherapies for Clostridium difficile infection. Clin. Exp. Gastroenterol. 2016, 9, 209–224. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Wang, E.Q.; Balthasar, J.P. Monoclonal antibody pharmacokinetics and pharmacodynamics. Clin. Pharmacol. Ther. 2008, 84, 548–558. [Google Scholar] [CrossRef]
- Norris, M.H.; Blackburn, J.K. Raxibacumab: A panacea for anthrax disease? Lancet Infect. Dis. 2020, 20, 886–887. [Google Scholar] [CrossRef]
- Mohamed, N.; Clagett, M.; Li, J.; Jones, S.; Pincus, S.; D’Alia, G.; Nardone, L.; Babin, M.; Spitalny, G.; Casey, L. A high-affinity monoclonal antibody to anthrax protective antigen passively protects rabbits before and after aerosolized Bacillus anthracis spore challenge. Infect. Immun. 2005, 73, 795–802. [Google Scholar] [CrossRef]
- Rounds, J.; Strain, J. Bezlotoxumab for Preventing Recurrent Clostridium difficile Infections. S. D. Med. 2017, 70, 422–423. [Google Scholar] [CrossRef]
- Jain, R.; Beckett, V.V.; Konstan, M.W.; Accurso, F.J.; Burns, J.L.; Mayer-Hamblett, N.; Milla, C.; VanDevante, D.R.; Chmiel, J.F. KB001-A, a novel anti-inflammatory, found to be safe and well-tolerated in cystic fibrosis patients infected with Pseudomonas aeruginosa. J. Cyst. Fibros. 2018, 17, 484–491. [Google Scholar] [CrossRef] [PubMed]
- Visan, L.; Rouleau, N.; Proust, E.; Peyrot, L.; Donadieu, A.; Ochs, M. Antibodies to PcpA and PhtD protect mice against Streptococcus pneumoniae by a macrophage- and complement-dependent mechanism. Hum. Vaccines Immunother. 2018, 14, 489–494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varshney, A.K.; Kuzmicheva, G.A.; Lin, J.; Sunley, K.M.; Bowling, R.A., Jr.; Kwan, T.Y.; Mays, H.R.; Rambhadran, A.; Zhang, Y.; Martin, R.L.; et al. A natural human monoclonal antibody targeting Staphylococcus Protein A protects against Staphylococcus aureus bacteremia. PLoS ONE 2018, 13, e0190537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, J.; Yi, F.; Tian, Q.; Dang, G.; Si, W.; Liu, S.; Yu, S. Targeting the gram-negative bacteria peptidoglycan synthase MraY as a new approach for monoclonal antibody anti-bacterial activity. Hum. Vaccines Immunother. 2017, 13, 2086–2091. [Google Scholar] [CrossRef] [PubMed]
- Motley, M.P.; Banerjee, K.; Fries, B.C. Monoclonal antibody-based therapies for bacterial infections. Curr. Opin. Infect. Dis. 2019, 32, 210–216. [Google Scholar] [CrossRef]
- Sousa, S.A.; Seixas, A.M.M.; Marques, J.M.M.; Leitão, J.H. Immunization and immunotherapy approaches against Pseudomonas aeruginosa and Burkholderia cepacia complex infections. Vaccines 2021, 9, 670. [Google Scholar] [CrossRef]
- Frank, D.W.; Vallis, A.; Wiener-Kronish, J.P.; Roy-Burman, A.; Spack, E.G.; Mullaney, B.P.; Megdoud, M.; Marks, J.D.; Fritz, R.; Sawa, T. Generation and characterization of a protective monoclonal antibody to Pseudomonas aeruginosa PcrV. J. Infect. Dis. 2002, 186, 64–73. [Google Scholar] [CrossRef] [Green Version]
- François, B.; Luyt, C.E.; Dugard, A.; Wolff, M.; Diehl, J.L.; Jaber, S.; Forel, J.M.; Garot, D.; Kipnis, E.; Mebazaa, A.; et al. Safety and pharmacokinetics of an anti-PcrV PEGylated monoclonal antibody fragment in mechanically ventilated patients colonized with Pseudomonas aeruginosa: A randomized, double-blind, placebo-controlled trial. Crit. Care Med. 2012, 40, 2320–2326. [Google Scholar] [CrossRef]
- Rollenske, T.; Szijarto, V.; Lukasiewicz, J.; Guachalla, L.M.; Stojkovic, K.; Hartl, K.; Stulik, L.; Kocher, S.; Lasitschka, F.; Al-Saeedi, M.; et al. Cross-specificity of protective human antibodies against Klebsiella pneumoniae LPS O-antigen. Nat. Immunol. 2018, 19, 617–624. [Google Scholar] [CrossRef]
