
Crossroads of Drug Abuse and HIV Infection: Neurotoxicity and CNS Reservoir


 and
and 
Abstract
1. Introduction
2. HIV Epidemic among People Who Abuse Drugs
3. Current Perspectives on HIV Infection in PWUDs
4. The Role of ART on CNS (Dys)Function
5. Mechanisms of HIV-Dependent Neurodegeneration
5.1. CNS as HIV Reservoir
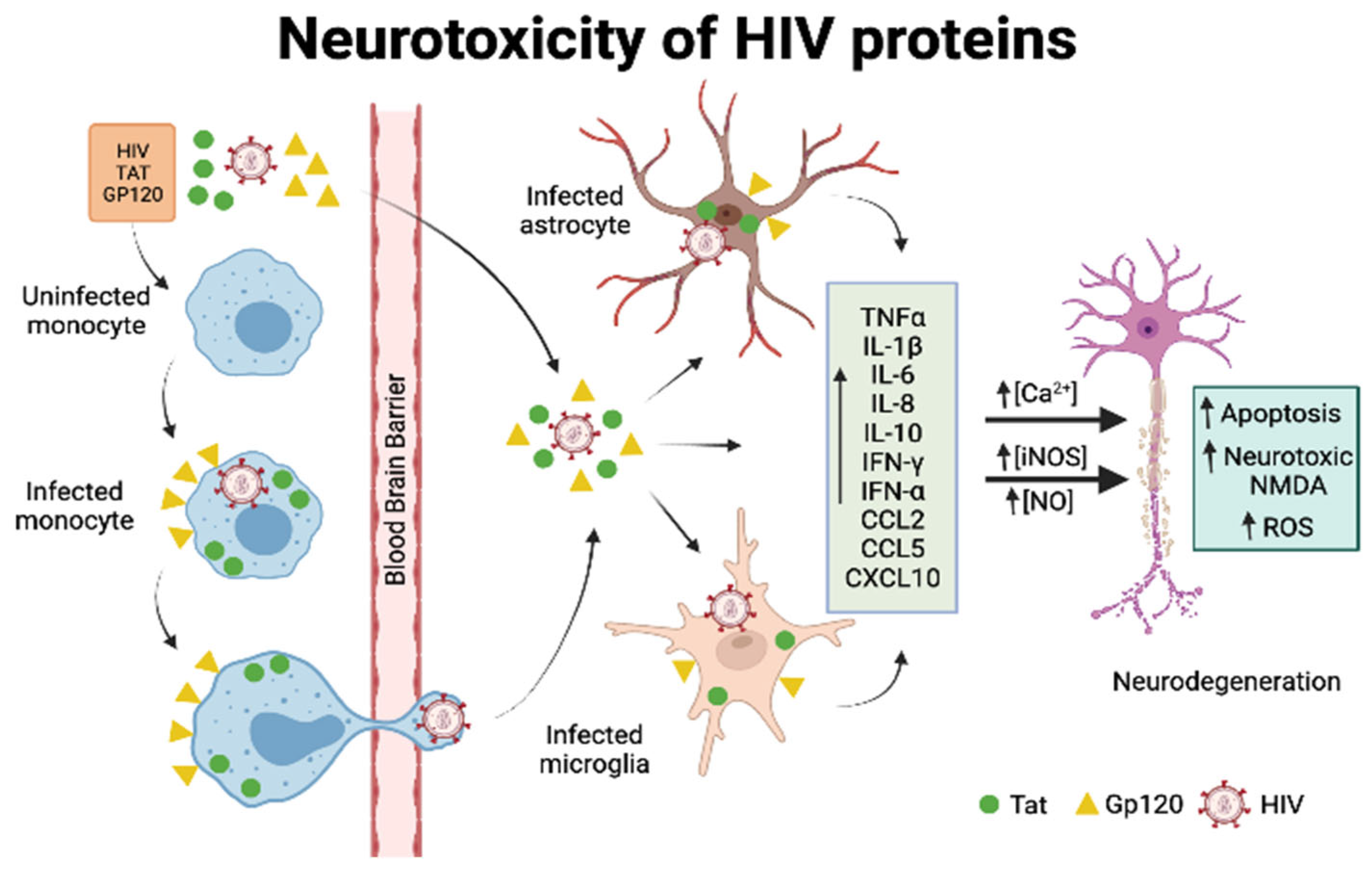
5.2. General Mechanisms of HIV-Mediated Neurotoxicity
5.3. Neurotoxicity of Viral Proteins gp120 and Tat
5.3.1. Gp120
5.3.2. Tat
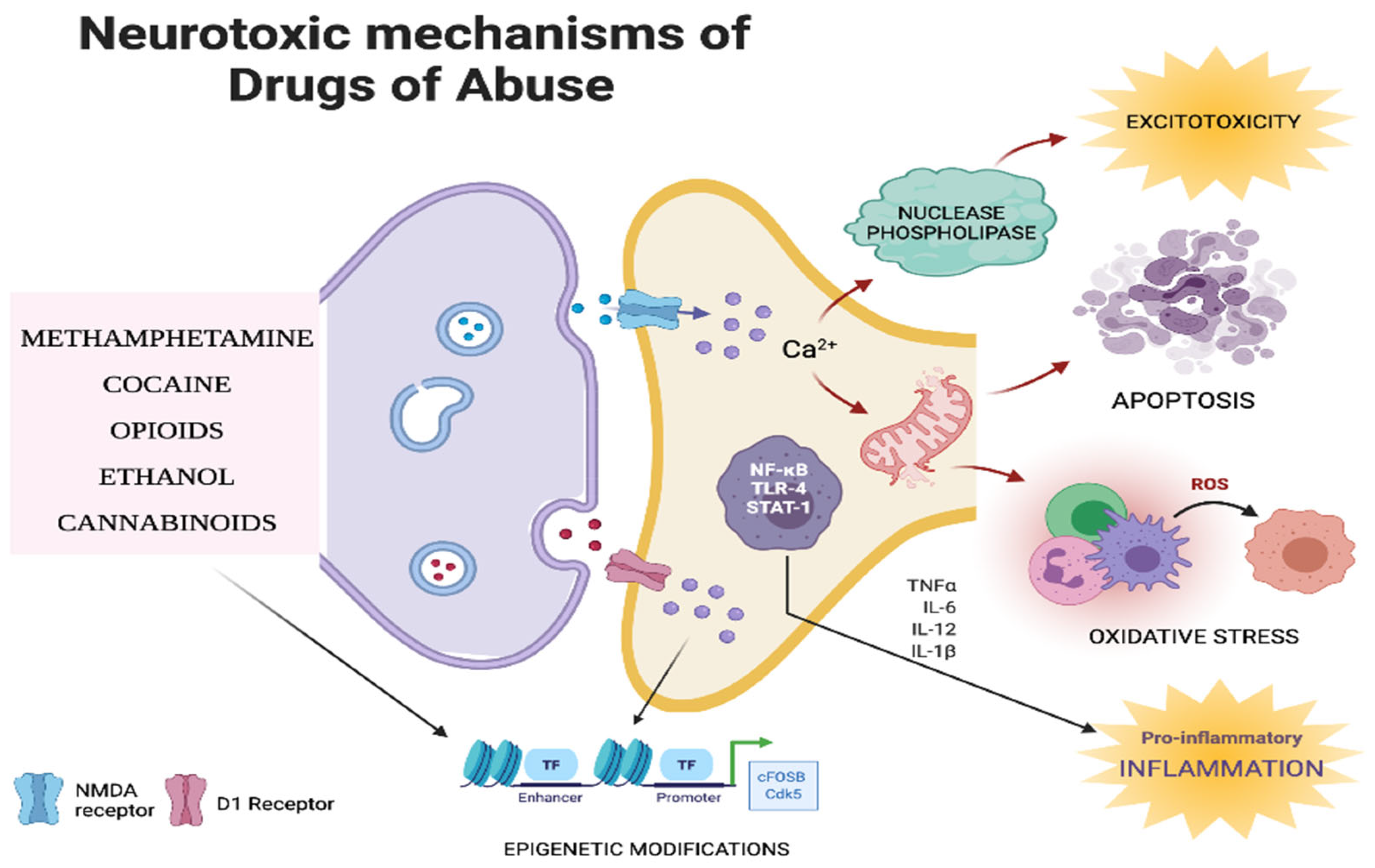
6. Neurotoxic Mechanisms of Drugs of Abuse
6.1. Inflammation
6.2. Oxidative Stress
6.3. Apoptosis
6.4. Excitotoxicity
6.5. Epigenetic Mechanisms
6.6. Other Biochemical Mechanisms
7. Effect of Major Drugs of Abuse on HIV-Dependent Neurodegeneration
7.1. Opioids
7.2. Cannabinoids
7.3. Cocaine
7.4. Methamphetamine
7.5. Ethanol
8. ART, Drugs of Abuse, and Hand
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- UNAIDS. Available online: https://www.unaids.org/en/resources/documents/2019/2019-UNAIDS-data (accessed on 10 June 2021).
- Tyagi, M.; Romerio, F. Models of HIV-1 persistence in the CD4+ T cell compartment: Past, present and future. Curr. HIV Res. 2011, 9, 579–587. [Google Scholar] [CrossRef]
- Tyagi, M.; Bukrinsky, M. Human immunodeficiency virus (HIV) latency: The major hurdle in HIV eradication. Mol. Med. 2012, 18, 1096–1108. [Google Scholar] [CrossRef]
- Hokello, J.; Sharma, A.L.; Dimri, M.; Tyagi, M. Insights into the HIV Latency and the Role of Cytokines. Pathogens 2019, 8, 137. [Google Scholar] [CrossRef]
- Sharma, A.L.; Hokello, J.; Sonti, S.; Zicari, S.; Sun, L.; Alqatawni, A.; Bukrinsky, M.; Simon, G.; Chauhan, A.; Daniel, R.; et al. CBF-1 Promotes the Establishment and Maintenance of HIV Latency by Recruiting Polycomb Repressive Complexes, PRC1 and PRC2, at HIV LTR. Viruses 2020, 12, 1040. [Google Scholar] [CrossRef]
- Zicari, S.; Sharma, A.L.; Sahu, G.; Dubrovsky, L.; Sun, L.; Yue, H.; Jada, T.; Ochem, A.; Simon, G.; Bukrinsky, M.; et al. DNA dependent protein kinase (DNA-PK) enhances HIV transcription by promoting RNA polymerase II activity and recruitment of transcription machinery at HIV LTR. Oncotarget 2020, 11, 699–726. [Google Scholar] [CrossRef]
- Alqatawni, A.; Sharma, A.L.; Attilus, B.; Tyagi, M.; Daniel, R. Shedding Light on the Role of Extracellular Vesicles in HIV Infection and Wound Healing. Viruses 2020, 12, 584. [Google Scholar] [CrossRef]
- Pomerantz, R.J. Reservoirs, sanctuaries, and residual disease: The hiding spots of HIV-1. HIV Clin. Trials 2003, 4, 137–143. [Google Scholar] [CrossRef]
- Sonti, S.; Sharma, A.L.; Tyagi, M. HIV-1 persistence in the CNS: Mechanisms of latency, pathogenesis and an update on eradication strategies. Virus Res. 2021, 303, 198523. [Google Scholar] [CrossRef]
- Tyagi, M.; Iordanskiy, S.; Ammosova, T.; Kumari, N.; Smith, K.; Breuer, D.; Ilatovskiy, A.V.; Kont, Y.S.; Ivanov, A.; Uren, A.; et al. Reactivation of latent HIV-1 provirus via targeting protein phosphatase-1. Retrovirology 2015, 12, 63. [Google Scholar] [CrossRef]
- Hokello, J.; Sharma, A.L.; Tyagi, M. Efficient Non-Epigenetic Activation of HIV Latency through the T-Cell Receptor Signalosome. Viruses 2020, 12, 868. [Google Scholar] [CrossRef]
- Hokello, J.; Lakhikumar Sharma, A.; Tyagi, M. AP-1 and NF-kappaB synergize to transcriptionally activate latent HIV upon T-cell receptor activation. FEBS Lett. 2021, 595, 577–594. [Google Scholar] [CrossRef] [PubMed]
- Hokello, J.; Sharma, A.L.; Tyagi, M. Combinatorial Use of Both Epigenetic and Non-Epigenetic Mechanisms to Efficiently Reactivate HIV Latency. Int. J. Mol. Sci. 2021, 22, 3697. [Google Scholar] [CrossRef] [PubMed]
- Proust, A.; Barat, C.; Leboeuf, M.; Drouin, J.; Gagnon, M.T.; Vanasse, F.; Tremblay, M.J. HIV-1 infection and latency-reversing agents bryostatin-1 and JQ1 disrupt amyloid beta homeostasis in human astrocytes. Glia 2020, 68, 2212–2227. [Google Scholar] [CrossRef] [PubMed]
- Lamers, S.L.; Salemi, M.; Galligan, D.C.; Morris, A.; Gray, R.; Fogel, G.; Zhao, L.; McGrath, M.S. Human immunodeficiency virus-1 evolutionary patterns associated with pathogenic processes in the brain. J. Neurovirol. 2010, 16, 230–241. [Google Scholar] [CrossRef]
- Borrajo Lopez, A.; Penedo, M.A.; Rivera-Baltanas, T.; Perez-Rodriguez, D.; Alonso-Crespo, D.; Fernandez-Pereira, C.; Olivares, J.M.; Agis-Balboa, R.C. Microglia: The Real Foe in HIV-1-Associated Neurocognitive Disorders? Biomedicines 2021, 9, 925. [Google Scholar] [CrossRef]
- Bougea, A.; Spantideas, N.; Galanis, P.; Gkekas, G.; Thomaides, T. Optimal treatment of HIV-associated neurocognitive disorders: Myths and reality. A critical review. Ther. Adv. Infect. Dis. 2019, 6, 2049936119838228. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, M.; Lu, Q.; Farrell, M.; Lappin, J.M.; Shi, J.; Lu, L.; Bao, Y. Global prevalence and burden of HIV-associated neurocognitive disorder: A meta-analysis. Neurology 2020, 95, e2610–e2621. [Google Scholar] [CrossRef]
- Underwood, J.; Robertson, K.R.; Winston, A. Could antiretroviral neurotoxicity play a role in the pathogenesis of cognitive impairment in treated HIV disease? AIDS 2015, 29, 253–261. [Google Scholar] [CrossRef]
- Hokello, J.; Sharma, A.L.; Tyagi, P.; Bhushan, A.; Tyagi, M. Human Immunodeficiency Virus Type-1 (HIV-1) Transcriptional Regulation, Latency and Therapy in the Central Nervous System. Vaccines 2021, 9, 1272. [Google Scholar] [CrossRef]
- Hokello, J.; Sharma, A.L.; Tyagi, M. An Update on the HIV DNA Vaccine Strategy. Vaccines 2021, 9, 605. [Google Scholar] [CrossRef]
- Sil, S.; Niu, F.; Chivero, E.T.; Singh, S.; Periyasamy, P.; Buch, S. Role of Inflammasomes in HIV-1 and Drug Abuse Mediated Neuroinflammaging. Cells 2020, 9, 1857. [Google Scholar] [CrossRef] [PubMed]
- Sahu, G.; Farley, K.; El-Hage, N.; Aiamkitsumrit, B.; Fassnacht, R.; Kashanchi, F.; Ochem, A.; Simon, G.L.; Karn, J.; Hauser, K.F.; et al. Cocaine promotes both initiation and elongation phase of HIV-1 transcription by activating NF-kappaB and MSK1 and inducing selective epigenetic modifications at HIV-1 LTR. Virology 2015, 483, 185–202. [Google Scholar] [CrossRef] [PubMed]
- Shapshak, P.; Duncan, R.; Nath, A.; Turchan, J.; Pandjassarame, K.; Rodriguez, H.; Duran, E.M.; Ziegler, F.; Amaro, E.; Lewis, A.; et al. Gene chromosomal organization and expression in cultured human neurons exposed to cocaine and HIV-1 proteins gp120 and tat: Drug abuse and NeuroAIDS. Front. Biosci. 2006, 11, 1774–1793. [Google Scholar] [CrossRef][Green Version]
- Tyagi, M.; Bukrinsky, M.; Simon, G.L. Mechanisms of HIV Transcriptional Regulation by Drugs of Abuse. Curr. HIV Res. 2016, 14, 442–454. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.I.; Reza, M.M.; Crowe, S.M.; Rahman, M.; Hellard, M.; Sarker, M.S.; Chowdhury, E.I.; Rana, A.; Sacks-Davis, R.; Banu, S.; et al. People who inject drugs in Bangladesh—The untold burden! Int. J. Infect. Dis. 2019, 83, 109–115. [Google Scholar] [CrossRef] [PubMed]
- Kostaki, E.; Magiorkinis, G.; Psichogiou, M.; Flampouris, A.; Iliopoulos, P.; Papachristou, E.; Daikos, G.L.; Bonovas, S.; Otelea, D.; Friedman, S.R.; et al. Detailed Molecular Surveillance of the HIV-1 Outbreak Among People who Inject Drugs (PWID) in Athens During a Period of Four Years. Curr. HIV Res. 2017, 15, 396–404. [Google Scholar] [CrossRef] [PubMed]
- Meacham, M.C.; Rudolph, A.E.; Strathdee, S.A.; Rusch, M.L.; Brouwer, K.C.; Patterson, T.L.; Vera, A.; Rangel, G.; Roesch, S.C. Polydrug Use and HIV Risk Among People Who Inject Heroin in Tijuana, Mexico: A Latent Class Analysis. Subst. Use Misuse 2015, 50, 1351–1359. [Google Scholar] [CrossRef]
- Niculescu, I.; Paraschiv, S.; Paraskevis, D.; Abagiu, A.; Batan, I.; Banica, L.; Otelea, D. Recent HIV-1 Outbreak Among Intravenous Drug Users in Romania: Evidence for Cocirculation of CRF14_BG and Subtype F1 Strains. AIDS Res. Hum. Retrovir. 2015, 31, 488–495. [Google Scholar] [CrossRef]
- Degenhardt, L.; Peacock, A.; Colledge, S.; Leung, J.; Grebely, J.; Vickerman, P.; Stone, J.; Cunningham, E.B.; Trickey, A.; Dumchev, K.; et al. Global prevalence of injecting drug use and sociodemographic characteristics and prevalence of HIV, HBV, and HCV in people who inject drugs: A multistage systematic review. Lancet Glob. Health 2017, 5, e1192–e1207. [Google Scholar] [CrossRef]
- CDC. Available online: https://www.cdc.gov/hiv/pdf/library/reports/surveillance/cdc-hiv-surveillance-report-2015-vol-27.pdf (accessed on 26 June 2021).
- Des Jarlais, D.C.; Kerr, T.; Carrieri, P.; Feelemyer, J.; Arasteh, K. HIV infection among persons who inject drugs: Ending old epidemics and addressing new outbreaks. AIDS 2016, 30, 815–826. [Google Scholar] [CrossRef]
- Booth, R.E.; Watters, J.K.; Chitwood, D.D. HIV risk-related sex behaviors among injection drug users, crack smokers, and injection drug users who smoke crack. Am. J. Public Health 1993, 83, 1144–1148. [Google Scholar] [CrossRef] [PubMed]
- Pechansky, F.; Woody, G.; Inciardi, J.; Surratt, H.; Kessler, F.; Von Diemen, L.; Bumaguin, D.B. HIV seroprevalence among drug users: An analysis of selected variables based on 10 years of data collection in Porto Alegre, Brazil. Drug Alcohol Depend. 2006, 82 (Suppl. 1), S109–S113. [Google Scholar] [CrossRef]
- Webber, M.P.; Schoenbaum, E.E.; Gourevitch, M.N.; Buono, D.; Klein, R.S. A prospective study of HIV disease progression in female and male drug users. AIDS 1999, 13, 257–262. [Google Scholar] [CrossRef] [PubMed]
- Siddiqui, N.S.; Brown, L.S., Jr.; Makuch, R.W. Short-term declines in CD4 levels associated with cocaine use in HIV-1 seropositive, minority injecting drug users. J. Natl. Med. Assoc. 1993, 85, 293–296. [Google Scholar] [PubMed]
