1. Introduction
In the two years since the emergence of the severe acute respiratory syndrome coronavirus 2 (SARS-CoV2) and the 2019 coronavirus disease (COVID-19) [
1], approximately 64.4% of the world’s population has received at least one dose of a COVID-19 vaccine [
2]. Currently, vaccine solutions remain inadequate, with only 14.5% of people in low-income countries having received at least one dose. Furthermore, the successive emergence of the SARS-CoV-2 variants around the globe nullifies the efficacy of most currently available vaccines, which are based on the ancestral virus strain. SARS-CoV-2 is likely to remain the greatest threat to public health worldwide for the foreseeable future and further vaccine development will be necessary to contain this ongoing threat.
Several platforms have been used to develop a vaccine against SARS-CoV-2. These include inactivated viruses, viral vectors (non-replicating viruses), nucleic acids (mRNA/DNA), and proteins [
3]. These various types of vaccines have different safety, efficacy, and production profiles. For example, inactivated vaccines are considered extremely safe, but offer a lower efficacy than vaccines based on viral vectors and nucleic acids [
4]. On the other hand, while viral vectors elicit robust humoral and cell-mediated immune responses against SARS-CoV-2, pre-existing immunity to their viral components is a drawback that may reduce the vaccination’s effectiveness [
5]. In addition, vaccines run the risk of reversion to high pathogenicity, and a biosafety level 3 (BSL3) facility is required to produce SARS-CoV-2-specific vaccines. Given the difficulties in handling live viruses, the requirement for a highly stringent biosafety environment, and the lack of cold chains, protein subunit vaccines are an alternative platform that offers ease of production and safety. Protein-based COVID-19 vaccines have been shown to induce robust neutralizing antibody and T cell responses [
6,
7], and a number of them have entered clinical trials [
8,
9,
10].
Virus-like particles (VLPs), a type of protein-based vaccine, are self-assembling viral protein structures consisting of multiple subunits that resemble native viruses [
11], thus mimicking the immunogenic properties of the natural virus [
12]. The size of VLPs is favorable for uptake by antigen-presenting cells leading to the induction of robust innate and adaptive immune responses [
13,
14,
15]. Although the co-administration of adjuvants with VLPs is not necessary due to the multimeric nature of the antigen, the use of adjuvants usually improves their immunogenicity. While VLPs are commonly produced using protein(s) from a single virus type, structural proteins from different viruses can also be used to form chimeric VLPs. This designation for chimeric VLPs is based primarily on the use of heterologous viral proteins that line the inner layer of the cellular plasma membrane to facilitate the budding and egress of the virus. Generally, the enveloped chimeric VLPs have repeating units of immunogenic glycoprotein embedded in the lipid layers and associated with the proteins derived from the different viruses to self-assemble. While several SARS-CoV-2 VLP vaccines based on the envelope (E), matrix (M), and spike (S) proteins have been produced on different expression platforms [
16,
17,
18,
19], there are few reports on the development of chimeric SARS-CoV-2 VLP vaccines. One report described chimeric MLV-Gag/S VLPs that could be produced on a large scale in proprietary HEK293 cells and induced strong immune responses and protection in Syrian golden hamsters [
20]. In another case, the transient expression of SARS-CoV-S glycoprotein and an influenza matrix (M1) in insect cells was reported, which self-assembled into VLPs capable of inducing neutralizing antibodies and protecting mice from lethal challenges [
21]. More recently, the same approach was used to display SARS-CoV-2 S [
22]. Although the vaccine could induce neutralizing antibodies in mice, its protective efficacy remains to be evaluated.
In this study, we report the development of a chimeric VLP-based COVID-19 vaccine and its production from a HEK293T cell line stably expressing SARS-CoV-2 spike fused with an influenza A transmembrane and the cytoplasmic tail of HA and the M1 protein. The intramuscular administration of a prime-boost regimen to mice demonstrated the strong induction of the humoral response, resulting in antibodies with neutralizing activity against various variants of concern as well as the strong systemic suppression of SARS-CoV-2 in the blood. Our studies here demonstrate that SARS-CoV-2-based VLPs can be developed as a potentially safe and effective vaccine against the SARS-CoV-2 infection, although additional refinements of the vaccine, especially its formulation or dosage, are required to achieve desirable results.
2. Materials and Methods
2.1. Cell Lines
Human embryonic kidney (HEK) 293T cells were purchased from ATCC® (CRL-3216); ATCC®, Manassas, VA, USA). Cells were maintained in Opti-MEMTM I reduced serum media (Thermofisher Scientific, Waltham, MA, USA) supplemented with 10% fetal bovine serum (FBS) (Thermofisher Scientific, Waltham, MA, USA). Vero E6 cells were cultured in EMEM containing 2% FBS, 1% L-glutamine, 1% penicillin-streptomycin (Thermofisher Scientific), 40 µg/mL gentamicin (Thermofisher Scientific), and 0.25 µg/mL fungizone (Thermofisher Scientific). All cells were maintained in a 5% CO2 incubator at 37 °C.
2.2. Plasmid Construction
The codon-optimized spike construct comprising of SARS-CoV-2 spike (Wuhan-Hu-1 (EPI_ISL_402125)) with the disruption of the furin cleavage site and deletions of the transmembrane and cytoplasmic domains, fused to the transmembrane and cytoplasmic tail of the HA protein from influenza A/Indonesia/5/2005 (H5N1) [
21] (SΔfurin-HAcyt), was synthesized and cloned into the pUbEm plasmid containing the Emerald GFP reporter gene, so-called pUbEm-S. The codon-optimized M gene of influenza A/Indonesia/5/2005 (H5N1) [
21] was synthesized and cloned into the pUB-mCherry plasmid, so-called pUbmC-M1.