- Doyle, C.R.; Moon, J.Y.; Daily, J.P.; Wang, T.; Pirofski, L.A. A capsular polysaccharidespecific antibody alters Streptococcus pneumoniae gene expression during nasopharyngeal colonization of mice. Infect. Immun. 2018, 86, e00300-18. [Google Scholar] [CrossRef] [Green Version]
- Lazar, H.; Horn, M.P.; Zuercher, A.W.; Imboden, M.A.; Durrer, P.; Seiberling, M.; Pokorny, R.; Hammer, C.; Lang, A.B. Pharmacokinetics and safety profile of the human anti-Pseudomonas aeruginosa serotype O11 immunoglobulin M monoclonal antibody KBPA-101 in healthy volunteers. Antimicrob. Agents Chemother. 2009, 53, 3442–3446. [Google Scholar] [CrossRef] [Green Version]
- Oleksiewicz, M.B.; Nagy, G.; Nagy, E. Anti-bacterial monoclonal antibodies: Back to the future? Arch. Biochem. Biophys. 2012, 526, 124–131. [Google Scholar] [CrossRef] [PubMed]
- Lu, Q.; Rouby, J.J.; Laterre, P.F.; Eggimann, P.; Dugard, A.; Giamarellos-Bourboulis, E.J.; Mercier, E.; Garbino, J.; Luyt, C.E.; Chastre, J.; et al. Pharmacokinetics and safety of panobacumab: Specific adjunctive immunotherapy in critical patients with nosocomial Pseudomonas aeruginosa O11 pneumonia. J. Antimicrob. Chemother. 2011, 66, 1110–1116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peck, M.; Rothenberg, M.E.; Deng, R.; Lewin-Koh, N.; She, G.; Kamath, A.V.; Carrasco-Triguero, M.; Saad, O.; Castro, A.; Teufel, L.; et al. A phase 1, randomized, single-ascending-dose study to investigate the safety, tolerability, and pharmacokinetics of DSTA4637S, an anti-Staphylococcus aureus thiomab antibody-antibiotic conjugate, in healthy volunteers. Antimicrob. Agents Chemother. 2019, 63, e02588-18. [Google Scholar] [CrossRef] [Green Version]
- Weems, J.J., Jr.; Steinberg, J.P.; Filler, S.; Baddley, J.W.; Corey, G.R.; Sampathkumar, P.; Winston, L.; John, J.F.; Kubin, C.J.; Talwani, R.; et al. Phase II, randomized, double-blind, multicenter study comparing the safety and pharmacokinetics of tefibazumab to placebo for treatment of Staphylococcus aureus bacteremia. Antimicrob. Agents Chemother. 2006, 50, 2751–2755. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tran, A.C.; Diogo, G.R.; Paul, M.J.; Copland, A.; Hart, P.; Mehta, N.; Irvine, E.B.; Mussá, T.; Drake, P.M.W.; Ivanyi, J.; et al. Mucosal Therapy of Multi-Drug Resistant Tuberculosis With IgA and Interferon-γ. Front. Immunol. 2020, 11, 582833. [Google Scholar] [CrossRef] [PubMed]
- Chalghoumi, R.; Beckers, Y.; Portetelle, D.; Théwis, A. Hen egg yolk antibodies (IgY), production and use for passive immunization against bacterial enteric infections in chicken: A review. Biotechnol. Agron. Soc. Environ. 2009, 13, 295–308. Available online: https://popups.uliege.be/1780-4507/index.php?id=4136 (accessed on 19 October 2022).
- Shimizu, M.; Nagashima, H.; Hashimoto, K.; Suzuki, T. Egg Yolk Antibody (Ig Y) Stability in Aqueous Solution with High Sugar Concentrations. J. Food Sci. 1994, 59, 763–765. [Google Scholar] [CrossRef]
- Klemperer, F. Ueber natürliche Immunität und ihre Verwerthung für die Immunisirungstherapie. Arch. Experiment. Pathol. Pharmakol. 1893, 31, 356–382. [Google Scholar] [CrossRef]
- Rose, M.E.; Orlans, E.; Buttress, N. Immunoglobulin classes in the hen’s egg: Their segregation in yolk and white. Eur. J. Immunol. 1974, 4, 521–523. [Google Scholar] [CrossRef]
- Schade, R.; Hlinak, A. Egg Yolk Antibodies, State of the Art and Future Prospects. ALTEX-Altern. Anim. Exp. 1996, 13, 5–9. Available online: http://www.ncbi.nlm.nih.gov/pubmed/11178463 (accessed on 19 October 2022).