- Arendt, V.; Guillorit, L.; Origer, A.; Sauvageot, N.; Vaillant, M.; Fischer, A.; Goedertz, H.; Francois, J.H.; Alexiev, I.; Staub, T.; et al. Injection of cocaine is associated with a recent HIV outbreak in people who inject drugs in Luxembourg. PLoS ONE 2019, 14, e0215570. [Google Scholar] [CrossRef]
- Levine, A.J.; Reynolds, S.; Cox, C.; Miller, E.N.; Sinsheimer, J.S.; Becker, J.T.; Martin, E.; Sacktor, N.; Ned Sacktor for the Neuropsychology Working Group of the Multicenter AIDS Cohort Study. The longitudinal and interactive effects of HIV status, stimulant use, and host genotype upon neurocognitive functioning. J. Neurovirol. 2014, 20, 243–257. [Google Scholar] [CrossRef]
- Holtz, T.H.; Pattanasin, S.; Chonwattana, W.; Tongtoyai, J.; Chaikummao, S.; Varangrat, A.; Mock, P.A. Longitudinal analysis of key HIV-risk behavior patterns and predictors in men who have sex with men, Bangkok, Thailand. Arch. Sex. Behav. 2015, 44, 341–348. [Google Scholar] [CrossRef]
- Rosinska, M.; Sieroslawski, J.; Wiessing, L. High regional variability of HIV, HCV and injecting risks among people who inject drugs in Poland: Comparing a cross-sectional bio-behavioural study with case-based surveillance. BMC Infect. Dis. 2015, 15, 83. [Google Scholar] [CrossRef]
- Chang, S.L.; Connaghan, K.P.; Wei, Y.; Li, M.D. NeuroHIV and use of addictive substances. Int. Rev. Neurobiol. 2014, 118, 403–440. [Google Scholar] [CrossRef]
- Huang, Y.F.; Yang, J.Y.; Nelson, K.E.; Kuo, H.S.; Lew-Ting, C.Y.; Yang, C.H.; Chen, C.H.; Chang, F.Y.; Liu, H.R. Changes in HIV incidence among people who inject drugs in Taiwan following introduction of a harm reduction program: A study of two cohorts. PLoS Med. 2014, 11, e1001625. [Google Scholar] [CrossRef]
- Shiau, S.; Arpadi, S.M.; Yin, M.T.; Martins, S.S. Patterns of drug use and HIV infection among adults in a nationally representative sample. Addict. Behav. 2017, 68, 39–44. [Google Scholar] [CrossRef] [PubMed]
- Hartzler, B.; Dombrowski, J.C.; Crane, H.M.; Eron, J.J.; Geng, E.H.; Christopher Mathews, W.; Mayer, K.H.; Moore, R.D.; Mugavero, M.J.; Napravnik, S.; et al. Prevalence and Predictors of Substance Use Disorders Among HIV Care Enrollees in the United States. AIDS Behav. 2017, 21, 1138–1148. [Google Scholar] [CrossRef]
- Duko, B.; Ayalew, M.; Ayano, G. The prevalence of alcohol use disorders among people living with HIV/AIDS: A systematic review and meta-analysis. Subst. Abuse Treat. Prev. Policy 2019, 14, 52. [Google Scholar] [CrossRef]
- Price, R.W.; Spudich, S. Antiretroviral therapy and central nervous system HIV type 1 infection. J. Infect. Dis. 2008, 197 (Suppl. 3), S294–S306. [Google Scholar] [CrossRef] [PubMed]
- Lanman, T.; Letendre, S.; Ma, Q.; Bang, A.; Ellis, R. CNS Neurotoxicity of Antiretrovirals. J. Neuroimmune Pharmacol. 2021, 16, 130–143. [Google Scholar] [CrossRef] [PubMed]
- Kohler, J.J.; Lewis, W. A brief overview of mechanisms of mitochondrial toxicity from NRTIs. Environ. Mol. Mutagen 2007, 48, 166–172. [Google Scholar] [CrossRef]
- Nooka, S.; Ghorpade, A. Organellar stress intersects the astrocyte endoplasmic reticulum, mitochondria and nucleolus in HIV associated neurodegeneration. Cell Death Dis. 2018, 9, 317. [Google Scholar] [CrossRef]
- Hung, K.M.; Chen, P.C.; Hsieh, H.C.; Calkins, M.J. Mitochondrial defects arise from nucleoside/nucleotide reverse transcriptase inhibitors in neurons: Potential contribution to HIV-associated neurocognitive disorders. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 406–413. [Google Scholar] [CrossRef]
- Blas-Garcia, A.; Polo, M.; Alegre, F.; Funes, H.A.; Martinez, E.; Apostolova, N.; Esplugues, J.V. Lack of mitochondrial toxicity of darunavir, raltegravir and rilpivirine in neurons and hepatocytes: A comparison with efavirenz. J. Antimicrob. Chemother. 2014, 69, 2995–3000. [Google Scholar] [CrossRef]
- Ciavatta, V.T.; Bichler, E.K.; Speigel, I.A.; Elder, C.C.; Teng, S.L.; Tyor, W.R.; Garcia, P.S. In vitro and Ex vivo Neurotoxic Effects of Efavirenz are Greater than Those of Other Common Antiretrovirals. Neurochem. Res. 2017, 42, 3220–3232. [Google Scholar] [CrossRef]
- Lv, Z.; Chu, Y.; Wang, Y. HIV protease inhibitors: A review of molecular selectivity and toxicity. HIV AIDS (Auckl.) 2015, 7, 95–104. [Google Scholar] [CrossRef] [PubMed]
- Patel, P.; Louie, S. Drug Interactions in HIV: Protease and Integrase Inhibitors. In Drug Interactions in Infectious Diseases: Antimicrobial Drug Interactions; Pai, M., Kiser, J., Gubbins, P., Rodvold, K., Eds.; Humana Press: Cham, Switzerland, 2018. [Google Scholar]
- Gray, J.; Young, B. Acute onset insomnia associated with the initiation of raltegravir: A report of two cases and literature review. AIDS Patient Care STDS 2009, 23, 689–690. [Google Scholar] [CrossRef] [PubMed]
- Ellis, R.J.; Deutsch, R.; Heaton, R.K.; Marcotte, T.D.; McCutchan, J.A.; Nelson, J.A.; Abramson, I.; Thal, L.J.; Atkinson, J.H.; Wallace, M.R.; et al. Neurocognitive impairment is an independent risk factor for death in HIV infection. San Diego HIV Neurobehavioral Research Center Group. Arch. Neurol. 1997, 54, 416–424. [Google Scholar] [CrossRef] [PubMed]
- Heaton, R.K.; Franklin, D.R.; Ellis, R.J.; McCutchan, J.A.; Letendre, S.L.; Leblanc, S.; Corkran, S.H.; Duarte, N.A.; Clifford, D.B.; Woods, S.P.; et al. HIV-associated neurocognitive disorders before and during the era of combination antiretroviral therapy: Differences in rates, nature, and predictors. J. Neurovirol. 2011, 17, 3–16. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Scarano, F.; Martin-Garcia, J. The neuropathogenesis of AIDS. Nat. Rev. Immunol. 2005, 5, 69–81. [Google Scholar] [CrossRef]
- Ketzler, S.; Weis, S.; Haug, H.; Budka, H. Loss of neurons in the frontal cortex in AIDS brains. Acta Neuropathol. 1990, 80, 92–94. [Google Scholar] [CrossRef] [PubMed]
- Reyes, M.G.; Faraldi, F.; Senseng, C.S.; Flowers, C.; Fariello, R. Nigral degeneration in acquired immune deficiency syndrome (AIDS). Acta Neuropathol. 1991, 82, 39–44. [Google Scholar] [CrossRef]
- Graus, F.; Ribalta, T.; Abos, J.; Alom, J.; Cruz-Sanchez, F.; Mallolas, J.; Miro, J.M.; Cardesa, A.; Tolosa, E. Subacute cerebellar syndrome as the first manifestation of AIDS dementia complex. Acta Neurol. Scand. 1990, 81, 118–120. [Google Scholar] [CrossRef]
- Sanchez, A.B.; Kaul, M. Neuronal Stress and Injury Caused by HIV-1, cART and Drug Abuse: Converging Contributions to HAND. Brain Sci. 2017, 7, 25. [Google Scholar] [CrossRef]
- Eisele, E.; Siliciano, R.F. Redefining the viral reservoirs that prevent HIV-1 eradication. Immunity 2012, 37, 377–388. [Google Scholar] [CrossRef]
- Gray, L.R.; Roche, M.; Flynn, J.K.; Wesselingh, S.L.; Gorry, P.R.; Churchill, M.J. Is the central nervous system a reservoir of HIV-1? Curr. Opin. HIV AIDS 2014, 9, 552–558. [Google Scholar] [CrossRef] [PubMed]
- Cosenza, M.A.; Zhao, M.L.; Si, Q.; Lee, S.C. Human brain parenchymal microglia express CD14 and CD45 and are productively infected by HIV-1 in HIV-1 encephalitis. Brain Pathol. 2002, 12, 442–455. [Google Scholar] [CrossRef] [PubMed]
- Churchill, M.J.; Gorry, P.R.; Cowley, D.; Lal, L.; Sonza, S.; Purcell, D.F.; Thompson, K.A.; Gabuzda, D.; McArthur, J.C.; Pardo, C.A.; et al. Use of laser capture microdissection to detect integrated HIV-1 DNA in macrophages and astrocytes from autopsy brain tissues. J. Neurovirol. 2006, 12, 146–152. [Google Scholar] [CrossRef] [PubMed]
- Churchill, M.J.; Wesselingh, S.L.; Cowley, D.; Pardo, C.A.; McArthur, J.C.; Brew, B.J.; Gorry, P.R. Extensive astrocyte infection is prominent in human immunodeficiency virus-associated dementia. Ann. Neurol. 2009, 66, 253–258. [Google Scholar] [CrossRef] [PubMed]
- Gianella, S.; Kosakovsky Pond, S.L.; Oliveira, M.F.; Scheffler, K.; Strain, M.C.; De la Torre, A.; Letendre, S.; Smith, D.M.; Ellis, R.J. Compartmentalized HIV rebound in the central nervous system after interruption of antiretroviral therapy. Virus Evol. 2016, 2, vew020. [Google Scholar] [CrossRef]
- Dahl, V.; Gisslen, M.; Hagberg, L.; Peterson, J.; Shao, W.; Spudich, S.; Price, R.W.; Palmer, S. An example of genetically distinct HIV type 1 variants in cerebrospinal fluid and plasma during suppressive therapy. J. Infect. Dis. 2014, 209, 1618–1622. [Google Scholar] [CrossRef]
- Sturdevant, C.B.; Dow, A.; Jabara, C.B.; Joseph, S.B.; Schnell, G.; Takamune, N.; Mallewa, M.; Heyderman, R.S.; Van Rie, A.; Swanstrom, R. Central nervous system compartmentalization of HIV-1 subtype C variants early and late in infection in young children. PLoS Pathog. 2012, 8, e1003094. [Google Scholar] [CrossRef]
- Meucci, O.; Fatatis, A.; Simen, A.A.; Bushell, T.J.; Gray, P.W.; Miller, R.J. Chemokines regulate hippocampal neuronal signaling and gp120 neurotoxicity. Proc. Natl. Acad. Sci. USA 1998, 95, 14500–14505. [Google Scholar] [CrossRef]
- Piller, S.C.; Jans, P.; Gage, P.W.; Jans, D.A. Extracellular HIV-1 virus protein R causes a large inward current and cell death in cultured hippocampal neurons: Implications for AIDS pathology. Proc. Natl. Acad. Sci. USA 1998, 95, 4595–4600. [Google Scholar] [CrossRef]
- Mattson, M.P.; Haughey, N.J.; Nath, A. Cell death in HIV dementia. Cell Death Differ. 2005, 12 (Suppl. 1), 893–904. [Google Scholar] [CrossRef]
- Lannuzel, A.; Lledo, P.M.; Lamghitnia, H.O.; Vincent, J.D.; Tardieu, M. HIV-1 envelope proteins gp120 and gp160 potentiate NMDA-induced [Ca2+]i increase, alter [Ca2+]i homeostasis and induce neurotoxicity in human embryonic neurons. Eur. J. Neurosci. 1995, 7, 2285–2293. [Google Scholar] [CrossRef] [PubMed]