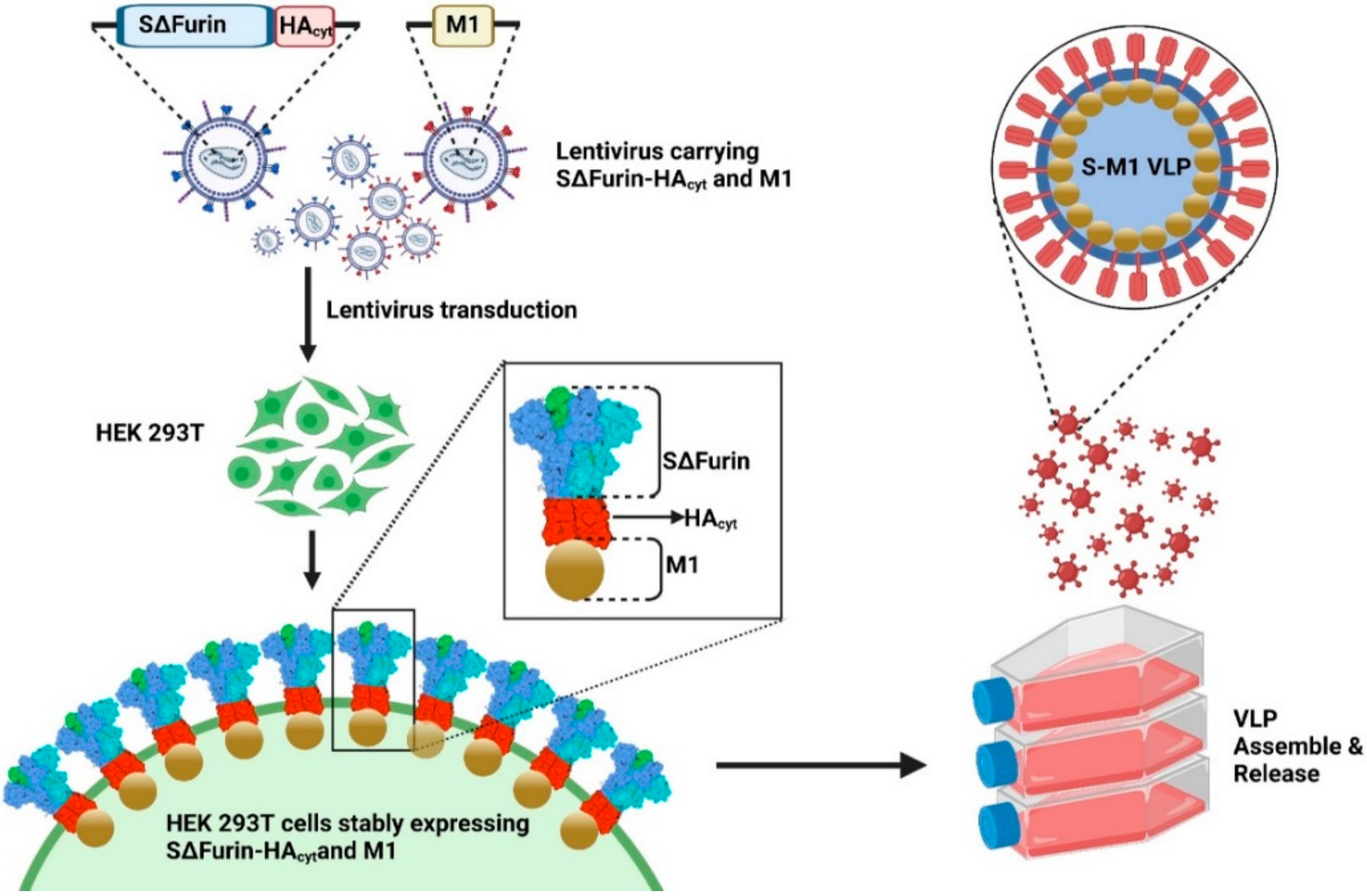
2.3. Establishment of a Stable Cell Line Producing Chimeric Virus-like Particles
To generate lentiviruses encoding S (Lenti-GFP-S) or M1 (Lenti-mCherry-M1) for cell transduction, HEK293T cells were seeded in a 6-well plate and transfected with 1.5 µg pUbEm-S or pUbmC-M1, 1.0 µg pCMVΔ8.91, and 0.5 µg pMD.G using Fugene HD (Promega, Madison, WI, USA) at a 3:1 ratio. Supernatants containing Lenti-mCherry-M1 or Lenti-GFP-S were harvested 3 days post-transfection and clarified by centrifugation.
To establish a stable cell line capable of continuously producing chimeric S-M1 virus-like particles (S-M1 VLPs), HEK293T cells seeded in a 6-well plate were transduced with Lenti-mCherry-M1 in the presence of 10 µg/mL polybrene. After 24 h, the media was replaced. Seventy-two hours post-transduction, cells were sorted for high fluorescence by flow-activated cell sorting (FACS) using the FACSAriaTM Fusion flow cytometer (BD Biosciences, CA, USA). The M1 positive cells were expanded, seeded in a 6-well plate, and further transduced with Lenti-GFP-S. The resulting cells expressing S and M1 were selected for green fluorescence of mEmerald GFP protein by FACS.
2.4. Expression and Purification of S-M1 VLPs
HEK293T cells expressing S-M1 VLPs (HEK293T-S-M1) were grown in Opti-MEMTM I reduced serum media (Thermofisher Scientific) in tissue-treated 15-cm2 dishes (5 dishes per production) for 72 h. The cell supernatant (up to 100 mL) was collected and concentrated through a 50 kDa Amicon® Ultra Centrifugal Filter Unit (Merck, Darmstadt, Germany) (final volume of 25 mL). The concentrated sample was subjected to ultracentrifugation through a 20% glycerol cushion at 135,000× g, 4 °C for 4 h. Cell pellet containing S-M1 VLPs was harvested and re-suspended with 500 µL of phosphate buffer saline (PBS). The S-M1 VLPs were quantified by dot-blot analysis against rabbit α-spike antibodies. The purified spike protein expressed in HEK293T cells was used as a standard.
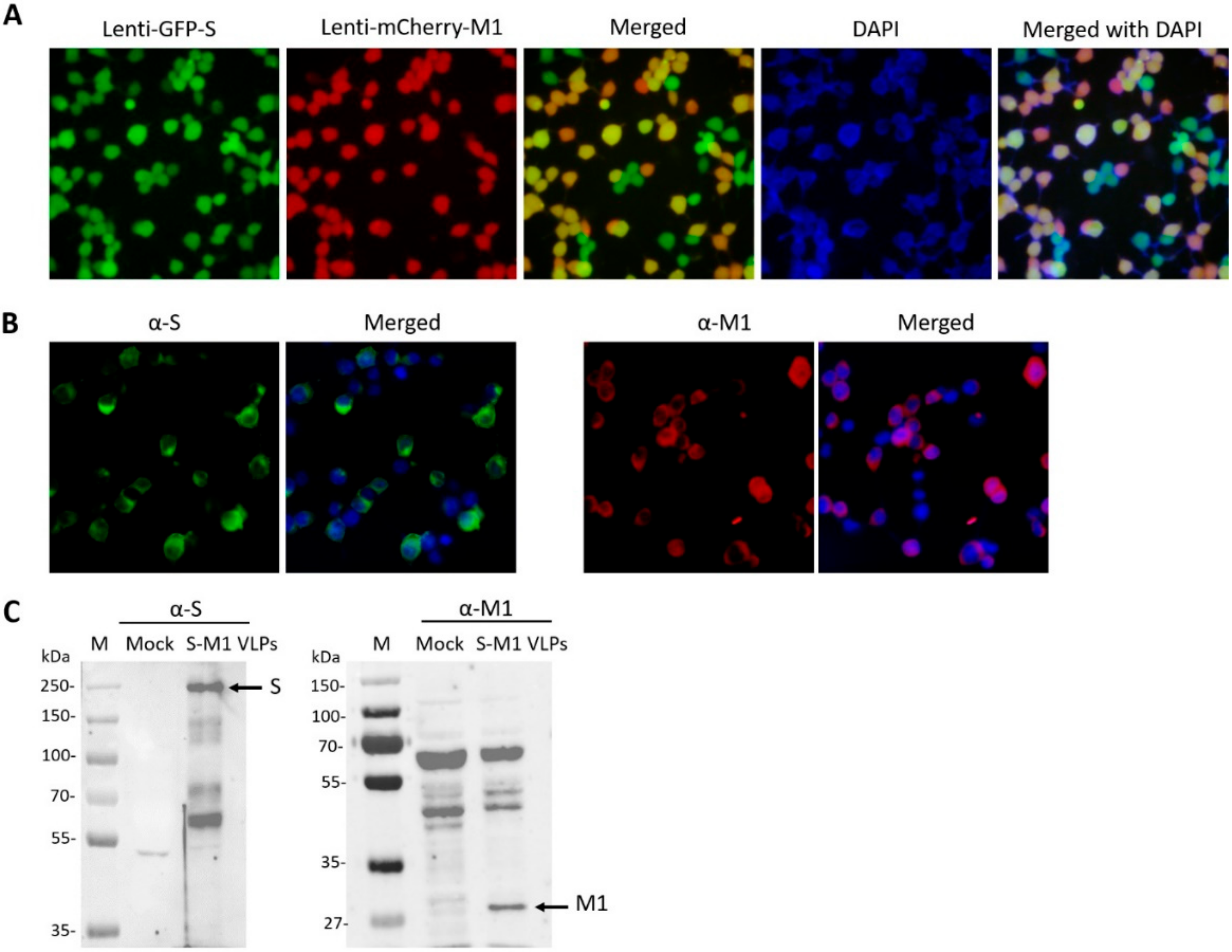
2.5. Immunofluorescence Assay
HEK293T cells expressing S-M1 VLPs were grown on coverslips in 6-well plates for 24 h. The cells were fixed with 4% paraformaldehyde at 4 °C for 20 min, blocked, and permeabilized with 3% bovine serum albumin (BSA) and 0.3% Triton-X for 30 min. The cells were then stained with mouse anti-S (Sino Biological, Beijing, China) and rabbit anti-influenza A M1 (Bio-Rad, Hercules, CA, USA) as primary antibodies for 1 h at room temperature. After incubation, cells were washed three times with PBST and then stained with goat α-rabbit Alexa Fluor® 488 (Abcam, Cambridge, UK) and goat α-mouse IgG Alexa Fluor® 647 (Abcam). After a wash step, cells were mounted with ProlongTM Gold Antifade Mountant with DAPI (Invitrogen, Carlsbad, CA, USA). Samples were visualized using FluoviewTM FV1000 confocal microscopy (Olympus, Tokyo, Japan).