- Akita, E.M.; Nakai, S. Immunoglobulins from Egg Yolk: Isolation and Purification. J. Food Sci. 1992, 57, 629–634. [Google Scholar] [CrossRef]
- Gadde, U.; Rathinam, T.; Lillehoj, H.S. Passive immunization with hyperimmune egg-yolk IgY as prophylaxis and therapy for poultry diseases-A review. Anim. Health Res. Rev. 2015, 16, 163–176. [Google Scholar] [CrossRef]
- Schade, R.; Calzado, E.G.; Sarmiento, R.; Chacana, P.A.; Porankiewicz-Asplund, J.; Terzolo, H.R. Chicken egg yolk antibodies (IgY-technology): A review of progress in production and use in research and human and veterinary medicine. ATLA Altern. Lab. Anim. 2005, 33, 129–154. [Google Scholar] [CrossRef]
- Shimizu, M.; Fitzsimmuns, R.C.; Nakai, S. Anti-E. coli lmmunoglobulin Y Isolated from Egg Yolk of Immunized Chickens as a Potential Food Ingredient. J. Food Sci. 1988, 53, 1360–1368. [Google Scholar] [CrossRef]
- Staak, C.; Schwarzkopf, C.; Behn, I.; Hommel, U.; Hlinak, A.; Schade, R.; Erhard, M. Isolation of IgY from Yolk. In Chicken Egg Yolk Antibodies, Production and Application; Schade, R., Behn, I., Erhard, M., Hlinak, A., Staak, C., Eds.; Springer: Berlin/Heidelberg, Germany, 2001. [Google Scholar] [CrossRef]
- Leiva, C.L.; Gallardo, M.J.; Casanova, N.; Terzolo, H.; Chacana, P. IgY-technology (egg yolk antibodies) in human medicine: A review of patents and clinical trials. Int. Immunopharmacol. 2020, 81, 106269. [Google Scholar] [CrossRef] [PubMed]
- Sesarman, A.; Mihai, S.; Chiriac, M.T.; Olaru, F.; Sitaru, A.G.; Thurman, J.M.; Zillikens, D.; Sitaru, C. Binding of avian IgY to type VII collagen does not activate complement and leucocytes and fails to induce subepidermal blistering in mice. Br. J. Dermatol. 2008, 158, 463–471. [Google Scholar] [CrossRef]
- Abbas, A.T.; El-Kafrawy, S.A.; Sohrab, S.S.; Azhar, E.I.A. IgY antibodies for the immunoprophylaxis and therapy of respiratory infections. Hum. Vaccines Immunother. 2019, 15, 264–275. [Google Scholar] [CrossRef] [Green Version]
- Lee, W.; Atif, A.S.; Tan, S.C.; Leow, C.H. Insights into the chicken IgY with emphasis on the generation and applications of chicken recombinant monoclonal antibodies. J. Immunol. Methods 2017, 447, 71–85. [Google Scholar] [CrossRef]
- Pereira, E.P.V.; van Tilburg, M.F.E.; Florean, O.P.T.; Guedes, M.I.F. Egg yolk antibodies (IgY)and their applications in human and veterinary health: A review. Int. Immunopharmacol. 2019, 73, 293–303. [Google Scholar] [CrossRef]
- Shi, H.; Zhu, J.; Zou, B.; Shi, L.; Du, L.; Long, Y.; Wang, H.; Xu, H.; Zhen, Y.; Sun, L. Effects of specific egg yolk immunoglobulin on pan-drug-resistant Acinetobacter baumannii. Biomed. Pharmacother. 2017, 95, 1734–1742. [Google Scholar] [CrossRef] [PubMed]
- Norouzi, F.; Behrouz, B.; Ranjbar, M.; Ranjbar, M.; Gargari, S.L.M. Immunotherapy with IgY Antibodies toward Outer Membrane Protein F Protects Burned Mice against Pseudomonas aeruginosa Infection. J. Immunol. Res. 2020, 2020, 7840631. [Google Scholar] [CrossRef] [PubMed]
- Henry, K.A.; MacKenzie, C.R. Antigen recognition by single-domain antibodies: Structural latitudes and constraints. In MAbs; Taylor & Francis: Oxford, UK, 2018; Volume 10, pp. 815–826. [Google Scholar] [CrossRef] [Green Version]
- Hudson, P.J.; Souriau, C. Engineered antibodies. Nat. Med. 2003, 9, 129–134. [Google Scholar] [CrossRef] [PubMed]
- Arbabi-Ghahroudi, M. Camelid Single-Domain Antibodies: Promises and Challenges as Lifesaving Treatments. Int. J. Mol. Sci. 2022, 23, 5009. [Google Scholar] [CrossRef] [PubMed]