- Garden, G.A.; Guo, W.; Jayadev, S.; Tun, C.; Balcaitis, S.; Choi, J.; Montine, T.J.; Moller, T.; Morrison, R.S. HIV associated neurodegeneration requires p53 in neurons and microglia. FASEB J. 2004, 18, 1141–1143. [Google Scholar] [CrossRef] [PubMed]
- Medders, K.E.; Sejbuk, N.E.; Maung, R.; Desai, M.K.; Kaul, M. Activation of p38 MAPK is required in monocytic and neuronal cells for HIV glycoprotein 120-induced neurotoxicity. J. Immunol. 2010, 185, 4883–4895. [Google Scholar] [CrossRef]
- Garden, G.A.; Budd, S.L.; Tsai, E.; Hanson, L.; Kaul, M.; D’Emilia, D.M.; Friedlander, R.M.; Yuan, J.; Masliah, E.; Lipton, S.A. Caspase cascades in human immunodeficiency virus-associated neurodegeneration. J. Neurosci. 2002, 22, 4015–4024. [Google Scholar] [CrossRef] [PubMed]
- Jordan-Sciutto, K.L.; Wang, G.; Murphey-Corb, M.; Wiley, C.A. Cell cycle proteins exhibit altered expression patterns in lentiviral-associated encephalitis. J. Neurosci. 2002, 22, 2185–2195. [Google Scholar] [CrossRef] [PubMed]
- Haughey, N.J.; Cutler, R.G.; Tamara, A.; McArthur, J.C.; Vargas, D.L.; Pardo, C.A.; Turchan, J.; Nath, A.; Mattson, M.P. Perturbation of sphingolipid metabolism and ceramide production in HIV-dementia. Ann. Neurol. 2004, 55, 257–267. [Google Scholar] [CrossRef]
- Kaul, M.; Garden, G.A.; Lipton, S.A. Pathways to neuronal injury and apoptosis in HIV-associated dementia. Nature 2001, 410, 988–994. [Google Scholar] [CrossRef]
- Genis, P.; Jett, M.; Bernton, E.W.; Boyle, T.; Gelbard, H.A.; Dzenko, K.; Keane, R.W.; Resnick, L.; Mizrachi, Y.; Volsky, D.J.; et al. Cytokines and arachidonic metabolites produced during human immunodeficiency virus (HIV)-infected macrophage-astroglia interactions: Implications for the neuropathogenesis of HIV disease. J. Exp. Med. 1992, 176, 1703–1718. [Google Scholar] [CrossRef]
- Bennett, B.A.; Rusyniak, D.E.; Hollingsworth, C.K. HIV-1 gp120-induced neurotoxicity to midbrain dopamine cultures. Brain Res. 1995, 705, 168–176. [Google Scholar] [CrossRef]
- Yeung, M.C.; Geertsma, F.; Liu, J.; Lau, A.S. Inhibition of HIV-1 gp120-induced apoptosis in neuroblastoma SK-N-SH cells by an antisense oligodeoxynucleotide against p53. AIDS 1998, 12, 349–354. [Google Scholar] [CrossRef]
- Bagetta, G.; Corasaniti, M.T.; Paoletti, A.M.; Berliocchi, L.; Nistico, R.; Giammarioli, A.M.; Malorni, W.; Finazzi-Agro, A. HIV-1 gp120-induced apoptosis in the rat neocortex involves enhanced expression of cyclo-oxygenase type 2 (COX-2). Biochem. Biophys. Res. Commun. 1998, 244, 819–824. [Google Scholar] [CrossRef] [PubMed]
- O’Donnell, L.A.; Agrawal, A.; Jordan-Sciutto, K.L.; Dichter, M.A.; Lynch, D.R.; Kolson, D.L. Human immunodeficiency virus (HIV)-induced neurotoxicity: Roles for the NMDA receptor subtypes. J. Neurosci. 2006, 26, 981–990. [Google Scholar] [CrossRef] [PubMed]
- Corasaniti, M.T.; Strongoli, M.C.; Piccirilli, S.; Nistico, R.; Costa, A.; Bilotta, A.; Turano, P.; Finazzi-Agro, A.; Bagetta, G. Apoptosis induced by gp120 in the neocortex of rat involves enhanced expression of cyclooxygenase type 2 and is prevented by NMDA receptor antagonists and by the 21-aminosteroid U-74389G. Biochem. Biophys. Res. Commun. 2000, 274, 664–669. [Google Scholar] [CrossRef] [PubMed]
- Lipton, S.A. Neuronal injury associated with HIV-1 and potential treatment with calcium-channel and NMDA antagonists. Dev. Neurosci. 1994, 16, 145–151. [Google Scholar] [CrossRef]
- Doble, A. The role of excitotoxicity in neurodegenerative disease: Implications for therapy. Pharmacol. Ther. 1999, 81, 163–221. [Google Scholar] [CrossRef]
- Ohagen, A.; Ghosh, S.; He, J.; Huang, K.; Chen, Y.; Yuan, M.; Osathanondh, R.; Gartner, S.; Shi, B.; Shaw, G.; et al. Apoptosis induced by infection of primary brain cultures with diverse human immunodeficiency virus type 1 isolates: Evidence for a role of the envelope. J. Virol. 1999, 73, 897–906. [Google Scholar] [CrossRef]
- Raber, J.; Toggas, S.M.; Lee, S.; Bloom, F.E.; Epstein, C.J.; Mucke, L. Central nervous system expression of HIV-1 Gp120 activates the hypothalamic-pituitary-adrenal axis: Evidence for involvement of NMDA receptors and nitric oxide synthase. Virology 1996, 226, 362–373. [Google Scholar] [CrossRef]
- Jana, A.; Pahan, K. Human immunodeficiency virus type 1 gp120 induces apoptosis in human primary neurons through redox-regulated activation of neutral sphingomyelinase. J. Neurosci. 2004, 24, 9531–9540. [Google Scholar] [CrossRef]
- Tyagi, M.; Pearson, R.J.; Karn, J. Establishment of HIV latency in primary CD4+ cells is due to epigenetic transcriptional silencing and P-TEFb restriction. J. Virol. 2010, 84, 6425–6437. [Google Scholar] [CrossRef]
- Kim, Y.K.; Bourgeois, C.F.; Pearson, R.; Tyagi, M.; West, M.J.; Wong, J.; Wu, S.Y.; Chiang, C.M.; Karn, J. Recruitment of TFIIH to the HIV LTR is a rate-limiting step in the emergence of HIV from latency. EMBO J. 2006, 25, 3596–3604. [Google Scholar] [CrossRef]
- Tyagi, M.; Karn, J. CBF-1 promotes transcriptional silencing during the establishment of HIV-1 latency. EMBO J. 2007, 26, 4985–4995. [Google Scholar] [CrossRef]
- Pearson, R.; Kim, Y.K.; Hokello, J.; Lassen, K.; Friedman, J.; Tyagi, M.; Karn, J. Epigenetic silencing of human immunodeficiency virus (HIV) transcription by formation of restrictive chromatin structures at the viral long terminal repeat drives the progressive entry of HIV into latency. J. Virol. 2008, 82, 12291–12303. [Google Scholar] [CrossRef] [PubMed]
- Marzio, G.; Tyagi, M.; Gutierrez, M.I.; Giacca, M. HIV-1 tat transactivator recruits p300 and CREB-binding protein histone acetyltransferases to the viral promoter. Proc. Natl. Acad. Sci. USA 1998, 95, 13519–13524. [Google Scholar] [CrossRef] [PubMed]
- Kiernan, R.E.; Vanhulle, C.; Schiltz, L.; Adam, E.; Xiao, H.; Maudoux, F.; Calomme, C.; Burny, A.; Nakatani, Y.; Jeang, K.T.; et al. HIV-1 tat transcriptional activity is regulated by acetylation. EMBO J. 1999, 18, 6106–6118. [Google Scholar] [CrossRef]
- Marino, J.; Maubert, M.E.; Mele, A.R.; Spector, C.; Wigdahl, B.; Nonnemacher, M.R. Functional impact of HIV-1 Tat on cells of the CNS and its role in HAND. Cell. Mol. Life Sci. 2020, 77, 5079–5099. [Google Scholar] [CrossRef]
- Nath, A.; Psooy, K.; Martin, C.; Knudsen, B.; Magnuson, D.S.; Haughey, N.; Geiger, J.D. Identification of a human immunodeficiency virus type 1 Tat epitope that is neuroexcitatory and neurotoxic. J. Virol. 1996, 70, 1475–1480. [Google Scholar] [CrossRef] [PubMed]
- Song, L.; Nath, A.; Geiger, J.D.; Moore, A.; Hochman, S. Human immunodeficiency virus type 1 Tat protein directly activates neuronal N-methyl-D-aspartate receptors at an allosteric zinc-sensitive site. J. Neurovirol. 2003, 9, 399–403. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Huang, Y.; Reid, R.; Steiner, J.; Malpica-Llanos, T.; Darden, T.A.; Shankar, S.K.; Mahadevan, A.; Satishchandra, P.; Nath, A. NMDA receptor activation by HIV-Tat protein is clade dependent. J. Neurosci. 2008, 28, 12190–12198. [Google Scholar] [CrossRef] [PubMed]
- Bruce-Keller, A.J.; Chauhan, A.; Dimayuga, F.O.; Gee, J.; Keller, J.N.; Nath, A. Synaptic transport of human immunodeficiency virus-Tat protein causes neurotoxicity and gliosis in rat brain. J. Neurosci. 2003, 23, 8417–8422. [Google Scholar] [CrossRef]
- McManus, C.M.; Weidenheim, K.; Woodman, S.E.; Nunez, J.; Hesselgesser, J.; Nath, A.; Berman, J.W. Chemokine and chemokine-receptor expression in human glial elements: Induction by the HIV protein, Tat, and chemokine autoregulation. Am. J. Pathol. 2000, 156, 1441–1453. [Google Scholar] [CrossRef]
- Rappaport, J.; Joseph, J.; Croul, S.; Alexander, G.; Del Valle, L.; Amini, S.; Khalili, K. Molecular pathway involved in HIV-1-induced CNS pathology: Role of viral regulatory protein, Tat. J. Leukoc. Biol. 1999, 65, 458–465. [Google Scholar] [CrossRef] [PubMed]
- Pei, L.; Lee, F.J.; Moszczynska, A.; Vukusic, B.; Liu, F. Regulation of dopamine D1 receptor function by physical interaction with the NMDA receptors. J. Neurosci. 2004, 24, 1149–1158. [Google Scholar] [CrossRef] [PubMed]
- Missale, C.; Fiorentini, C.; Busi, C.; Collo, G.; Spano, P.F. The NMDA/D1 receptor complex as a new target in drug development. Curr. Top. Med. Chem. 2006, 6, 801–808. [Google Scholar] [CrossRef] [PubMed]
- Silvers, J.M.; Aksenova, M.V.; Aksenov, M.Y.; Mactutus, C.F.; Booze, R.M. Neurotoxicity of HIV-1 Tat protein: Involvement of D1 dopamine receptor. Neurotoxicology 2007, 28, 1184–1190. [Google Scholar] [CrossRef]
- Aksenova, M.V.; Silvers, J.M.; Aksenov, M.Y.; Nath, A.; Ray, P.D.; Mactutus, C.F.; Booze, R.M. HIV-1 Tat neurotoxicity in primary cultures of rat midbrain fetal neurons: Changes in dopamine transporter binding and immunoreactivity. Neurosci. Lett. 2006, 395, 235–239. [Google Scholar] [CrossRef]
- Wallace, D.R.; Dodson, S.; Nath, A.; Booze, R.M. Estrogen attenuates gp120- and tat1-72-induced oxidative stress and prevents loss of dopamine transporter function. Synapse 2006, 59, 51–60. [Google Scholar] [CrossRef]
- Aksenov, M.Y.; Hasselrot, U.; Bansal, A.K.; Wu, G.; Nath, A.; Anderson, C.; Mactutus, C.F.; Booze, R.M. Oxidative damage induced by the injection of HIV-1 Tat protein in the rat striatum. Neurosci. Lett. 2001, 305, 5–8. [Google Scholar] [CrossRef]
- Kruman, I.I.; Nath, A.; Mattson, M.P. HIV-1 protein Tat induces apoptosis of hippocampal neurons by a mechanism involving caspase activation, calcium overload, and oxidative stress. Exp. Neurol. 1998, 154, 276–288. [Google Scholar] [CrossRef]
- Haughey, N.J.; Holden, C.P.; Nath, A.; Geiger, J.D. Involvement of inositol 1,4,5-trisphosphate-regulated stores of intracellular calcium in calcium dysregulation and neuron cell death caused by HIV-1 protein tat. J. Neurochem. 1999, 73, 1363–1374. [Google Scholar] [CrossRef]