2.6. Western Blot Analysis
The presence of S-M1 VLPs in stably expressed S-M1 cells and purified S-M1 VLPs was assessed by Western blot analysis. Cells were lysed using the Pierce™ mammalian cell lysis buffer (Thermofisher Scientific). Protein samples were loaded onto polyacrylamide gels and transferred to nitrocellulose membranes. Membranes were probed with rabbit α-S (Sino Biological) and α-influenza A M1 (Bio-Rad). Goat α-rabbit IgG-HRP (KPL, Milford, MA, USA) and α-mouse IgG-HRP antibodies were used as secondary antibodies for chemiluminescence detection with the ChemiDoc™ XRS+ imager (Bio-Rad).
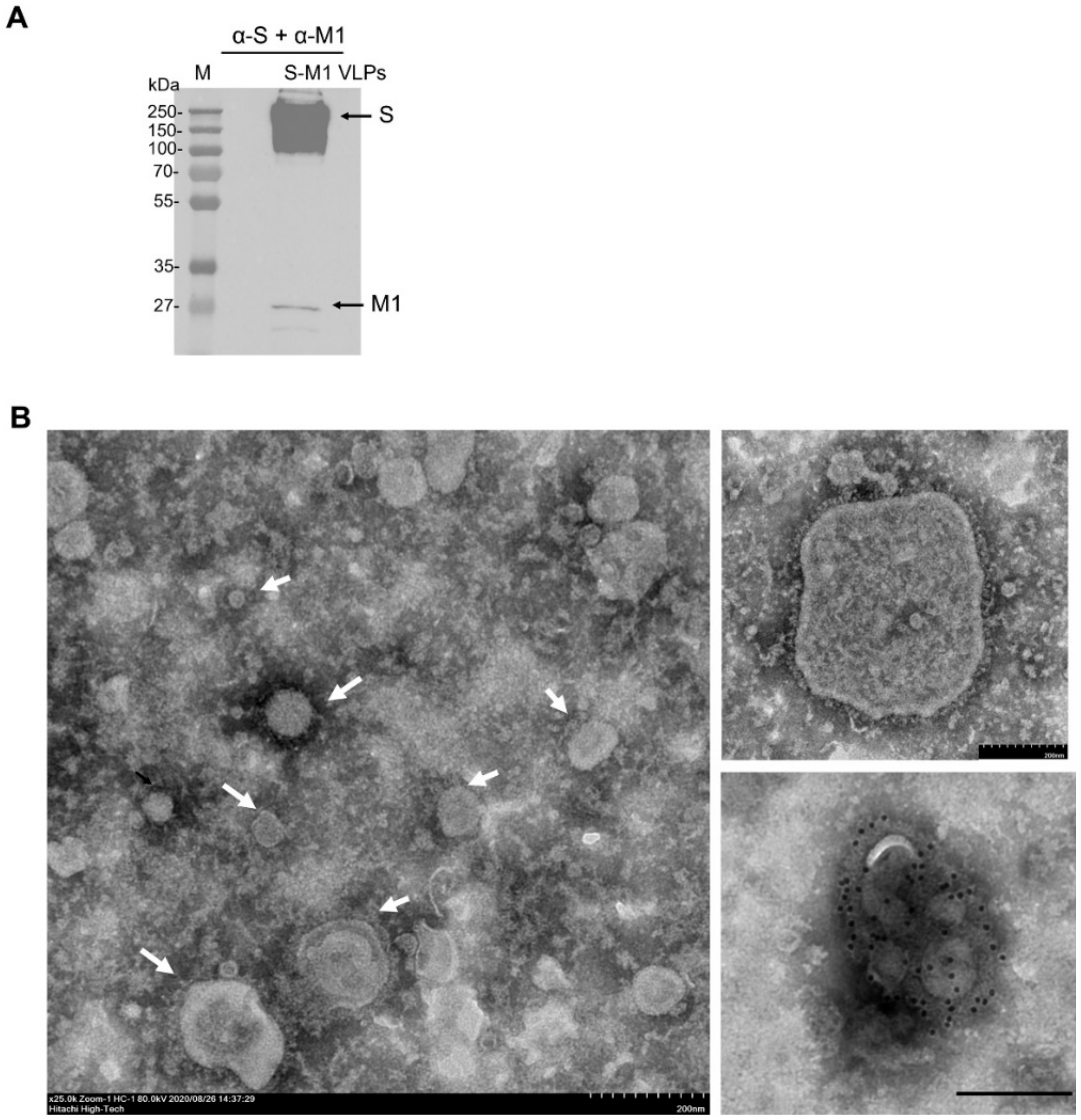
2.7. Transmission Electron Microscopy
Five microliters of the S-M1 VLPs in PBS were negatively stained with 3% phosphotungstic acid solution (PTA) for 30 sec. The S-M1 VLPs/PTA mixture was incubated on a carbon-coated copper grid (EMS, PA, USA) for 1 min. For immunogold labeling, the S-M1 VLPs/PTA mixture was adsorbed onto a carbon-coated copper grid for 30 min and then blocked with 1% bovine serum albumin (BSA) for 1 h. The grids were then incubated with rabbit α-spike antibody (Sino Biological) at a dilution of 1:30 in PBS for 1 h and rinsed six times with PBS, followed by incubation with goat α-rabbit IgG antibodies (conjugated with 10-nm gold particles) at a dilution of 1:100 in PBS for 1 h. After washing, the samples were fixed in 2% glutaraldehyde for 5 min and negatively stained with 3% PTA. The grid was kept in a vacuum incubator before observation with a transmission electron microscope (Hitachi H7700, Tokyo, Japan) at 80 kV.