- Juma, S.N.; Gong, X.; Hu, S.; Lv, Z.; Shao, J.; Liu, L.; Chen, G. Shark new antigen receptor (IgNAR): Structure, characteristics and potential biomedical applications. Cells 2021, 10, 1140. [Google Scholar] [CrossRef]
- De Genst, E.; Saerens, D.; Muyldermans, S.; Conrath, K. Antibody repertoire development in camelids. Dev. Comp. Immunol. 2006, 30, 187–198. [Google Scholar] [CrossRef]
- De Genst, E.; Silence, K.; Decanniere, K.; Conrath, K.; Loris, R.; Kinne, J.; Muyldermans, S.; Wyns, L. Molecular basis for the preferential cleft recognition by dromedary heavy-chain antibodies. Proc. Natl. Acad. Sci. USA 2006, 103, 4586–4591. [Google Scholar] [CrossRef] [Green Version]
- Stanfield, R.L.; Dooley, H.; Verdino, P.; Flajnik, M.F.; Wilson, I.A. Maturation of Shark Single-domain (IgNAR) Antibodies: Evidence for Induced-fit Binding. J. Mol. Biol. 2007, 367, 358–372. [Google Scholar] [CrossRef]
- Kirchhofer, A.; Helma, J.; Schmidthals, K.; Frauer, C.; Cui, S.; Karcher, A.; Pellis, M.; Muyldermans, S.; Casas-Delucchi, C.S.; Cardoso, M.C.; et al. Modulation of protein properties in living cells using nanobodies. Nat. Struct. Mol. Biol. 2010, 17, 133–139. [Google Scholar] [CrossRef]
- Holliger, P.; Hudson, P.J. Engineered antibody fragments and the rise of single domains. Nat. Biotechnol. 2005, 23, 1126–1136. [Google Scholar] [CrossRef]
- Yang, E.Y.; Shah, K. Nanobodies: Next Generation of Cancer Diagnostics and Therapeutics. Front. Oncol. 2020, 10, 1182. [Google Scholar] [CrossRef] [PubMed]
- Arbabi-Ghahroudi, M.; Tanha, J.; MacKenzie, R. Prokaryotic expression of antibodies. Cancer Metastasis Rev. 2005, 24, 501–519. [Google Scholar] [CrossRef]
- Klarenbeek, A.; El Mazouari, K.; Desmyter, A.; Blanchetot, C.; Hultberg, A.; de Jonge, N.; Roovers, R.C.; Cambillau, C.; Spinelli, S.; Del-Favero, J.; et al. Camelid Ig V genes reveal significant human homology not seen in therapeutic target genes, providing for a powerful therapeutic antibody platform. In MAbs; Taylor & Francis: Oxford, UK, 2015; Volume 7, pp. 693–706. [Google Scholar] [CrossRef]
- Kim, D.Y.; Hussack, G.; Kandalaft, H.; Tanha, J. Mutational approaches to improve the biophysical properties of human single-domain antibodies. Biochim. Biophys. Acta-Proteins Proteom. 2014, 1844, 1983–2001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vincke, C.; Loris, R.; Saerens, D.; Martinez-Rodriguez, S.; Muyldermans, S.; Conrath, K. General strategy to humanize a camelid single-domain antibody and identification of a universal humanized nanobody scaffold. J. Biol. Chem. 2009, 284, 3273–3284. [Google Scholar] [CrossRef] [Green Version]
- Conrath, K.; Vincke, C.; Stijlemans, B.; Schymkowitz, J.; Decanniere, K.; Wyns, L.; Muyldermans, S.; Loris, R. Antigen binding and solubility effects upon the veneering of a camel VHH in framework-2 to mimic a VH. J. Mol. Biol. 2005, 350, 112–125. [Google Scholar] [CrossRef] [PubMed]
- Wells, J.M.; Mercenier, A. Mucosal delivery of therapeutic and prophylactic molecules using lactic acid bacteria. Nat. Rev. Microbiol. 2008, 6, 349–362. [Google Scholar] [CrossRef]
- Andersen, K.K.; Strokappe, N.M.; Hultberg, A.; Truusalu, K.; Smidt, I.; Mikelsaar, R.H.; Mikelsaar, M.; Verrips, T.; Hammarström, L.; Marcotte, H. Neutralization of Clostridium difficile toxin B mediated by engineered lactobacilli that produce single-domain antibodies. Infect. Immun. 2016, 84, 395–406. [Google Scholar] [CrossRef] [Green Version]
- Scully, M.; Cataland, S.R.; Peyvandi, F.; Coppo, P.; Knöbl, P.; Kremer Hovinga, J.A.; Metjian, A.; de la Rubia, J.; Pavenski, K.; Callewaert, F.; et al. Caplacizumab Treatment for Acquired Thrombotic Thrombocytopenic Purpura. N. Engl. J. Med. 2019, 380, 335–346. [Google Scholar] [CrossRef]