- New, D.R.; Maggirwar, S.B.; Epstein, L.G.; Dewhurst, S.; Gelbard, H.A. HIV-1 Tat induces neuronal death via tumor necrosis factor-alpha and activation of non-N-methyl-D-aspartate receptors by a NFkappaB-independent mechanism. J. Biol. Chem. 1998, 273, 17852–17858. [Google Scholar] [CrossRef]
- Polazzi, E.; Levi, G.; Minghetti, L. Human immunodeficiency virus type 1 Tat protein stimulates inducible nitric oxide synthase expression and nitric oxide production in microglial cultures. J. Neuropathol. Exp. Neurol. 1999, 58, 825–831. [Google Scholar] [CrossRef]
- Mohammad Ahmadi Soleimani, S.; Ekhtiari, H.; Cadet, J.L. Drug-induced neurotoxicity in addiction medicine: From prevention to harm reduction. Prog. Brain Res. 2016, 223, 19–41. [Google Scholar] [CrossRef] [PubMed]
- Frank, M.G.; Weber, M.D.; Watkins, L.R.; Maier, S.F. Stress sounds the alarmin: The role of the danger-associated molecular pattern HMGB1 in stress-induced neuroinflammatory priming. Brain Behav. Immun. 2015, 48, 1–7. [Google Scholar] [CrossRef]
- Salter, M.W.; Stevens, B. Microglia emerge as central players in brain disease. Nat. Med. 2017, 23, 1018–1027. [Google Scholar] [CrossRef]
- Bylicky, M.A.; Mueller, G.P.; Day, R.M. Mechanisms of Endogenous Neuroprotective Effects of Astrocytes in Brain Injury. Oxid Med. Cell Longev. 2018, 2018, 6501031. [Google Scholar] [CrossRef]
- Hodes, G.E.; Kana, V.; Menard, C.; Merad, M.; Russo, S.J. Neuroimmune mechanisms of depression. Nat. Neurosci. 2015, 18, 1386–1393. [Google Scholar] [CrossRef] [PubMed]
- Felger, J.C.; Miller, A.H. Cytokine effects on the basal ganglia and dopamine function: The subcortical source of inflammatory malaise. Front. Neuroendocrinol. 2012, 33, 315–327. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.; Taylor, J.R.; Wolf, M.E.; Shaham, Y. Circuit and Synaptic Plasticity Mechanisms of Drug Relapse. J. Neurosci. 2017, 37, 10867–10876. [Google Scholar] [CrossRef]
- Birben, E.; Sahiner, U.M.; Sackesen, C.; Erzurum, S.; Kalayci, O. Oxidative stress and antioxidant defense. World Allergy Organ. J. 2012, 5, 9–19. [Google Scholar] [CrossRef]
- Lyras, L.; Cairns, N.J.; Jenner, A.; Jenner, P.; Halliwell, B. An assessment of oxidative damage to proteins, lipids, and DNA in brain from patients with Alzheimer’s disease. J. Neurochem. 1997, 68, 2061–2069. [Google Scholar] [CrossRef]
- Poli, G.; Leonarduzzi, G.; Biasi, F.; Chiarpotto, E. Oxidative stress and cell signalling. Curr. Med. Chem. 2004, 11, 1163–1182. [Google Scholar] [CrossRef] [PubMed]
- Bannon, M.J. The dopamine transporter: Role in neurotoxicity and human disease. Toxicol. Appl. Pharmacol. 2005, 204, 355–360. [Google Scholar] [CrossRef] [PubMed]
- Graham, D.G.; Tiffany, S.M.; Bell, W.R., Jr.; Gutknecht, W.F. Autoxidation versus covalent binding of quinones as the mechanism of toxicity of dopamine, 6-hydroxydopamine, and related compounds toward C1300 neuroblastoma cells in vitro. Mol. Pharmacol. 1978, 14, 644–653. [Google Scholar]
- Lyles, J.; Cadet, J.L. Methylenedioxymethamphetamine (MDMA, Ecstasy) neurotoxicity: Cellular and molecular mechanisms. Brain Res. Brain Res. Rev. 2003, 42, 155–168. [Google Scholar] [CrossRef]
- Yamamoto, B.K.; Bankson, M.G. Amphetamine neurotoxicity: Cause and consequence of oxidative stress. Crit. Rev. Neurobiol. 2005, 17, 87–117. [Google Scholar] [CrossRef] [PubMed]
- Dietrich, J.B.; Mangeol, A.; Revel, M.O.; Burgun, C.; Aunis, D.; Zwiller, J. Acute or repeated cocaine administration generates reactive oxygen species and induces antioxidant enzyme activity in dopaminergic rat brain structures. Neuropharmacology 2005, 48, 965–974. [Google Scholar] [CrossRef]
- Macedo, D.S.; de Vasconcelos, S.M.; dos Santos, R.S.; Aguiar, L.M.; Lima, V.T.; Viana, G.S.; de Sousa, F.C. Cocaine alters catalase activity in prefrontal cortex and striatum of mice. Neurosci. Lett. 2005, 387, 53–56. [Google Scholar] [CrossRef]
- Lipton, J.W.; Gyawali, S.; Borys, E.D.; Koprich, J.B.; Ptaszny, M.; McGuire, S.O. Prenatal cocaine administration increases glutathione and alpha-tocopherol oxidation in fetal rat brain. Brain Res. Dev. Brain Res. 2003, 147, 77–84. [Google Scholar] [CrossRef]
- Qiusheng, Z.; Yuntao, Z.; Rongliang, Z.; Dean, G.; Changling, L. Effects of verbascoside and luteolin on oxidative damage in brain of heroin treated mice. Pharmazie 2005, 60, 539–543. [Google Scholar]
- Xu, B.; Wang, Z.; Li, G.; Li, B.; Lin, H.; Zheng, R.; Zheng, Q. Heroin-administered mice involved in oxidative stress and exogenous antioxidant-alleviated withdrawal syndrome. Basic Clin. Pharmacol. Toxicol. 2006, 99, 153–161. [Google Scholar] [CrossRef]
- Alberts, B.; Johnson, A.; Lewis, J.; Raff, M.; Roberts, K.; Walter, P. Programmed Cell Death (Apoptosis), 4th ed.; Garland Science: New York, NY, USA, 2002. [Google Scholar]
- Raychaudhuri, S. A minimal model of signaling network elucidates cell-to-cell stochastic variability in apoptosis. PLoS ONE 2010, 5, e11930. [Google Scholar] [CrossRef] [PubMed]
- Jayanthi, S.; Deng, X.; Ladenheim, B.; McCoy, M.T.; Cluster, A.; Cai, N.S.; Cadet, J.L. Calcineurin/NFAT-induced up-regulation of the Fas ligand/Fas death pathway is involved in methamphetamine-induced neuronal apoptosis. Proc. Natl. Acad. Sci. USA 2005, 102, 868–873. [Google Scholar] [CrossRef]
- Krasnova, I.N.; Ladenheim, B.; Cadet, J.L. Amphetamine induces apoptosis of medium spiny striatal projection neurons via the mitochondria-dependent pathway. FASEB J. 2005, 19, 851–853. [Google Scholar] [CrossRef]
- Dey, S.; Mactutus, C.F.; Booze, R.M.; Snow, D.M. Cocaine exposure in vitro induces apoptosis in fetal locus coeruleus neurons by altering the Bax/Bcl-2 ratio and through caspase-3 apoptotic signaling. Neuroscience 2007, 144, 509–521. [Google Scholar] [CrossRef]
- Imam, S.Z.; Duhart, H.M.; Skinner, J.T.; Ali, S.F. Cocaine induces a differential dose-dependent alteration in the expression profile of immediate early genes, transcription factors, and caspases in PC12 cells: A possible mechanism of neurotoxic damage in cocaine addiction. Ann. N. Y. Acad. Sci. 2005, 1053, 482–490. [Google Scholar] [CrossRef] [PubMed]
- Poon, H.F.; Abdullah, L.; Mullan, M.A.; Mullan, M.J.; Crawford, F.C. Cocaine-induced oxidative stress precedes cell death in human neuronal progenitor cells. Neurochem. Int. 2007, 50, 69–73. [Google Scholar] [CrossRef] [PubMed]
- Cunha-Oliveira, T.; Rego, A.C.; Garrido, J.; Borges, F.; Macedo, T.; Oliveira, C.R. Street heroin induces mitochondrial dysfunction and apoptosis in rat cortical neurons. J. Neurochem. 2007, 101, 543–554. [Google Scholar] [CrossRef]
- Oliveira, M.T.; Rego, A.C.; Macedo, T.R.; Oliveira, C.R. Drugs of abuse induce apoptotic features in PC12 cells. Ann. N. Y. Acad. Sci. 2003, 1010, 667–670. [Google Scholar] [CrossRef]
- Dong, X.X.; Wang, Y.; Qin, Z.H. Molecular mechanisms of excitotoxicity and their relevance to pathogenesis of neurodegenerative diseases. Acta Pharmacol. Sin. 2009, 30, 379–387. [Google Scholar] [CrossRef]
- Dani, A.; Huang, B.; Bergan, J.; Dulac, C.; Zhuang, X. Superresolution imaging of chemical synapses in the brain. Neuron 2010, 68, 843–856. [Google Scholar] [CrossRef]
- Chávez-Castillo, M.; Rojas, M.; Bautista, J. Excitotoxicity: An Organized Crime at The Cellular Level. Arch. Med. 2017, 8, 193. [Google Scholar] [CrossRef]
- Bronner, F. Extracellular and intracellular regulation of calcium homeostasis. Sci. World J. 2001, 1, 919–925. [Google Scholar] [CrossRef] [PubMed]
- Pivovarova, N.B.; Andrews, S.B. Calcium-dependent mitochondrial function and dysfunction in neurons. FEBS J. 2010, 277, 3622–3636. [Google Scholar] [CrossRef] [PubMed]
- Watkins, J.C.; Jane, D.E. The glutamate story. Br. J. Pharmacol. 2006, 147 (Suppl. 1), S100–S108. [Google Scholar] [CrossRef] [PubMed]
- Novelli, A.; Reilly, J.A.; Lysko, P.G.; Henneberry, R.C. Glutamate becomes neurotoxic via the N-methyl-D-aspartate receptor when intracellular energy levels are reduced. Brain Res. 1988, 451, 205–212. [Google Scholar] [CrossRef]
- Mao, J.; Sung, B.; Ji, R.R.; Lim, G. Neuronal apoptosis associated with morphine tolerance: Evidence for an opioid-induced neurotoxic mechanism. J. Neurosci. 2002, 22, 7650–7661. [Google Scholar] [CrossRef]
- Reid, M.S.; Hsu, K., Jr.; Berger, S.P. Cocaine and amphetamine preferentially stimulate glutamate release in the limbic system: Studies on the involvement of dopamine. Synapse 1997, 27, 95–105. [Google Scholar] [CrossRef]
- Williams, J.M.; Steketee, J.D. Cocaine increases medial prefrontal cortical glutamate overflow in cocaine-sensitized rats: A time course study. Eur. J. Neurosci. 2004, 20, 1639–1646. [Google Scholar] [CrossRef]
- Wolf, M.E.; Xue, C.J.; Li, Y.; Wavak, D. Amphetamine increases glutamate efflux in the rat ventral tegmental area by a mechanism involving glutamate transporters and reactive oxygen species. J. Neurochem. 2000, 75, 1634–1644. [Google Scholar] [CrossRef]
- Langlais, P.J.; Mair, R.G. Protective effects of the glutamate antagonist MK-801 on pyrithiamine-induced lesions and amino acid changes in rat brain. J. Neurosci. 1990, 10, 1664–1674. [Google Scholar] [CrossRef]
- Jaenisch, R.; Bird, A. Epigenetic regulation of gene expression: How the genome integrates intrinsic and environmental signals. Nat. Genet. 2003, 33, 245–254. [Google Scholar] [CrossRef]
- Nestler, E.J.; Luscher, C. The Molecular Basis of Drug Addiction: Linking Epigenetic to Synaptic and Circuit Mechanisms. Neuron 2019, 102, 48–59. [Google Scholar] [CrossRef] [PubMed]
- Tyagi, M.; Weber, J.; Bukrinsky, M.; Simon, G.L. The effects of cocaine on HIV transcription. J. Neurovirol. 2016, 22, 261–274. [Google Scholar] [CrossRef] [PubMed]