2.8. Immunization in BALB/c Mice
To determine the immunogenicity of the vaccine in a mouse model, 8-week-old female BALB/c mice were purchased from Nomura Siam International Co., Ltd. (Bangkok, Thailand). Mice were divided into 4 groups (n = 5/group) and received the vaccine using a prime-boost regimen (3-week intervals). Mice were injected intramuscularly with the S-M1 VLPs with or without alum adjuvant. At 3 weeks after boosting, mice were euthanized for collection of serum and spleen samples. Sera were subjected to ELISA to determine IgG levels. Spleens were processed for splenocyte isolation and subjected to a mouse IFN gamma ELISpot assay (CTL, Shaker Heights, OH, USA) according to the manufacturer’s protocol.
2.9. Quantification of Specific Anti-SARS-CoV-2 Spike IgG by ELISA
SARS-CoV-2 S RBD-specific antibody responses were determined using an in-house anti-IgA/IgG SARS-CoV-2 S RBD ELISA. Briefly, 96-well high-binding ELISA plates were coated with 0.1 µg of SARS-CoV-2 S RBD per well and incubated overnight at 4 °C. Coated plates were washed six times (6×) with phosphate buffer saline containing 0.1% Tween 20 (PBST) before blocking with 5% skim milk in PBST (blocking solution) and then incubated at room temperature (RT) for 1 h. Sera were two-fold diluted with blocking solution before addition of 100 µL into duplicate wells. After incubation for 1 h at RT, plates were washed (6×) with PBST before adding 100 µL of HRP-conjugated goat α-human IgA (1:10,000) or HRP-conjugated goat α-human IgG (1:40,000), and incubating the plates at RT for 1 h. Plates were washed (6×) with PBST before adding 100 µL of TMB substrate and incubating at RT for 15–20 min. Enzyme activity was stopped by adding 50 µL of 2 M sulfuric acid and OD values at 450 nm were immediately measured using a plate reader. Values equal or higher than the cut-off value (≥3-fold of the mean OD450 of the negative control) were considered positive for IgA or IgG antibodies. Using this criterion, the IgA assay performance had 85.7% sensitivity and 97.3% specificity while the IgG assay exhibited 96.0% sensitivity and 96.7% specificity.
2.10. SARS-CoV-2 Microneutralization
A CPE-based microneutralization assay (MN) with colorimetric readout was used to determine neutralizing (NT) antibodies against live SARS-CoV-2 Wuhan-Hu-1 strain virus. Cell (CC) and virus control (VC) wells were included. Sera were heat-inactivated at 56 °C for 30 min and two-fold serially diluted starting from 1:10. Sera were mixed with 100 TCID50 of SARS-CoV-2 virus and incubated for 1 h at 37 °C in a 5% CO2 incubator. Then, the serum–virus mixtures were inoculated in duplicate onto Vero E6 cells seeded in 96-well plates and incubated for 3 days at 37 °C, and 5% CO2. Cells were fixed with methanol/acetone and blocked with 1X PBS containing 2% bovine serum albumin (BSA) and 0.05% Tween 20. Cells were then stained with SARS-CoV/SARS-CoV-2 nucleocapsid mAb (Sino Biological) as the primary antibody and HRP-conjugated goat α-rabbit polyclonal antibody (Dako, Denmark A/S). After the washing steps, TMB substrate was added (KPL) into each well and the reaction was stopped with 1N HCl. Absorbance was measured at 450 nm by an ELISA microplate reader. The percentage of virus infectivity in VC and samples was calculated based on the OD of CC using the formula infectivity (%) = (OD of CC − OD of the sample) × 100. The MN50 titer was calculated by determining the half-maximal inhibitory dilution. All procedures were performed in a BSL-3 laboratory following a standard neutralization assay.
2.11. SARS-CoV-2 Pseudotyped Virus Neutralization Assay
Lentiviral vectors carrying the firefly luciferase reporter were pseudotyped with the full-length S of various SARS-CoV-2 variants of concern (VOC), namely alpha, beta, gamma, delta, and omicron, using methods as described previously [
23,
24]. Sera were heat-inactivated at 56 °C for 30 min, incubated with 1 × 10
4 relative light units (RLU)/mL of the pseudotyped virus at 37 °C for 1 h, then diluted (two-fold serial dilutions) in DMEM supplemented with 10% FBS. The mixtures were added to individual wells of opaque, white 96-well microplates. HEK 293T cells expressing ACE2 and TMPRSS2 cells (1 × 10
4 cells) were suspended in 50 µL DMEM supplemented with 10% FBS, and then added to each well. Plates were incubated at 37 °C, 5% CO
2 for 48 h, and a readout was performed by removing supernatants and adding 25 µL of Bright-Glo
TM luciferase substrate (Promega) to each well. Entry of pseudotyped viruses, as quantified by firefly luciferase signal, was measured using the Synergy HTX plate reader (BioTek, Winusky, VT, USA). Values were normalized against signals from no-serum controls. The PVNT50 value was calculated by determining the half-maximal inhibitory dilution.
2.12. SARS-CoV-2 Challenge Test in K18-hACE2 Mice and Sample Collection
Female K18-hACE2 mice (6 weeks old) strain 2B6.Cg-Tg (K18-ACE2)2Prlmn/J were purchased from InVivos (Singapore) and maintained in micro-isolator cages in an ABSL-2 facility before the SARS-CoV-2 challenge or in an ABSL-3 facility after the challenge. Mice had nesting and shelter facilities as part of their cage environment and ad libitum access to commercial pelleted diets and chlorinated water.