- Wang, J.; Kang, G.; Yuan, H.; Cao, X.; Huang, H.; de Marco, A. Research Progress and Applications of Multivalent, Multispecific and Modified Nanobodies for Disease Treatment. Front. Immunol. 2022, 12, 838082. [Google Scholar] [CrossRef]
- Yang, Z.; Schmidt, D.; Liu, W.; Li, S.; Shi, L.; Sheng, J.; Chen, K.; Yu, H.; Tremblay, J.M.; Chen, X.; et al. A novel multivalent, single-domain antibody targeting TcdA and TcdB prevents fulminant Clostridium difficile infection in mice. J. Infect. Dis. 2014, 210, 964–972. [Google Scholar] [CrossRef] [Green Version]
- Babcock, G.J.; Broering, T.J.; Hernandez, H.J.; Mandell, R.B.; Donahue, K.; Boatright, N.; Stack, A.M.; Lowy, I.; Graziano, R.; Molrine, D.; et al. Human monoclonal antibodies directed against toxins A and B prevent Clostridium difficile-induced mortality in hamsters. Infect. Immun. 2006, 74, 6339–6347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Albanese, A.; Sacerdoti, F.; Seyahian, E.A.; Amaral, M.M.; Fiorentino, G.; Fernandez Brando, R.; Vilte, D.A.; Mercado, E.C.; Palermo, M.S.; Cataldi, A.; et al. Immunization of pregnant cows with Shiga toxin-2 induces high levels of specific colostral antibodies and lactoferrin able to neutralize E. coli O157:H7 pathogenicity. Vaccine 2018, 36, 1728–1735. [Google Scholar] [CrossRef] [PubMed]
- Garimano, N.; Diaz Vergara, L.I.; Kim, A.D.; Badin, E.E.; Sodero, S.; Bernal, A.M.; Gonzalez, D.D.; Amaral, M.M.; Lespinard, A.R.; Porporatto, C.; et al. Preservation of protective capacity of hyperimmune anti-Stx2 bovine colostrum against enterohemorrhagic Escherichia coli O157:H7 pathogenicity after pasteurization and spray-drying processes. J. Dairy Sci. 2021, 104, 5229–5238. [Google Scholar] [CrossRef] [PubMed]
- Smith, R.; Coast, J. The true cost of antimicrobial resistance. BMJ 2013, 346, f1493. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Bacteria | Antibody Format | Immunogen | Clinical Trial | Results | References |
|---|---|---|---|---|---|
| Gram-positive bacteria | |||||
| C. difficile | Polyclonal | Anti-toxin B goat antibodies | Preclinical | Hamsters challenged with toxin B showed survival rates of 98% | [106] |
| Monoclonal | Bexlotoxumab (Enterotoxin B) | FDA approved | Approved for reduction of recurrence of infection | [52] | |
| IgY | IM-01 (Toxin A and B and spore preparation) | Phase II (NCT04121169) Randomized, parallel assignment, open-label trial | Clinical improvement and no relapse of infection | [79] | |
| Single-domain antibodies | Tetravalent and bispecific tandem linked molecule of four VhH against toxin A and B | Preclinical | Protect mice from a lethal systemic challenge of a mixture of both toxins and reverse infection in mice | [105] | |
| Single-domain antibodies | 4 VhHs from lamas against toxin B expressed on the surface of Lactobacillus | Preclinical | Delayed death of the hamsters challenged | [102] | |
| S. aureus | Polyclonal | Altastaph (capsular polysaccharides) | Phase II (NCT00063089) double-blind, placebo-controlled trial | Not powered to show efficacy, safety profile suggests that Altastaph may be an effective adjunct to antibiotics | [42] |
| Polyclonal | Veronate® (surface adhesin) | Phase III trial (NCT00113191) double-blind, comparing the safety and efficacy versus placebo | Exhibited no effect in reduction of S. aureus prevention of late-onset sepsis in very low birth weight infants | [43] | |