- Koob, G.; Kreek, M.J. Stress, dysregulation of drug reward pathways, and the transition to drug dependence. Am. J. Psychiatry 2007, 164, 1149–1159. [Google Scholar] [CrossRef] [PubMed]
- Kelley, A.E.; Berridge, K.C. The neuroscience of natural rewards: Relevance to addictive drugs. J. Neurosci. 2002, 22, 3306–3311. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Choi, K.H.; Renthal, W.; Tsankova, N.M.; Theobald, D.E.; Truong, H.T.; Russo, S.J.; Laplant, Q.; Sasaki, T.S.; Whistler, K.N.; et al. Chromatin remodeling is a key mechanism underlying cocaine-induced plasticity in striatum. Neuron 2005, 48, 303–314. [Google Scholar] [CrossRef]
- Botia, B.; Legastelois, R.; Alaux-Cantin, S.; Naassila, M. Expression of ethanol-induced behavioral sensitization is associated with alteration of chromatin remodeling in mice. PLoS ONE 2012, 7, e47527. [Google Scholar] [CrossRef]
- Renthal, W.; Carle, T.L.; Maze, I.; Covington, H.E., 3rd; Truong, H.T.; Alibhai, I.; Kumar, A.; Montgomery, R.L.; Olson, E.N.; Nestler, E.J. Delta FosB mediates epigenetic desensitization of the c-fos gene after chronic amphetamine exposure. J. Neurosci. 2008, 28, 7344–7349. [Google Scholar] [CrossRef]
- Hamilton, P.J.; Lim, C.J.; Nestler, E.J.; Heller, E.A. Neuroepigenetic Editing. Methods Mol. Biol. 2018, 1767, 113–136. [Google Scholar] [CrossRef]
- Heller, E.A.; Cates, H.M.; Pena, C.J.; Sun, H.; Shao, N.; Feng, J.; Golden, S.A.; Herman, J.P.; Walsh, J.J.; Mazei-Robison, M.; et al. Locus-specific epigenetic remodeling controls addiction- and depression-related behaviors. Nat. Neurosci. 2014, 17, 1720–1727. [Google Scholar] [CrossRef]
- Heller, E.A.; Hamilton, P.J.; Burek, D.D.; Lombroso, S.I.; Pena, C.J.; Neve, R.L.; Nestler, E.J. Targeted Epigenetic Remodeling of the Cdk5 Gene in Nucleus Accumbens Regulates Cocaine- and Stress-Evoked Behavior. J. Neurosci. 2016, 36, 4690–4697. [Google Scholar] [CrossRef] [PubMed]
- Kalant, H.; Kalant, O.J. Death in amphetamine users: Causes and rates. Can. Med. Assoc. J. 1975, 112, 299–304. [Google Scholar] [PubMed]
- Sandoval, V.; Hanson, G.R.; Fleckenstein, A.E. Methamphetamine decreases mouse striatal dopamine transporter activity: Roles of hyperthermia and dopamine. Eur. J. Pharmacol. 2000, 409, 265–271. [Google Scholar] [CrossRef]
- Lin, P.S.; Quamo, S.; Ho, K.C.; Gladding, J. Hyperthermia enhances the cytotoxic effects of reactive oxygen species to Chinese hamster cells and bovine endothelial cells in vitro. Radiat. Res. 1991, 126, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Jayanthi, S.; Deng, X.; Noailles, P.A.; Ladenheim, B.; Cadet, J.L. Methamphetamine induces neuronal apoptosis via cross-talks between endoplasmic reticulum and mitochondria-dependent death cascades. FASEB J. 2004, 18, 238–251. [Google Scholar] [CrossRef]
- Sharma, H.S.; Olsson, Y.; Dey, P.K. Changes in blood-brain barrier and cerebral blood flow following elevation of circulating serotonin level in anesthetized rats. Brain Res. 1990, 517, 215–223. [Google Scholar] [CrossRef]
- Sharma, H.S. Methods to produce hyperthermia-induced brain dysfunction. Prog. Brain Res. 2007, 162, 173–199. [Google Scholar] [CrossRef]
- Levy, A.D.; Baumann, M.H.; Van de Kar, L.D. Monoaminergic regulation of neuroendocrine function and its modification by cocaine. Front. Neuroendocrinol. 1994, 15, 85–156. [Google Scholar] [CrossRef]
- Cunningham, K.A.; Paris, J.M.; Goeders, N.E. Chronic cocaine enhances serotonin autoregulation and serotonin uptake binding. Synapse 1992, 11, 112–123. [Google Scholar] [CrossRef]
- Bose, J.; Hedden, S.L.; Lipari, R.N.; Park-Lee, E.; Porter, J.D.; Pemberton, M.R. Key Substance Use and Mental Health Indicators in the United States: Results from the 2017 National Survey on Drug Use and Health. Subst. Abus. Ment. Health Serv. Adm. 2016, 16, 2–25. [Google Scholar]
- Dutta, R.; Roy, S. Mechanism(s) involved in opioid drug abuse modulation of HAND. Curr. HIV Res. 2012, 10, 469–477. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Young, A.M.; Havens, J.R. Transition from first illicit drug use to first injection drug use among rural Appalachian drug users: A cross-sectional comparison and retrospective survival analysis. Addiction 2012, 107, 587–596. [Google Scholar] [CrossRef] [PubMed]
- Mateu-Gelabert, P.; Guarino, H.; Jessell, L.; Teper, A. Injection and sexual HIV/HCV risk behaviors associated with nonmedical use of prescription opioids among young adults in New York City. J. Subst. Abuse Treat. 2015, 48, 13–20. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, C.O. Opioids and HIV Infection: From Pain Management to Addiction Treatment. Top. Antivir. Med. 2018, 25, 143–146. [Google Scholar]
- Weisberg, D.F.; Gordon, K.S.; Barry, D.T.; Becker, W.C.; Crystal, S.; Edelman, E.J.; Gaither, J.; Gordon, A.J.; Goulet, J.; Kerns, R.D.; et al. Long-term Prescription of Opioids and/or Benzodiazepines and Mortality Among HIV-Infected and Uninfected Patients. J. Acquir. Immune Defic. Syndr. 2015, 69, 223–233. [Google Scholar] [CrossRef]
- Filipczak-Bryniarska, I.; Nowak, B.; Sikora, E.; Nazimek, K.; Woron, J.; Wordliczek, J.; Bryniarski, K. The influence of opioids on the humoral and cell-mediated immune responses in mice. The role of macrophages. Pharmacol. Rep. 2012, 64, 1200–1215. [Google Scholar] [CrossRef]
- Dave, R.S.; Khalili, K. Morphine treatment of human monocyte-derived macrophages induces differential miRNA and protein expression: Impact on inflammation and oxidative stress in the central nervous system. J. Cell Biochem. 2010, 110, 834–845. [Google Scholar] [CrossRef]
- Reynolds, J.L.; Law, W.C.; Mahajan, S.D.; Aalinkeel, R.; Nair, B.; Sykes, D.E.; Mammen, M.J.; Yong, K.T.; Hui, R.; Prasad, P.N.; et al. Morphine and galectin-1 modulate HIV-1 infection of human monocyte-derived macrophages. J. Immunol. 2012, 188, 3757–3765. [Google Scholar] [CrossRef]
- Guo, C.J.; Li, Y.; Tian, S.; Wang, X.; Douglas, S.D.; Ho, W.Z. Morphine enhances HIV infection of human blood mononuclear phagocytes through modulation of beta-chemokines and CCR5 receptor. J. Investig. Med. 2002, 50, 435–442. [Google Scholar] [CrossRef]
- Kim, S.; Hahn, Y.K.; Podhaizer, E.M.; McLane, V.D.; Zou, S.; Hauser, K.F.; Knapp, P.E. A central role for glial CCR5 in directing the neuropathological interactions of HIV-1 Tat and opiates. J. Neuroinflamm. 2018, 15, 285. [Google Scholar] [CrossRef]
- Ozawa, T.; Nakagawa, T.; Shige, K.; Minami, M.; Satoh, M. Changes in the expression of glial glutamate transporters in the rat brain accompanied with morphine dependence and naloxone-precipitated withdrawal. Brain Res. 2001, 905, 254–258. [Google Scholar] [CrossRef]
- Rodriguez, M.; Lapierre, J.; Ojha, C.R.; Estrada-Bueno, H.; Dever, S.M.; Gewirtz, D.A.; Kashanchi, F.; El-Hage, N. Importance of Autophagy in Mediating Human Immunodeficiency Virus (HIV) and Morphine-Induced Metabolic Dysfunction and Inflammation in Human Astrocytes. Viruses 2017, 9, 201. [Google Scholar] [CrossRef] [PubMed]
- El-Hage, N.; Bruce-Keller, A.J.; Yakovleva, T.; Bazov, I.; Bakalkin, G.; Knapp, P.E.; Hauser, K.F. Morphine exacerbates HIV-1 Tat-induced cytokine production in astrocytes through convergent effects on [Ca(2+)](i), NF-kappaB trafficking and transcription. PLoS ONE 2008, 3, e4093. [Google Scholar] [CrossRef] [PubMed]
- Mahajan, S.D.; Aalinkeel, R.; Sykes, D.E.; Reynolds, J.L.; Bindukumar, B.; Fernandez, S.F.; Chawda, R.; Shanahan, T.C.; Schwartz, S.A. Tight junction regulation by morphine and HIV-1 tat modulates blood-brain barrier permeability. J. Clin. Immunol. 2008, 28, 528–541. [Google Scholar] [CrossRef]
- Yousif, S.; Saubamea, B.; Cisternino, S.; Marie-Claire, C.; Dauchy, S.; Scherrmann, J.M.; Decleves, X. Effect of chronic exposure to morphine on the rat blood-brain barrier: Focus on the P-glycoprotein. J. Neurochem. 2008, 107, 647–657. [Google Scholar] [CrossRef]
- Coley, J.S.; Calderon, T.M.; Gaskill, P.J.; Eugenin, E.A.; Berman, J.W. Dopamine increases CD14+CD16+ monocyte migration and adhesion in the context of substance abuse and HIV neuropathogenesis. PLoS ONE 2015, 10, e0117450. [Google Scholar] [CrossRef]
- Calderon, T.M.; Williams, D.W.; Lopez, L.; Eugenin, E.A.; Cheney, L.; Gaskill, P.J.; Veenstra, M.; Anastos, K.; Morgello, S.; Berman, J.W. Dopamine Increases CD14(+)CD16(+) Monocyte Transmigration across the Blood Brain Barrier: Implications for Substance Abuse and HIV Neuropathogenesis. J. Neuroimmune Pharmacol. 2017, 12, 353–370. [Google Scholar] [CrossRef]
- Gaskill, P.J.; Calderon, T.M.; Luers, A.J.; Eugenin, E.A.; Javitch, J.A.; Berman, J.W. Human immunodeficiency virus (HIV) infection of human macrophages is increased by dopamine: A bridge between HIV-associated neurologic disorders and drug abuse. Am. J. Pathol. 2009, 175, 1148–1159. [Google Scholar] [CrossRef]
- Gaskill, P.J.; Yano, H.H.; Kalpana, G.V.; Javitch, J.A.; Berman, J.W. Dopamine receptor activation increases HIV entry into primary human macrophages. PLoS ONE 2014, 9, e108232. [Google Scholar] [CrossRef]
- Iuvone, T.; Capasso, A.; D’Acquisto, F.; Carnuccio, R. Opioids inhibit the induction of nitric oxide synthase in J774 macrophages. Biochem. Biophys. Res. Commun. 1995, 212, 975–980. [Google Scholar] [CrossRef]
- Turchan-Cholewo, J.; Dimayuga, F.O.; Gupta, S.; Keller, J.N.; Knapp, P.E.; Hauser, K.F.; Bruce-Keller, A.J. Morphine and HIV-Tat increase microglial-free radical production and oxidative stress: Possible role in cytokine regulation. J. Neurochem. 2009, 108, 202–215. [Google Scholar] [CrossRef] [PubMed]