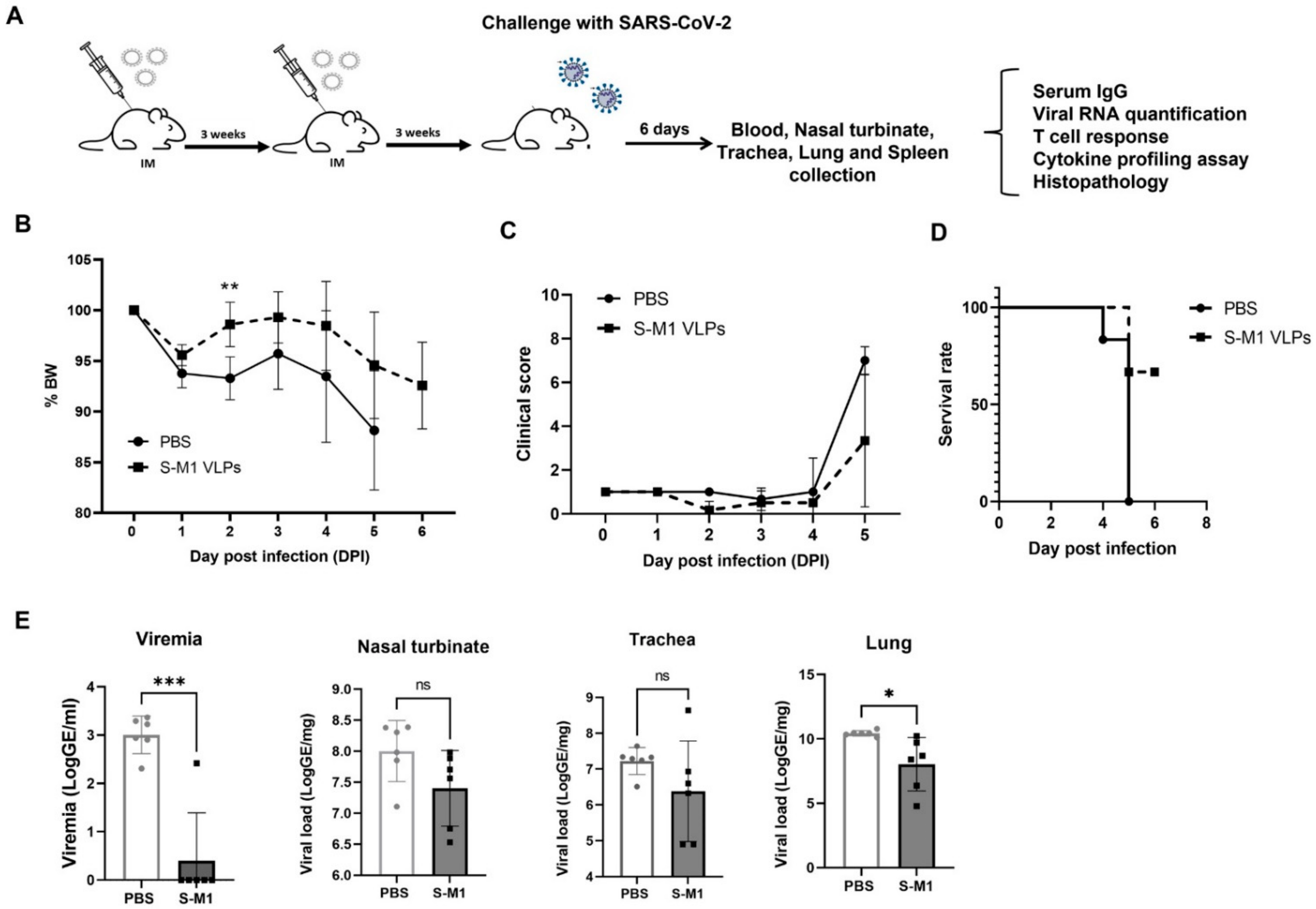
Twelve mice were included and randomly divided into two groups (n = 6). Mice were either intramuscularly (IM) administered S-M1 VLPs or PBS in a three-week interval prime-boost schedule. Three weeks following the second immunization (day 42), all mice were intranasally (IN) challenged with 2 × 104 PFU/ 50 μL of the SARS-CoV-2 Wuhan reference strain hCoV-19/Hong Kong/VM20001061/2020. Observations for clinical signs of disease including body weight, inappetence, and behavioral anomalies were performed daily post-infection. Animals were humanely euthanized by CO2 inhalation upon meeting euthanasia criteria or at pre-determined endpoints, following institutional and AVMA guidelines (2020 edition).
At the study endpoint, animals were euthanized. Serum samples were analyzed for viremia (qRT-PCR) and SARS-CoV-2-specific antibody responses (qualitative IgG & IgA spike-RBD). Major organs were collected for viral RNA determination, histopathology, and SARS-CoV-2-specific-in situ hybridization. SARS-CoV-2-specific cellular immune responses were determined at the study endpoint in splenocytes by the ELISpot assay and supernatants of stimulated cells were used for cytokine profiling.
2.13. Virus Titration
Titers of viable virus in tracheal lavage specimens were determined using a 50% tissue culture infectious dose assay (TCID
50). All procedures were performed in a BSL-3 laboratory following a standard neutralization assay. Tracheal lavage samples were serially diluted 5-fold (starting from undiluted to 10
−5) with Vero E6 culture media, before inoculation onto Vero E6 cells in 96-well plates. Cell control (CC) wells were included in every single plate. After incubation for 1 h, the infectious medium was removed before replenishing with Vero E6 culture media and cultured for 5 days at 37 °C in a 5% CO
2 incubator. Cells were stained with 0.02% neutral red in PBS, pH 7.2 ± 0.1, and incubated for 1 h at room temperature (RT). Then, cells were washed twice with 1× PBS before adding lysis solution (50% absolute ethanol and 1% acetic acid in distilled water). After incubation for 15 min at RT, absorbance (OD) values at 540 nm were determined using the SpectraMax microplate reader (Molecular Devices, CA, USA). The cut-off value was calculated by dividing the mean OD of CC wells by two. Any test well with OD values less than the cut-off was scored as positive for viral growth. TCID
50 value for each sample was calculated using the Reed–Muench method [
25].
2.14. Viral RNA Quantification by Real-Time RT-PCR
Viral RNA in serum (140 µL) and tissue (20–30 mg) samples was extracted using the QIAamp viral RNA mini kit (QIAGEN, Hilden, Germany) or the RNeasy Mini kit (QIAGEN) according to the manufacturer’s instructions. A total volume of 50 µL of viral RNA was obtained for each sample. Five microliters of each RNA sample were used for RT-qPCR targeting the N1 region of the SARS-CoV-2 nucleocapsid (N) gene, using the US-CDC procedure [
26] and in vitro SARS-CoV-2 RNA transcripts (IVTs) as standards. The RT-qPCR was performed using the SuperScript III Platinum One-Step Quantitative RT-PCR System (Invitrogen) with the US-CDC 2019-nCoV-N1 combined primer and probe mix. A one-step RT-PCR consisting of a 30-min RT step at 50 °C, 5 min of Taq polymerase inhibitor inactivation at 95 °C, followed by 45 at 95 °C for 15 s and 58 °C for 30 s was performed on an ABI 7500 Fast Real-Time PCR system (Applied Biosystems, MA, USA). Three internal controls included a no-template control (NTC), a negative extraction control (NEC), and a positive extraction control (PEC). Serial dilutions of IVT RNA standards were run along with test samples in each experiment. The number of copies of viral RNA per sample was derived from standard curves generated from serial dilutions of IVTs (5, 50, 5 × 10
2, 5 × 10
3, 5 × 10
4, and 5 × 10
5 RNA copies or genomic equivalent (GE) per reaction). The GE per ml of virus in a serum sample was calculated using the formula: the number of copies per reaction × 10,000 × the volume of a serum sample used (µL) for extraction. The GE per gram of virus in a tissue sample was calculated using the formula: number of copies per reaction × 10,000 × weight of tissue sample (mg) used for extraction.
2.15. Splenocyte Isolation
Spleens were collected from euthanized mice and placed in 50-mL conical tubes containing 5 mL of RPMI media with 10% heat-inactivated FBS supplemented with L-glutamine and penicillin-streptomycin antibiotics to prepare single-cell suspensions. Each spleen was placed on a Petri dish, minced into small pieces using a surgical blade, and then mashed against the surface of a 70-µm cell strainer over a 50-mL conical tube. Red blood cell contaminants were removed by incubation with Gibco ACK lysing buffer (Thermofisher Scientific) for 5 min at room temperature. Cell suspensions were washed by centrifugation at 300× g for 5 min and resuspended in RPMI containing 10% FBS for counting before adjusting to the desired cell quantity. All procedures were performed in a BSL-3 laboratory.