| Monoclonal | DSTA4637S (human anti-S. aureus IgG1 allied with a novel rifamycin-class antibiotic) | Phase I (NCT02596399) randomized, double- blind, placebo-controlled, single-ascending-dose | Safety and pharmacokinetic profile favorable for development of new therapeutic | [66] | |
| Monoclonal | Tefibazumab (surface-expressed adhesion protein clumping factor A) | PhaseII (NCT00198302) randomized, double-blind, placebo-controlled clinical trial with the objective of S. aureus bacteremia treatment. | Well tolerated, with safety profile similar to other monoclonal antibodies. Further trials are necessary for dose range and efficacy | [67] | |
| B. anthracis | Monoclonal | Raxibacumab (Toxin) | FDA approved | Approved for treatment of anthrax inhalation as a result of B. anthraci. | [50] |
| Monoclonal | Obiltoxaximab (Toxin) | FDA approved | Approved for treatment of anthrax inhalation as a result of B. anthraci. | [51] | |
| M. tuberculosis | Monoclonal | Human monoclonal IgA 2E9 and Interferon-γ | Preclinical | Reduction of 50-fold of lung bacterial load when applied at the time of infection | [68] |
| Gram-negative bacteria | |||||
| E. coli | Polyclonal | Hyperimmune anti-Stx2 bovine colostrum | Preclinical | Prevention of 100% of the lethality caused by E. coli O157:H7 in a weaned mice model | [107,108] |
| B. cenocepacia | Polyclonal | Goat anti-OmpA-like protein | Preclinical | In vitro greatly impairs the ability to adhere and invade human epithelial cells | [41] |
| P. aeruginosa | Monoclonal | MAb 166 (Murine monoclonal antibody to PcrV) | Preclinical | Protective when intraperitoneally transferred to mice | [59] |
| Monoclonal | KB001-A (anti-PcrV PEGylated mouse Mab | Phase II (NCT01695343) double-blind, placebo-controlled trial | Prevents ventilator-associated pneumonia, the efficacy was low in patients suffering from CF | [53,60] | |
| Monoclonal | Panobacumab (IgM targeting the O-antigen of serotype O11) | Phase II (NCT00851435) safety and PK in patients with hospital acquired pneumonia | Improve clinical outcome in a shorter time. | [65] | |
| IgY | PsAer-IgY (anti-pseudomonas antibodies) | Phase III (NCT01455675) randomized, parallel assignment, double-blind trial | Good toleration profile, lacked a clear demonstration of a therapeutic benefit in CF patients | [79] | |
| IgY | Anti-OprF antibodies | Preclinical | Increased survival rates, in burned mice infected with the bacteria | [85] | |
| A. baumannii | IgY | Specific anti-A. baumannii antibodies | Preclinical | Reduced mortality in BALB/c mice with an induced acute pneumonia after intraperitoneal injection with specific IgYs | [84] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Seixas, A.M.M.; Sousa, S.A.; Leitão, J.H. Antibody-Based Immunotherapies as a Tool for Tackling Multidrug-Resistant Bacterial Infections. Vaccines 2022, 10, 1789. https://doi.org/10.3390/vaccines10111789
Seixas AMM, Sousa SA, Leitão JH. Antibody-Based Immunotherapies as a Tool for Tackling Multidrug-Resistant Bacterial Infections. Vaccines. 2022; 10(11):1789. https://doi.org/10.3390/vaccines10111789
Chicago/Turabian StyleSeixas, António M. M., Sílvia A. Sousa, and Jorge H. Leitão. 2022. "Antibody-Based Immunotherapies as a Tool for Tackling Multidrug-Resistant Bacterial Infections" Vaccines 10, no. 11: 1789. https://doi.org/10.3390/vaccines10111789
APA StyleSeixas, A. M. M., Sousa, S. A., & Leitão, J. H. (2022). Antibody-Based Immunotherapies as a Tool for Tackling Multidrug-Resistant Bacterial Infections. Vaccines, 10(11), 1789. https://doi.org/10.3390/vaccines10111789