- Riss, G.L.; Chang, D.I.; Wevers, C.; Westendorf, A.M.; Buer, J.; Scherbaum, N.; Hansen, W. Opioid maintenance therapy restores CD4+ T cell function by normalizing CD4+CD25(high) regulatory T cell frequencies in heroin user. Brain Behav. Immun. 2012, 26, 972–978. [Google Scholar] [CrossRef]
- Sharp, B.M.; McAllen, K.; Gekker, G.; Shahabi, N.A.; Peterson, P.K. Immunofluorescence detection of delta opioid receptors (DOR) on human peripheral blood CD4+ T cells and DOR-dependent suppression of HIV-1 expression. J. Immunol. 2001, 167, 1097–1102. [Google Scholar] [CrossRef] [PubMed]
- Peterson, P.K.; Gekker, G.; Lokensgard, J.R.; Bidlack, J.M.; Chang, A.C.; Fang, X.; Portoghese, P.S. Kappa-opioid receptor agonist suppression of HIV-1 expression in CD4+ lymphocytes. Biochem. Pharmacol. 2001, 61, 1145–1151. [Google Scholar] [CrossRef]
- Plein, L.M.; Rittner, H.L. Opioids and the immune system—Friend or foe. Br. J. Pharmacol. 2018, 175, 2717–2725. [Google Scholar] [CrossRef] [PubMed]
- Desai, N.; Burns, L.; Gong, Y.; Zhi, K.; Kumar, A.; Summers, N.; Kumar, S.; Cory, T.J. An update on drug-drug interactions between antiretroviral therapies and drugs of abuse in HIV systems. Expert Opin. Drug Metab. Toxicol. 2020, 16, 1005–1018. [Google Scholar] [CrossRef]
- Feng, X.Q.; Zhu, L.L.; Zhou, Q. Opioid analgesics-related pharmacokinetic drug interactions: From the perspectives of evidence based on randomized controlled trials and clinical risk management. J. Pain Res. 2017, 10, 1225–1239. [Google Scholar] [CrossRef]
- Wynn, G.H.; Cozza, K.L.; Zapor, M.J.; Wortmann, G.W.; Armstrong, S.C. Med-psych drug-drug interactions update. Antiretrovirals, part III: Antiretrovirals and drugs of abuse. Psychosomatics 2005, 46, 79–87. [Google Scholar] [CrossRef]
- Carliner, H.; Brown, Q.L.; Sarvet, A.L.; Hasin, D.S. Cannabis use, attitudes, and legal status in the U.S.: A review. Prev. Med. 2017, 104, 13–23. [Google Scholar] [CrossRef]
- Lorenzetti, V.; Lubman, D.I.; Whittle, S.; Solowij, N.; Yucel, M. Structural MRI findings in long-term cannabis users: What do we know? Subst. Use Misuse 2010, 45, 1787–1808. [Google Scholar] [CrossRef]
- Cristiani, S.A.; Pukay-Martin, N.D.; Bornstein, R.A. Marijuana use and cognitive function in HIV-infected people. J. Neuropsychiatry Clin. Neurosci. 2004, 16, 330–335. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Shin, A.H.; Thayer, S.A. Activation of cannabinoid type 2 receptors inhibits HIV-1 envelope glycoprotein gp120-induced synapse loss. Mol. Pharmacol. 2011, 80, 357–366. [Google Scholar] [CrossRef] [PubMed]
- Yi, Y.; Lee, C.; Liu, Q.H.; Freedman, B.D.; Collman, R.G. Chemokine receptor utilization and macrophage signaling by human immunodeficiency virus type 1 gp120: Implications for neuropathogenesis. J. Neurovirol. 2004, 10 (Suppl. 1), 91–96. [Google Scholar] [CrossRef] [PubMed]
- Galve-Roperh, I.; Aguado, T.; Palazuelos, J.; Guzman, M. Mechanisms of control of neuron survival by the endocannabinoid system. Curr. Pharm. Des. 2008, 14, 2279–2288. [Google Scholar] [CrossRef]
- Benito, C.; Kim, W.K.; Chavarria, I.; Hillard, C.J.; Mackie, K.; Tolon, R.M.; Williams, K.; Romero, J. A glial endogenous cannabinoid system is upregulated in the brains of macaques with simian immunodeficiency virus-induced encephalitis. J. Neurosci. 2005, 25, 2530–2536. [Google Scholar] [CrossRef]
- Cosenza-Nashat, M.A.; Bauman, A.; Zhao, M.L.; Morgello, S.; Suh, H.S.; Lee, S.C. Cannabinoid receptor expression in HIV encephalitis and HIV-associated neuropathologic comorbidities. Neuropathol. Appl. Neurobiol. 2011, 37, 464–483. [Google Scholar] [CrossRef]
- Klegeris, A.; Bissonnette, C.J.; McGeer, P.L. Reduction of human monocytic cell neurotoxicity and cytokine secretion by ligands of the cannabinoid-type CB2 receptor. Br. J. Pharmacol. 2003, 139, 775–786. [Google Scholar] [CrossRef]
- Avraham, H.K.; Jiang, S.; Fu, Y.; Rockenstein, E.; Makriyannis, A.; Zvonok, A.; Masliah, E.; Avraham, S. The cannabinoid CB(2) receptor agonist AM1241 enhances neurogenesis in GFAP/Gp120 transgenic mice displaying deficits in neurogenesis. Br. J. Pharmacol. 2014, 171, 468–479. [Google Scholar] [CrossRef]
- Guzman, M.; Sanchez, C.; Galve-Roperh, I. Cannabinoids and cell fate. Pharmacol. Ther. 2002, 95, 175–184. [Google Scholar] [CrossRef]
- Mechoulam, R.; Panikashvili, D.; Shohami, E. Cannabinoids and brain injury: Therapeutic implications. Trends Mol. Med. 2002, 8, 58–61. [Google Scholar] [CrossRef]
- Minagar, A.; Shapshak, P.; Fujimura, R.; Ownby, R.; Heyes, M.; Eisdorfer, C. The role of macrophage/microglia and astrocytes in the pathogenesis of three neurologic disorders: HIV-associated dementia, Alzheimer disease, and multiple sclerosis. J. Neurol. Sci. 2002, 202, 13–23. [Google Scholar] [CrossRef]
- Weber, A.; Ni, J.; Ling, K.H.; Acheampong, A.; Tang-Liu, D.D.; Burk, R.; Cravatt, B.F.; Woodward, D. Formation of prostamides from anandamide in FAAH knockout mice analyzed by HPLC with tandem mass spectrometry. J. Lipid Res. 2004, 45, 757–763. [Google Scholar] [CrossRef] [PubMed]
- Maccarrone, M.; Piccirilli, S.; Battista, N.; Del Duca, C.; Nappi, G.; Corasaniti, M.T.; Finazzi-Agro, A.; Bagetta, G. Enhanced anandamide degradation is associated with neuronal apoptosis induced by the HIV-1 coat glycoprotein gp120 in the rat neocortex. J. Neurochem. 2004, 89, 1293–1300. [Google Scholar] [CrossRef] [PubMed]
- Howlett, A.C.; Barth, F.; Bonner, T.I.; Cabral, G.; Casellas, P.; Devane, W.A.; Felder, C.C.; Herkenham, M.; Mackie, K.; Martin, B.R.; et al. International Union of Pharmacology. XXVII. Classification of cannabinoid receptors. Pharmacol. Rev. 2002, 54, 161–202. [Google Scholar] [CrossRef]
- Ranganathan, M.; D’Souza, D.C. The acute effects of cannabinoids on memory in humans: A review. Psychopharmacology 2006, 188, 425–444. [Google Scholar] [CrossRef]
- Volkow, N.D.; Baler, R.D.; Compton, W.M.; Weiss, S.R. Adverse health effects of marijuana use. N. Engl. J. Med. 2014, 370, 2219–2227. [Google Scholar] [CrossRef]
- Lucas, C.J.; Galettis, P.; Schneider, J. The pharmacokinetics and the pharmacodynamics of cannabinoids. Br. J. Clin. Pharmacol. 2018, 84, 2477–2482. [Google Scholar] [CrossRef]
- Kosel, B.W.; Aweeka, F.T.; Benowitz, N.L.; Shade, S.B.; Hilton, J.F.; Lizak, P.S.; Abrams, D.I. The effects of cannabinoids on the pharmacokinetics of indinavir and nelfinavir. AIDS 2002, 16, 543–550. [Google Scholar] [CrossRef]
- Gannon, B.M.; Reichard, E.E.; Fantegrossi, W.E. Psychostimulant Abuse and HIV Infection: Cocaine, methamphetamine, and “bath salts” cathinone analogues. Curr. Addict. Rep. 2014, 1, 237–242. [Google Scholar] [CrossRef][Green Version]
- Fardin, S.; Gholami, S.; Werner, T.; Kenney, T.; Silverman, R.; Metzger, D.; Alavi, A.; Tebas, P. Deteriorating effects of cocaine abuse on brain metabolic function of HIV infected patients. J. Nucl. Med. 2016, 57, 1849. [Google Scholar]
- Baldwin, G.C.; Tashkin, D.P.; Buckley, D.M.; Park, A.N.; Dubinett, S.M.; Roth, M.D. Marijuana and cocaine impair alveolar macrophage function and cytokine production. Am. J. Respir. Crit. Care Med. 1997, 156, 1606–1613. [Google Scholar] [CrossRef] [PubMed]
- Cao, L.; Walker, M.P.; Vaidya, N.K.; Fu, M.; Kumar, S.; Kumar, A. Cocaine-Mediated Autophagy in Astrocytes Involves Sigma 1 Receptor, PI3K, mTOR, Atg5/7, Beclin-1 and Induces Type II Programed Cell Death. Mol. Neurobiol. 2016, 53, 4417–4430. [Google Scholar] [CrossRef] [PubMed]
- Peterson, P.K.; Gekker, G.; Chao, C.C.; Schut, R.; Molitor, T.W.; Balfour, H.H., Jr. Cocaine potentiates HIV-1 replication in human peripheral blood mononuclear cell cocultures. Involvement of transforming growth factor-beta. J. Immunol. 1991, 146, 81–84. [Google Scholar] [PubMed]
- Dhillon, N.K.; Williams, R.; Peng, F.; Tsai, Y.J.; Dhillon, S.; Nicolay, B.; Gadgil, M.; Kumar, A.; Buch, S.J. Cocaine-mediated enhancement of virus replication in macrophages: Implications for human immunodeficiency virus-associated dementia. J. Neurovirol. 2007, 13, 483–495. [Google Scholar] [CrossRef]
- Nath, A.; Maragos, W.F.; Avison, M.J.; Schmitt, F.A.; Berger, J.R. Acceleration of HIV dementia with methamphetamine and cocaine. J. Neurovirol. 2001, 7, 66–71. [Google Scholar] [CrossRef]
- Roth, M.D.; Tashkin, D.P.; Choi, R.; Jamieson, B.D.; Zack, J.A.; Baldwin, G.C. Cocaine enhances human immunodeficiency virus replication in a model of severe combined immunodeficient mice implanted with human peripheral blood leukocytes. J. Infect. Dis. 2002, 185, 701–705. [Google Scholar] [CrossRef]
- Swepson, C.; Ranjan, A.; Balasubramaniam, M.; Pandhare, J.; Dash, C. Cocaine Enhances HIV-1 Transcription in Macrophages by Inducing p38 MAPK Phosphorylation. Front. Microbiol. 2016, 7, 823. [Google Scholar] [CrossRef]
- Roth, M.D.; Whittaker, K.M.; Choi, R.; Tashkin, D.P.; Baldwin, G.C. Cocaine and sigma-1 receptors modulate HIV infection, chemokine receptors, and the HPA axis in the huPBL-SCID model. J. Leukoc. Biol. 2005, 78, 1198–1203. [Google Scholar] [CrossRef]
- Nair, M.P.; Mahajan, S.D.; Schwartz, S.A.; Reynolds, J.; Whitney, R.; Bernstein, Z.; Chawda, R.P.; Sykes, D.; Hewitt, R.; Hsiao, C.B. Cocaine modulates dendritic cell-specific C type intercellular adhesion molecule-3-grabbing nonintegrin expression by dendritic cells in HIV-1 patients. J. Immunol. 2005, 174, 6617–6626. [Google Scholar] [CrossRef]
- Turchan, J.; Anderson, C.; Hauser, K.F.; Sun, Q.; Zhang, J.; Liu, Y.; Wise, P.M.; Kruman, I.; Maragos, W.; Mattson, M.P.; et al. Estrogen protects against the synergistic toxicity by HIV proteins, methamphetamine and cocaine. BMC Neurosci. 2001, 2, 3. [Google Scholar] [CrossRef]