2.16. IFN-γ ELISpot Assay
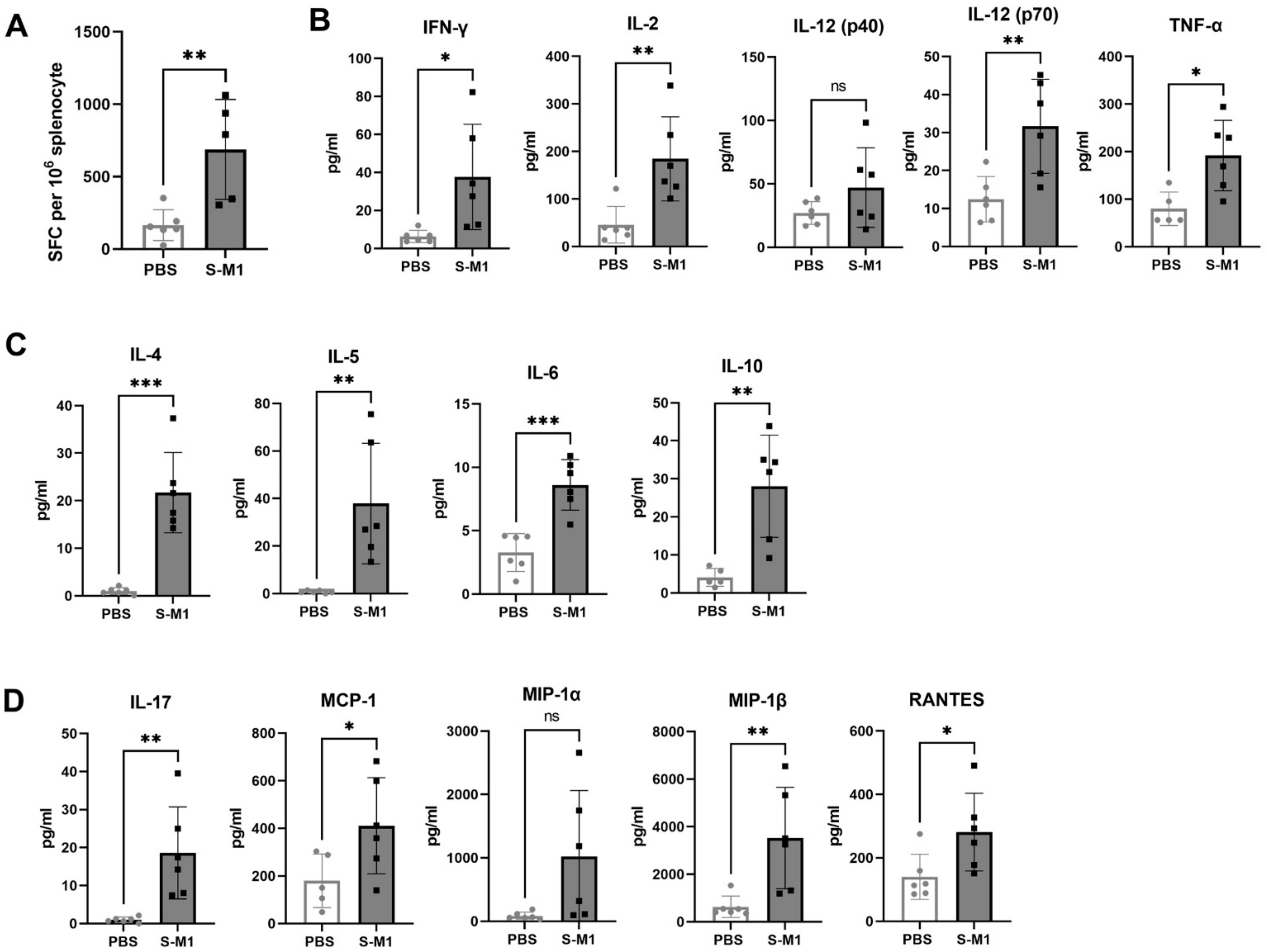
To assess SARS-CoV-2-specific cellular immune responses in splenocytes, an ex vivo IFN-γ ELISpot assay was performed using an ELISpot Mouse IFN-γ ELISpot PLUS kit (ALP) (Mabtech, Sweden) in a BSL-3 laboratory according to the manufacturer’s protocol. Briefly, 96-well-PVDF membrane plates pre-coated with monoclonal antibody clone AN18 were washed and then blocked with RPMI media containing 10% FBS, supplemented with L-glutamine and penicillin-streptavidin antibiotics for at least 30 min at 37 °C. The media was discarded and 8 × 105 splenocytes isolated from the spleen were stimulated with 2 µg/mL PepMix™ SARS-CoV-2 S peptide pools based on the Wuhan-Hu-1 strain (JPT Peptide Technologies, Berlin, Germany) for 18–20 h at 37 °C in a 5% CO2 incubator. Phytohemagglutinin (PHA) at a final concentration of 10 µg/mL was used as a positive control and media alone was used as a negative control. After the incubation period was complete, culture supernatants were collected for cytokine profiling and IFN-γ-secreting cells were detected by incubation of biotinylated detection antibody (clone R4-6A2) at a 1:1000 dilution for 2 h at room temperature. Then, the plates were washed and further incubated with streptavidin-conjugated alkaline phosphatase at a 1:1000 dilution for 1 h at room temperature. After further washing, BCIP/NBT-plus substrate was added. Spot numbers were counted by an automated ELISpot reader with Smart Count™ ImmunoSpot 6 software (S6 Universal FluoroSpot, Cellular Technology Limited, Shaker Heights, OH, USA). Results were reported as spot-forming cells (SFC) per one million cells. The non-specific background was subtracted from the experimental readings.
2.17. Cytokine Profiling Bio-Plex Assay
Culture supernatants were collected from the ELISpot assay plate and inactivated with Triton-X 100 at a final concentration of 0.5% for 30 min at room temperature. Cytokine profiling was performed using the Bio-Plex Pro Mouse Cytokine 23-plex Assay (Bio-Rad) based on magnetic beads, and the final reaction was detected using the Bio-Plex 200 analyzer. The 23 cytokines consisted of IL-1α, IL-1β, IL-2, IL-3, IL-4, IL-5, IL-6, IL-9, IL-10, IL-12 (p40), IL-12 (p70), IL-13, IL-17A, eotaxin, G-CSF, GM-CSF, IFN-γ, KC, MCP-1 (MCAF), MIP-1α, MIP-1β, RANTES, and TNF.