- Nath, A.; Hauser, K.F.; Wojna, V.; Booze, R.M.; Maragos, W.; Prendergast, M.; Cass, W.; Turchan, J.T. Molecular basis for interactions of HIV and drugs of abuse. J. Acquir. Immune Defic. Syndr. 2002, 31 (Suppl. 2), S62–S69. [Google Scholar] [CrossRef] [PubMed]
- Koutsilieri, E.; Gotz, M.E.; Sopper, S.; Sauer, U.; Demuth, M.; ter Meulen, V.; Riederer, P. Regulation of glutathione and cell toxicity following exposure to neurotropic substances and human immunodeficiency virus-1 in vitro. J. Neurovirol. 1997, 3, 342–349. [Google Scholar] [CrossRef] [PubMed]
- Nath, A.; Anderson, C.; Jones, M.; Maragos, W.; Booze, R.; Mactutus, C.; Bell, J.; Hauser, K.F.; Mattson, M. Neurotoxicity and dysfunction of dopaminergic systems associated with AIDS dementia. J. Psychopharmacol. 2000, 14, 222–227. [Google Scholar] [CrossRef] [PubMed]
- Yao, H.; Allen, J.E.; Zhu, X.; Callen, S.; Buch, S. Cocaine and human immunodeficiency virus type 1 gp120 mediate neurotoxicity through overlapping signaling pathways. J. Neurovirol. 2009, 15, 164–175. [Google Scholar] [CrossRef][Green Version]
- Aksenov, M.Y.; Aksenova, M.V.; Nath, A.; Ray, P.D.; Mactutus, C.F.; Booze, R.M. Cocaine-mediated enhancement of Tat toxicity in rat hippocampal cell cultures: The role of oxidative stress and D1 dopamine receptor. Neurotoxicology 2006, 27, 217–228. [Google Scholar] [CrossRef]
- Doke, M.; Jeganathan, V.; McLaughlin, J.P.; Samikkannu, T. HIV-1 Tat and cocaine impact mitochondrial epigenetics: Effects on DNA methylation. Epigenetics 2020, 16, 980–999. [Google Scholar] [CrossRef]
- Yao, H.; Bethel-Brown, C.; Buch, S. Cocaine Exposure Results in Formation of Dendritic Varicosity in Rat Primary Hippocampal Neurons. Am. J. Infect. Dis. 2009, 5, 26–30. [Google Scholar] [CrossRef]
- Bagetta, G.; Piccirilli, S.; Del Duca, C.; Morrone, L.A.; Rombola, L.; Nappi, G.; De Alba, J.; Knowles, R.G.; Corasaniti, M.T. Inducible nitric oxide synthase is involved in the mechanisms of cocaine enhanced neuronal apoptosis induced by HIV-1 gp120 in the neocortex of rat. Neurosci. Lett. 2004, 356, 183–186. [Google Scholar] [CrossRef]
- Mohseni Ahooyi, T.; Shekarabi, M.; Torkzaban, B.; Langford, T.D.; Burdo, T.H.; Gordon, J.; Datta, P.K.; Amini, S.; Khalili, K. Dysregulation of Neuronal Cholesterol Homeostasis upon Exposure to HIV-1 Tat and Cocaine Revealed by RNA-Sequencing. Sci. Rep. 2018, 8, 16300. [Google Scholar] [CrossRef]
- Lukas, S.E.; Sholar, M.; Lundahl, L.H.; Lamas, X.; Kouri, E.; Wines, J.D.; Kragie, L.; Mendelson, J.H. Sex differences in plasma cocaine levels and subjective effects after acute cocaine administration in human volunteers. Psychopharmacology 1996, 125, 346–354. [Google Scholar] [CrossRef]
- Cone, E.J. Pharmacokinetics and pharmacodynamics of cocaine. J. Anal Toxicol. 1995, 19, 459–478. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Wang, Y.; Li, Q.; Zhong, Y.; Chen, L.; Du, Y.; He, J.; Liao, L.; Xiong, K.; Yi, C.X.; et al. The Main Molecular Mechanisms Underlying Methamphetamine- Induced Neurotoxicity and Implications for Pharmacological Treatment. Front. Mol. Neurosci. 2018, 11, 186. [Google Scholar] [CrossRef] [PubMed]
- The, L. Opioids and methamphetamine: A tale of two crises. Lancet 2018, 391, 713. [Google Scholar] [CrossRef]
- Zuckerman, M.D.; Boyer, E.W. HIV and club drugs in emerging adulthood. Curr. Opin. Pediatr. 2012, 24, 219–224. [Google Scholar] [CrossRef]
- Cadet, J.L.; Krasnova, I.N. Molecular bases of methamphetamine-induced neurodegeneration. Int. Rev. Neurobiol. 2009, 88, 101–119. [Google Scholar] [CrossRef]
- Cadet, J.L.; Bisagno, V.; Milroy, C.M. Neuropathology of substance use disorders. Acta Neuropathol. 2014, 127, 91–107. [Google Scholar] [CrossRef]
- Buttner, A. Review: The neuropathology of drug abuse. Neuropathol Appl. Neurobiol. 2011, 37, 118–134. [Google Scholar] [CrossRef]
- Theodore, S.; Cass, W.A.; Nath, A.; Maragos, W.F. Progress in understanding basal ganglia dysfunction as a common target for methamphetamine abuse and HIV-1 neurodegeneration. Curr. HIV Res. 2007, 5, 301–313. [Google Scholar] [CrossRef]
- Nath, A. Human immunodeficiency virus-associated neurocognitive disorder: Pathophysiology in relation to drug addiction. Ann. N. Y. Acad. Sci. 2010, 1187, 122–128. [Google Scholar] [CrossRef]
- Kaushal, N.; Matsumoto, R.R. Role of sigma receptors in methamphetamine-induced neurotoxicity. Curr. Neuropharmacol. 2011, 9, 54–57. [Google Scholar] [CrossRef]
- Quinton, M.S.; Yamamoto, B.K. Causes and consequences of methamphetamine and MDMA toxicity. AAPS J. 2006, 8, E337–E347. [Google Scholar] [CrossRef] [PubMed]
- Theodore, S.; Cass, W.A.; Maragos, W.F. Methamphetamine and human immunodeficiency virus protein Tat synergize to destroy dopaminergic terminals in the rat striatum. Neuroscience 2006, 137, 925–935. [Google Scholar] [CrossRef] [PubMed]
- Miyatake, M.; Narita, M.; Shibasaki, M.; Nakamura, A.; Suzuki, T. Glutamatergic neurotransmission and protein kinase C play a role in neuron-glia communication during the development of methamphetamine-induced psychological dependence. Eur. J. Neurosci. 2005, 22, 1476–1488. [Google Scholar] [CrossRef] [PubMed]
- Hardingham, G.E.; Fukunaga, Y.; Bading, H. Extrasynaptic NMDARs oppose synaptic NMDARs by triggering CREB shut-off and cell death pathways. Nat. Neurosci. 2002, 5, 405–414. [Google Scholar] [CrossRef] [PubMed]
- Czub, S.; Koutsilieri, E.; Sopper, S.; Czub, M.; Stahl-Hennig, C.; Muller, J.G.; Pedersen, V.; Gsell, W.; Heeney, J.L.; Gerlach, M.; et al. Enhancement of central nervous system pathology in early simian immunodeficiency virus infection by dopaminergic drugs. Acta Neuropathol. 2001, 101, 85–91. [Google Scholar] [CrossRef]
- Hu, S.; Sheng, W.S.; Lokensgard, J.R.; Peterson, P.K.; Rock, R.B. Preferential sensitivity of human dopaminergic neurons to gp120-induced oxidative damage. J. Neurovirol. 2009, 15, 401–410. [Google Scholar] [CrossRef]
- Maragos, W.F.; Young, K.L.; Turchan, J.T.; Guseva, M.; Pauly, J.R.; Nath, A.; Cass, W.A. Human immunodeficiency virus-1 Tat protein and methamphetamine interact synergistically to impair striatal dopaminergic function. J. Neurochem. 2002, 83, 955–963. [Google Scholar] [CrossRef]
- Theodore, S.; Cass, W.A.; Dwoskin, L.P.; Maragos, W.F. HIV-1 protein Tat inhibits vesicular monoamine transporter-2 activity in rat striatum. Synapse 2012, 66, 755–757. [Google Scholar] [CrossRef]
- Zhu, J.; Ananthan, S.; Mactutus, C.F.; Booze, R.M. Recombinant human immunodeficiency virus-1 transactivator of transcription1-86 allosterically modulates dopamine transporter activity. Synapse 2011, 65, 1251–1254. [Google Scholar] [CrossRef]
- Alvarez-Carbonell, D.; Ye, F.; Ramanath, N.; Garcia-Mesa, Y.; Knapp, P.E.; Hauser, K.F.; Karn, J. Cross-talk between microglia and neurons regulates HIV latency. PLoS Pathog. 2019, 15, e1008249. [Google Scholar] [CrossRef]
- Pal, D.; Kwatra, D.; Minocha, M.; Paturi, D.K.; Budda, B.; Mitra, A.K. Efflux transporters- and cytochrome P-450-mediated interactions between drugs of abuse and antiretrovirals. Life Sci. 2011, 88, 959–971. [Google Scholar] [CrossRef] [PubMed]
- Nookala, A.R.; Li, J.; Ande, A.; Wang, L.; Vaidya, N.K.; Li, W.; Kumar, S.; Kumar, A. Effect of Methamphetamine on Spectral Binding, Ligand Docking and Metabolism of Anti-HIV Drugs with CYP3A4. PLoS ONE 2016, 11, e0146529. [Google Scholar] [CrossRef] [PubMed]
- Ikonomidou, C.; Bittigau, P.; Ishimaru, M.J.; Wozniak, D.F.; Koch, C.; Genz, K.; Price, M.T.; Stefovska, V.; Horster, F.; Tenkova, T.; et al. Ethanol-induced apoptotic neurodegeneration and fetal alcohol syndrome. Science 2000, 287, 1056–1060. [Google Scholar] [CrossRef] [PubMed]
- Pawlak, R.; Skrzypiec, A.; Sulkowski, S.; Buczko, W. Ethanol-induced neurotoxicity is counterbalanced by increased cell proliferation in mouse dentate gyrus. Neurosci. Lett. 2002, 327, 83–86. [Google Scholar] [CrossRef]
- Ikegami, Y.; Goodenough, S.; Inoue, Y.; Dodd, P.R.; Wilce, P.A.; Matsumoto, I. Increased TUNEL positive cells in human alcoholic brains. Neurosci. Lett. 2003, 349, 201–205. [Google Scholar] [CrossRef]
- Dingle, G.A.; Oei, T.P. Is alcohol a cofactor of HIV and AIDS? Evidence from immunological and behavioral studies. Psychol. Bull. 1997, 122, 56–71. [Google Scholar] [CrossRef]
- Meyerhoff, D.J. Effects of alcohol and HIV infection on the central nervous system. Alcohol Res. Health 2001, 25, 288–298. [Google Scholar]
- Friedman, H.; Newton, C.; Klein, T.W. Microbial infections, immunomodulation, and drugs of abuse. Clin. Microbiol. Rev. 2003, 16, 209–219. [Google Scholar] [CrossRef]
- Alak, J.I.; Shahbazian, M.; Huang, D.S.; Wang, Y.; Darban, H.; Jenkins, E.M.; Watson, R.R. Alcohol and murine acquired immunodeficiency syndrome suppression of resistance to Cryptosporidium parvum infection during modulation of cytokine production. Alcohol. Clin. Exp. Res. 1993, 17, 539–544. [Google Scholar] [CrossRef]
- Sepulveda, R.T.; Jiang, S.; Besselsen, D.G.; Watson, R.R. Alcohol consumption during murine acquired immunodeficiency syndrome accentuates heart pathology due to coxsackievirus. Alcohol Alcohol. 2002, 37, 157–163. [Google Scholar] [CrossRef][Green Version]
- Acheampong, E.; Mukhtar, M.; Parveen, Z.; Ngoubilly, N.; Ahmad, N.; Patel, C.; Pomerantz, R.J. Ethanol strongly potentiates apoptosis induced by HIV-1 proteins in primary human brain microvascular endothelial cells. Virology 2002, 304, 222–234. [Google Scholar] [CrossRef] [PubMed]
- Dong, Q.; Kelkar, S.; Xiao, Y.; Joshi-Barve, S.; McClain, C.J.; Barve, S.S. Ethanol enhances TNF-alpha-inducible NFkappaB activation and HIV-1-LTR transcription in CD4+ Jurkat T lymphocytes. J. Lab. Clin. Med. 2000, 136, 333–343. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; George, I.; Sperber, K. Effect of ethanol on monocytic function in human immunodeficiency virus type 1 infection. Clin. Diagn. Lab. Immunol. 1998, 5, 790–798. [Google Scholar] [CrossRef]
- Chen, W.; Tang, Z.; Fortina, P.; Patel, P.; Addya, S.; Surrey, S.; Acheampong, E.A.; Mukhtar, M.; Pomerantz, R.J. Ethanol potentiates HIV-1 gp120-induced apoptosis in human neurons via both the death receptor and NMDA receptor pathways. Virology 2005, 334, 59–73. [Google Scholar] [CrossRef] [PubMed]
- Prakash, O.; Zhang, P.; Xie, M.; Ali, M.; Zhou, P.; Coleman, R.; Stoltz, D.A.; Bagby, G.J.; Shellito, J.E.; Nelson, S. The human immunodeficiency virus type I Tat protein potentiates ethanol-induced neutrophil functional impairment in transgenic mice. Alcohol. Clin. Exp. Res. 1998, 22, 2043–2049. [Google Scholar] [CrossRef] [PubMed]
- Marra, C.M.; Zhao, Y.; Clifford, D.B.; Letendre, S.; Evans, S.; Henry, K.; Ellis, R.J.; Rodriguez, B.; Coombs, R.W.; Schifitto, G.; et al. Impact of combination antiretroviral therapy on cerebrospinal fluid HIV RNA and neurocognitive performance. AIDS 2009, 23, 1359–1366. [Google Scholar] [CrossRef] [PubMed]
- Smurzynski, M.; Wu, K.; Letendre, S.; Robertson, K.; Bosch, R.J.; Clifford, D.B.; Evans, S.; Collier, A.C.; Taylor, M.; Ellis, R. Effects of central nervous system antiretroviral penetration on cognitive functioning in the ALLRT cohort. AIDS 2011, 25, 357–365. [Google Scholar] [CrossRef]
- Vittinghoff, E.; Hessol, N.A.; Bacchetti, P.; Fusaro, R.E.; Holmberg, S.D.; Buchbinder, S.P. Cofactors for HIV disease progression in a cohort of homosexual and bisexual men. J. Acquir. Immune Defic. Syndr. 2001, 27, 308–314. [Google Scholar] [CrossRef]
- Baum, M.K.; Rafie, C.; Lai, S.; Sales, S.; Page, B.; Campa, A. Crack-cocaine use accelerates HIV disease progression in a cohort of HIV-positive drug users. J. Acquir. Immune Defic. Syndr. 2009, 50, 93–99. [Google Scholar] [CrossRef]
- Hinkin, C.H.; Barclay, T.R.; Castellon, S.A.; Levine, A.J.; Durvasula, R.S.; Marion, S.D.; Myers, H.F.; Longshore, D. Drug use and medication adherence among HIV-1 infected individuals. AIDS Behav. 2007, 11, 185–194. [Google Scholar] [CrossRef]
- Hicks, P.L.; Mulvey, K.P.; Chander, G.; Fleishman, J.A.; Josephs, J.S.; Korthuis, P.T.; Hellinger, J.; Gaist, P.; Gebo, K.A.; Network, H.I.V.R. The impact of illicit drug use and substance abuse treatment on adherence to HAART. AIDS Care 2007, 19, 1134–1140. [Google Scholar] [CrossRef] [PubMed]
- Meyer, M.R.; Maurer, H.H. Absorption, distribution, metabolism and excretion pharmacogenomics of drugs of abuse. Pharmacogenomics 2011, 12, 215–233. [Google Scholar] [CrossRef] [PubMed]
- Anzenbacher, P.; Anzenbacherova, E. Cytochromes P450 and metabolism of xenobiotics. Cell. Mol. Life Sci. 2001, 58, 737–747. [Google Scholar] [CrossRef] [PubMed]
- Walubo, A. The role of cytochrome P450 in antiretroviral drug interactions. Expert Opin. Drug Metab. Toxicol. 2007, 3, 583–598. [Google Scholar] [CrossRef]
- Kumar, S.; Rao, P.S.; Earla, R.; Kumar, A. Drug-drug interactions between anti-retroviral therapies and drugs of abuse in HIV systems. Expert Opin. Drug Metab. Toxicol. 2015, 11, 343–355. [Google Scholar] [CrossRef] [PubMed]
- Lucas, G.M.; Griswold, M.; Gebo, K.A.; Keruly, J.; Chaisson, R.E.; Moore, R.D. Illicit drug use and HIV-1 disease progression: A longitudinal study in the era of highly active antiretroviral therapy. Am. J. Epidemiol. 2006, 163, 412–420. [Google Scholar] [CrossRef] [PubMed]
- Ferris, M.J.; Mactutus, C.F.; Booze, R.M. Neurotoxic profiles of HIV, psychostimulant drugs of abuse, and their concerted effect on the brain: Current status of dopamine system vulnerability in NeuroAIDS. Neurosci. Biobehav. Rev. 2008, 32, 883–909. [Google Scholar] [CrossRef]
- Ciccarone, D. Stimulant abuse: Pharmacology, cocaine, methamphetamine, treatment, attempts at pharmacotherapy. Prim. Care 2011, 38, 41–58. [Google Scholar] [CrossRef]
- Borgmann, K.; Ghorpade, A. HIV-1, methamphetamine and astrocytes at neuroinflammatory Crossroads. Front. Microbiol. 2015, 6, 1143. [Google Scholar] [CrossRef]
- Ramesh, G.; MacLean, A.G.; Philipp, M.T. Cytokines and chemokines at the crossroads of neuroinflammation, neurodegeneration, and neuropathic pain. Mediators Inflamm. 2013, 2013, 480739. [Google Scholar] [CrossRef]
- Atluri, V.S.; Hidalgo, M.; Samikkannu, T.; Kurapati, K.R.; Jayant, R.D.; Sagar, V.; Nair, M.P. Effect of human immunodeficiency virus on blood-brain barrier integrity and function: An update. Front. Cell Neurosci. 2015, 9, 212. [Google Scholar] [CrossRef] [PubMed]
- Stoops, W.W.; Rush, C.R. Agonist replacement for stimulant dependence: A review of clinical research. Curr. Pharm. Des. 2013, 19, 7026–7035. [Google Scholar] [CrossRef] [PubMed]
- Ingersoll, K.S.; Cohen, J. The impact of medication regimen factors on adherence to chronic treatment: A review of literature. J. Behav. Med. 2008, 31, 213–224. [Google Scholar] [CrossRef] [PubMed]
- Atluri, V.S.; Jayant, R.D.; Pilakka-Kanthikeel, S.; Garcia, G.; Samikkannu, T.; Yndart, A.; Kaushik, A.; Nair, M. Development of TIMP1 magnetic nanoformulation for regulation of synaptic plasticity in HIV-1 infection. Int. J. Nanomed. 2016, 11, 4287–4298. [Google Scholar] [CrossRef] [PubMed]
- Ding, H.; Sagar, V.; Agudelo, M.; Pilakka-Kanthikeel, S.; Atluri, V.S.; Raymond, A.; Samikkannu, T.; Nair, M.P. Enhanced blood-brain barrier transmigration using a novel transferrin embedded fluorescent magneto-liposome nanoformulation. Nanotechnology 2014, 25, 055101. [Google Scholar] [CrossRef]
- Jayant, R.D.; Atluri, V.S.; Agudelo, M.; Sagar, V.; Kaushik, A.; Nair, M. Sustained-release nanoART formulation for the treatment of neuroAIDS. Int. J. Nanomed. 2015, 10, 1077–1093. [Google Scholar] [CrossRef]
- Kaushik, A.; Jayant, R.D.; Nikkhah-Moshaie, R.; Bhardwaj, V.; Roy, U.; Huang, Z.; Ruiz, A.; Yndart, A.; Atluri, V.; El-Hage, N.; et al. Magnetically guided central nervous system delivery and toxicity evaluation of magneto-electric nanocarriers. Sci. Rep. 2016, 6, 25309. [Google Scholar] [CrossRef]
- Jayant, R.D.; Atluri, V.S.R.; Tiwari, S.; Pilakka-Kanthikeel, S.; Kaushik, A.; Yndart, A.; Nair, M. Novel nanoformulation to mitigate co-effects of drugs of abuse and HIV-1 infection: Towards the treatment of NeuroAIDS. J. Neurovirol. 2017, 23, 603–614. [Google Scholar] [CrossRef]
- Stolbach, A.; Paziana, K.; Heverling, H.; Pham, P. A Review of the Toxicity of HIV Medications II: Interactions with Drugs and Complementary and Alternative Medicine Products. J. Med. Toxicol. 2015, 11, 326–341. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Drug | Receptor Involved in Mediating Neurotoxicity | Neurotransmitter System Disrupted | Effect on Viral Proteins/HIV Replication | Mechanism of Neurotoxicity | Interaction with ART |
|---|---|---|---|---|---|
| Opioids | MOR: Mu (μ) opioid receptor KOR: Kappa (κ) opioid receptor DOR: Delta (δ) opioid receptor | Dopaminergic system | Potentiates viral replication through: Increased expression of galectin 1 Inhibition of interferon Increased expression of CCR5 | Disrupt the integrity of BBB Oxidative stress Neuroinflammation through cytokine secretion | Compete with antiretrovirals for cytochrome P450 enzymes (CYP3A4, CYP2D6, CYP2C19, CYP2C9, and CYP2D67) |
| Cannabinoids | CB1 receptor CB2 receptor NMDA receptor | Endocannabinoid system Glutamatergic system | Gp120 increases the expression of FAAH, the enzyme that metabolizes the neuroprotective endocannabinoid, anandamide | Apoptosis Neuroinflammation | Compete with antiretrovirals for cytochrome P450 enzymes (CYP3A4 and CYP2C19) |
| Cocaine | Dopamine (Serotonin) (Norepinephrine) | Dopaminergic system | The combination of GP120 and cocaine increases the production of ROS and iNOS expression Cocaine augments Tat-mediated mitochondrial depolarization and production of ROS | Oxidative stress Epigenetic modifications Apoptosis | Compete with antiretrovirals for cytochrome P450 enzymes (CYP3A4 and CYP3A5) |
| Methamphetamine | Dopamine Serotonin Norepinephrine NMDA receptor | Dopaminergic system Glutamatergic system | Methamphetamine and Tat synergistically decrease dopamine reserves by binding to VMAT and DAT | Disrupt the integrity of BBB Impairing glial signaling Excitotoxicity | Compete with antiretrovirals for cytochrome P450 enzymes (CYP2D6 and CYP3A4) |
| Ethanol | NMDA receptor GABA receptor | Glutamatergic system GABAergic system | Stimulates HIV transcription through TNF secretion The combination of ethanol and Tat increases cytokine production | Apoptosis Ethanol exerts direct neurotoxicity | Compete with antiretrovirals for cytochrome P450 enzymes (CYP2E1 and CYP3A4) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sonti, S.; Tyagi, K.; Pande, A.; Daniel, R.; Sharma, A.L.; Tyagi, M. Crossroads of Drug Abuse and HIV Infection: Neurotoxicity and CNS Reservoir. Vaccines 2022, 10, 202. https://doi.org/10.3390/vaccines10020202
Sonti S, Tyagi K, Pande A, Daniel R, Sharma AL, Tyagi M. Crossroads of Drug Abuse and HIV Infection: Neurotoxicity and CNS Reservoir. Vaccines. 2022; 10(2):202. https://doi.org/10.3390/vaccines10020202
Chicago/Turabian StyleSonti, Shilpa, Kratika Tyagi, Amit Pande, Rene Daniel, Adhikarimayum Lakhikumar Sharma, and Mudit Tyagi. 2022. "Crossroads of Drug Abuse and HIV Infection: Neurotoxicity and CNS Reservoir" Vaccines 10, no. 2: 202. https://doi.org/10.3390/vaccines10020202
APA StyleSonti, S., Tyagi, K., Pande, A., Daniel, R., Sharma, A. L., & Tyagi, M. (2022). Crossroads of Drug Abuse and HIV Infection: Neurotoxicity and CNS Reservoir. Vaccines, 10(2), 202. https://doi.org/10.3390/vaccines10020202