2.18. Histopathology
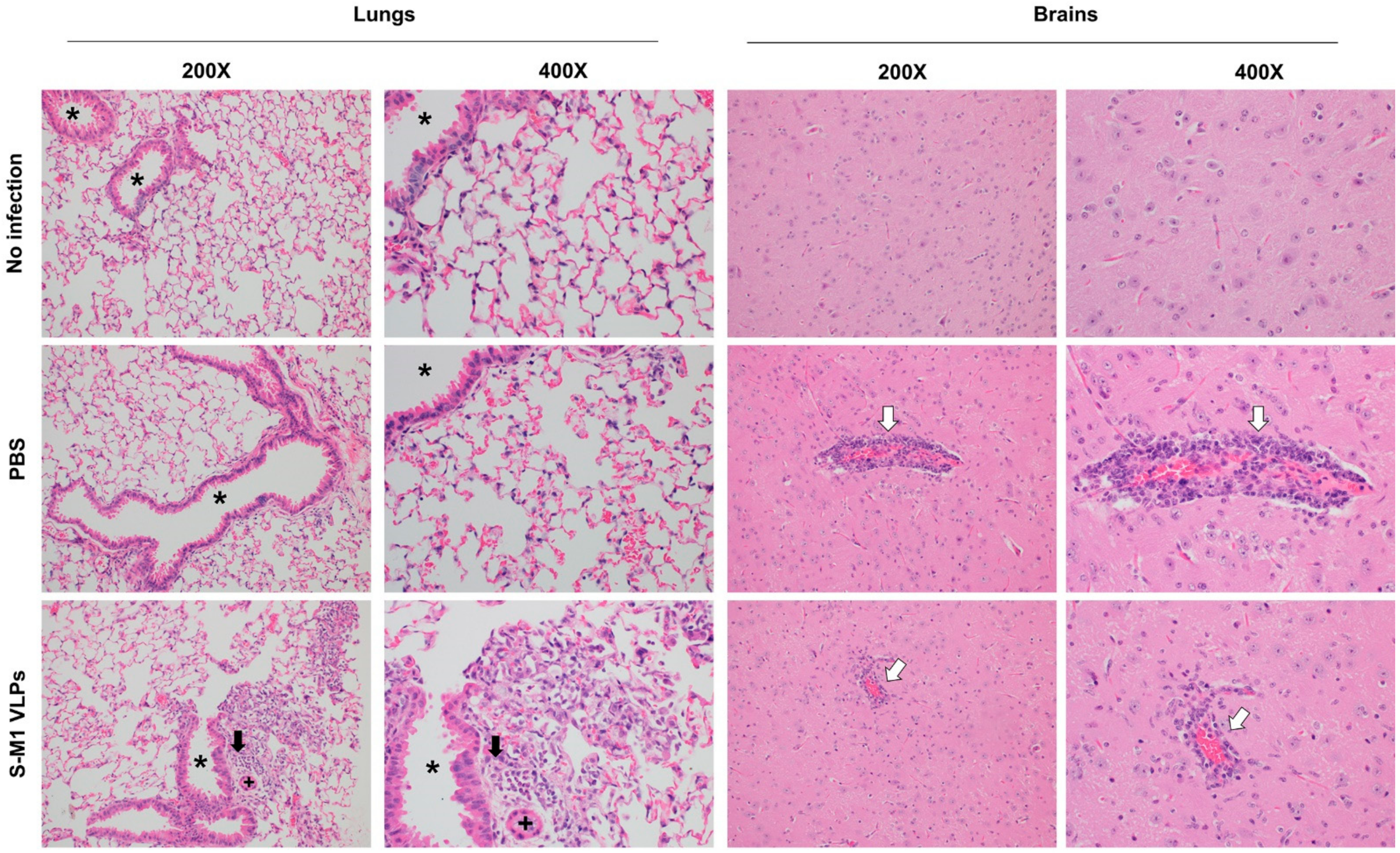
Necropsies of mice were performed in an ABSL3 laboratory. Major organs harvested postmortem for histopathologic analysis included lung, lymphoid tissue (lymph nodes, spleen, and thymus), brain, nasal turbinates, and adrenal glands. All tissues were immediately fixed in 10% neutral buffered formalin for at least 48 h, then paraffin-embedded, and sectioned at 4 µm thickness. Embedded tissue sections were then de-paraffinized, and rehydrated through a series of xylene substitutes and isopropanol. Tissue sections were stained with hematoxylin-eosin (H&E) and analyzed by a board-certified veterinary pathologist.
2.19. In-Situ Hybridization for SARS-CoV-2 RNA
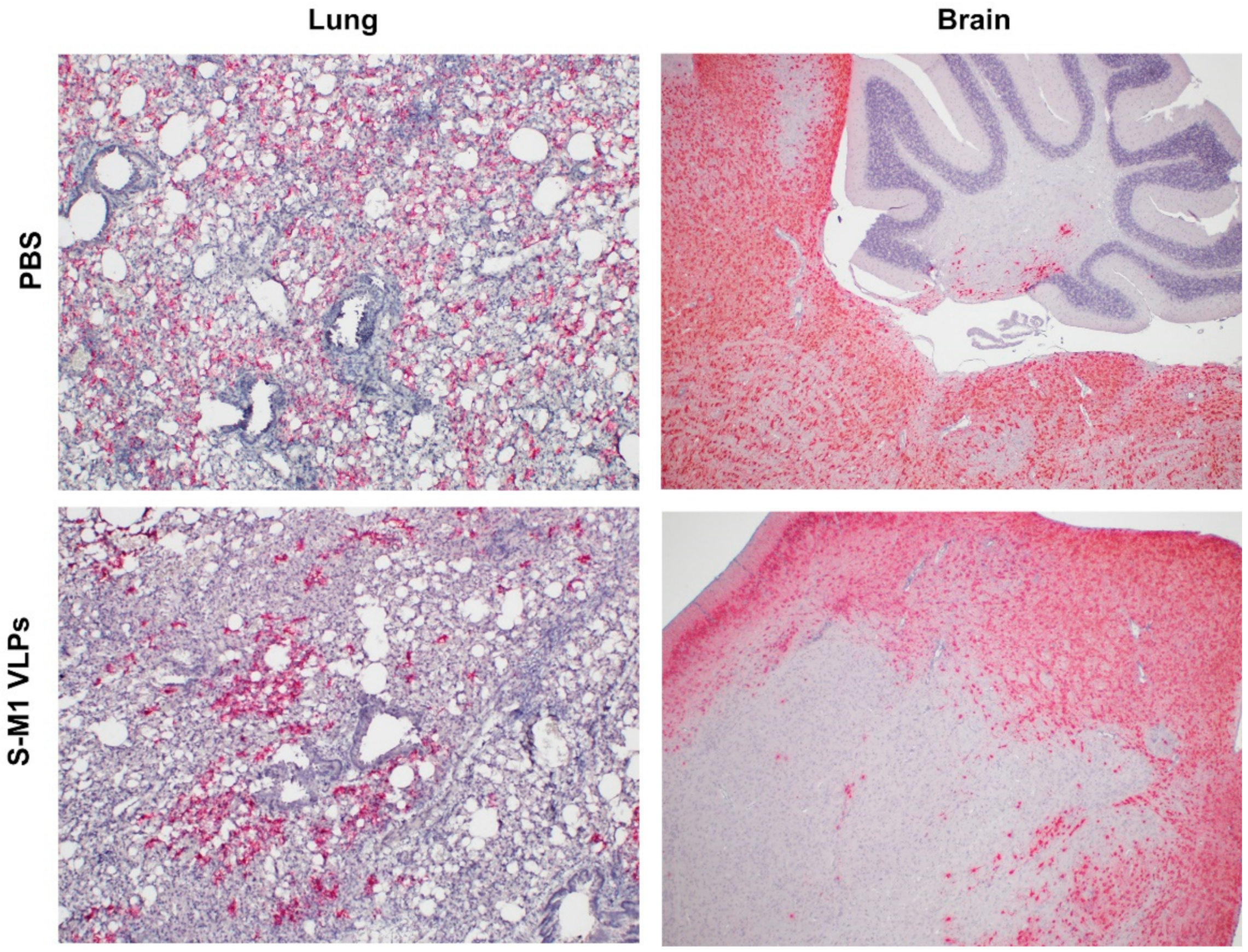
To detect the presence of SARS-CoV-2 RNA in mouse tissue samples, formalin-fixed paraffin-embedded (FFPE) lung and brain tissues were examined using the RNAscope® in situ hybridization (ISH) assay. The V-nCoV2019-S RNAscope® probe (Advanced Cell Diagnostics, Newark, CA, USA), specific for the SARS-CoV-2 S gene based on isolate Wuhan-Hu-1 (Genbank accession number NC_045512.2, targeting region 21631-23303), was used. The RNAscope® ISH assay was performed using an RNAscope 2.5 HD Red Detection Kit (Advanced Cell Diagnostics) as follows. FFPE tissue slides were de-paraffinized and treated with hydrogen peroxide for 10 min at room temperature, followed by target retrieval in 1× target retrieval solution in a steamer at least 99 °C for 15 min. Slides were then incubated with Protease Plus (Advanced Cell Diagnostics) for 30 min at 40 °C in a HybEZ™ oven (Advanced Cell Diagnostics) and subsequently incubated with the SARS-CoV-2 specific probe for 2 h at 40 °C in the HybEZ™ oven. The signal was amplified using a specific set of amplifiers (AMP1-6), as recommended by the manufacturer, and was detected using a Fast Red solution for 5 min at room temperature. Slides were counterstained with 50% Gill hematoxylin III (Sigma Aldrich, MO, USA) for 2 min and extensively washed with tap water. Next, the slides were dehydrated in a 60 °C drying oven until completely dry and then immersed in xylene before mounting with mounting medium.
2.20. Statistical Analyses
Differences in means between groups were analyzed by the one-way method ANOVA using GraphPad Prism 5.0 (GraphPad Software Inc., San Diego, CA, USA). p values of <0.05 were considered statistically significant. All data are presented as means ± standard error of means (SEM), unless otherwise stated.
2.21. Ethics Statement
Immunogenicity testing in BALB/c mice was conducted following the guidelines of the Institutional Animal Care and Use Committee (IACUC) of the National Center for Genetic Engineering and Biotechnology, Thailand (BT-Animal 22/2563). The SARS-CoV-2 challenge study in K18-hACE2 mice was performed under an approved Institutional Animal Care and Use Committee (IACUC) protocol at the Armed Forces Research Institute of Medical Sciences (AFRIMS), Thailand, and AAALAC International-accredited facility. The IACUC protocol number was PN21-03. The animal research was conducted in compliance with Thai laws, the Animals for Scientific Purposes Act, B.E. 2558 (A.D. 2015), the Animal Welfare Act, and all applicable U.S. Department of Agriculture, Office of Laboratory Animal Welfare, and U.S. Department of Defense guidelines. All animal research adhered to the Guide for the Care and Use of Laboratory Animals, NRC Publication (8th Edition) [
27].
4. Discussion
In this study, we successfully developed a cell line that stably and continuously generates chimeric VLPs based on the influenza virus M1 protein and SARS-CoV-2 S fused with the influenza virus HA transmembrane domain and the cytoplasmic tail. This approach was able to generate VLPs containing the S on their surface in their uncleaved form. When immunized in mice, these chimeric VLPs elicited robust humoral immune responses that were able to neutralize the ancestral strain of SARS-CoV-2 and maintain varying degrees of neutralizing activity against the alpha, beta, gamma, and delta variants, but not against omicron. Cellular immunity was also induced, along with balanced Th1/Th2 responses. While there was a slight trend toward viral load reduction in the lower respiratory tract, the effect of vaccination on the viremia suppression was considerable. The vaccine also improved the survival of mice after a SARS-CoV-2 challenge. However, the histological findings indicated that the S-M1 VLPs elicited immune responses that only partially protected the mice from severe disease, but not from viral invasion of the brain and other organs.
Recently, an influenza virus-based SARS-CoV-2 VLP construct was reported, with the vaccine produced in SF9 cells by the co-transfection of baculovirus plasmids expressing S and M1 proteins. In this study, mice receiving the VLPs elicited neutralizing antibodies; however, the vaccine efficacy was not evaluated [
22]. While we have also used influenza virus M1 as the structural basis for VLP self-assembly, our development of an S-M1-expressing cell line that constitutively releases VLPs into the cell culture supernatant will greatly facilitate cost-effective and large-scale VLP production. It has been shown that target antigens must be present on the surface of VLPs in a high density to induce a high-titer antibody response. As shown by α-spike immunogold labeling (
Figure 3B), the membrane-associated SARS-CoV-2 S protein was densely localized on the S-M1 VLP membrane. As shown in
Figure 4A,B, mice immunized with chimeric S-M1 VLPs elicited strong anti-S antibody responses. In addition, from the presence of IFN-γ-secreting S-specific T cells, our S-M1 VLPs could elicit robust cell-mediated immune responses. While previous studies reported that VLP epitopes could be presented to dendritic cells by both MHC I and II [
13], leading to the activation of helper and cytotoxic T cells, whether S-M1 VLPs can elicit antigen-specific cytotoxic T lymphocyte (CTL) responses remains to be tested. Furthermore, although many VLPs do not require co-administration of an adjuvant due to their inherent property of displaying multimeric antigens, the addition of alum to our S-M1 VLP increased its efficacy and enhanced its adaptive immune responses.
The influenza M1 protein, via interactions with lipid membranes, cytoplasmic tails of transmembrane glycoproteins, and M1–M1 interactions, helps mediate virus budding from the cell surface [
30]. The M1 anchoring of the HAcyt-fused SARS-CoV-2 S likely provided a three-dimensional architecture similar to the native SARS-CoV-2 S trimer, thereby promoting a strong neutralizing antibody in the vaccinated mice. The use of other viral proteins, such as the VLP base, may organize the trimeric S in different distributions and densities, and allow modulation of the immune response against the S protein.
Both humoral immunoglobulin (IgA and IgG) and T cell responses were demonstrated to contribute to protective immune responses against the SARS-CoV-2 infection [
31,
32]. While sterilizing immunity against SARS-CoV-2 appears to be mediated mainly by neutralizing antibodies, cell-mediated immunity, particularly CTL responses, may play a crucial role in reducing the severity of symptoms, in the elimination of infected cells, and in long-term memory. Although intramuscular administration of S-M1 VLPs elicited a strong neutralizing antibody, the vaccine-induced T cell response needed to be improved to determine how long the immune responses would last.
Despite having high serum-neutralizing antibody titers, mice immunized with S-M1 VLPs were just as susceptible to the SARS-CoV-2 infection via the respiratory mucosa as their control counterparts. Compared to the control mice, all vaccinated mice displayed only mild to moderate clinical symptoms, lower viral titers in the lungs, and, most importantly, an undetectable virus in the bloodstream. Despite complete viremia suppression, significant lesions were detected in multiple body systems, indicating a systemic spread. Furthermore, due to the intramuscular administration, our vaccine did not produce detectable S-specific mucosal secretory IgA antibodies. We believe that novel VLP formulations that allow mucosal administration, particularly via the upper respiratory tract, could improve the S-M1 VLP-based vaccine’s protective efficacy against SARS-CoV-2.
Currently, many technical and practical challenges associated with large-scale production limit the use of VLPs to small-scale research, although their immunogenicity has been demonstrated in many preclinical studies. In particular, VLPs that are structurally composed of multiple protein components encounter difficulties in upscaling to obtain highly purified VLPs. Combinations of different purification methods such as size exclusion chromatography and ion exchange chromatography could be used. However, these methods would inevitably increase the complexity and cost of S-M1 VLP production. Factors such as storage temperature, pH, and stabilizer/buffer could also affect the integrity and stability of VLP particles, thus determining the efficacy of S-M1 VLPs. While we have attempted to take advantage of the HEK293T cell line that constitutively expresses both SARS-CoV-2 S and influenza M1 proteins, this mammalian cell-based S-M1 VLP production system requires further process optimization to achieve a high purity and yield which is suitable for large-scale production.
Unlike many subunit vaccines, which generally lack the native danger signals that are critical for stimulating immune responses, VLPs possess not only inherent multimeric structures, but also various pathogen-associated molecular patterns (PAMPs) that resemble wild-type viruses. By combining many surface PAMPs with highly repetitive epitopes on the particles, VLPs have unique features that confer superior immunogenic properties. While these unique properties make VLPs a potential COVID-19 vaccine platform from an immunological perspective, much work remains to be done to translate this immunogenicity into robust and long-lasting protection.


 ,
, 









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}