
The Role of Natural Killer Cells in Oncolytic Virotherapy: Friends or Foes?


Abstract
1. Introduction
2. Immune Response to Cancer
3. Cancer Immunotherapies
4. Oncolytic Virotherapy
4.1. Introduction to Oncolytic Viruses
4.2. Oncolytic Viruses for Cancer Immunotherapy
4.3. Genetic Engineering in OVT
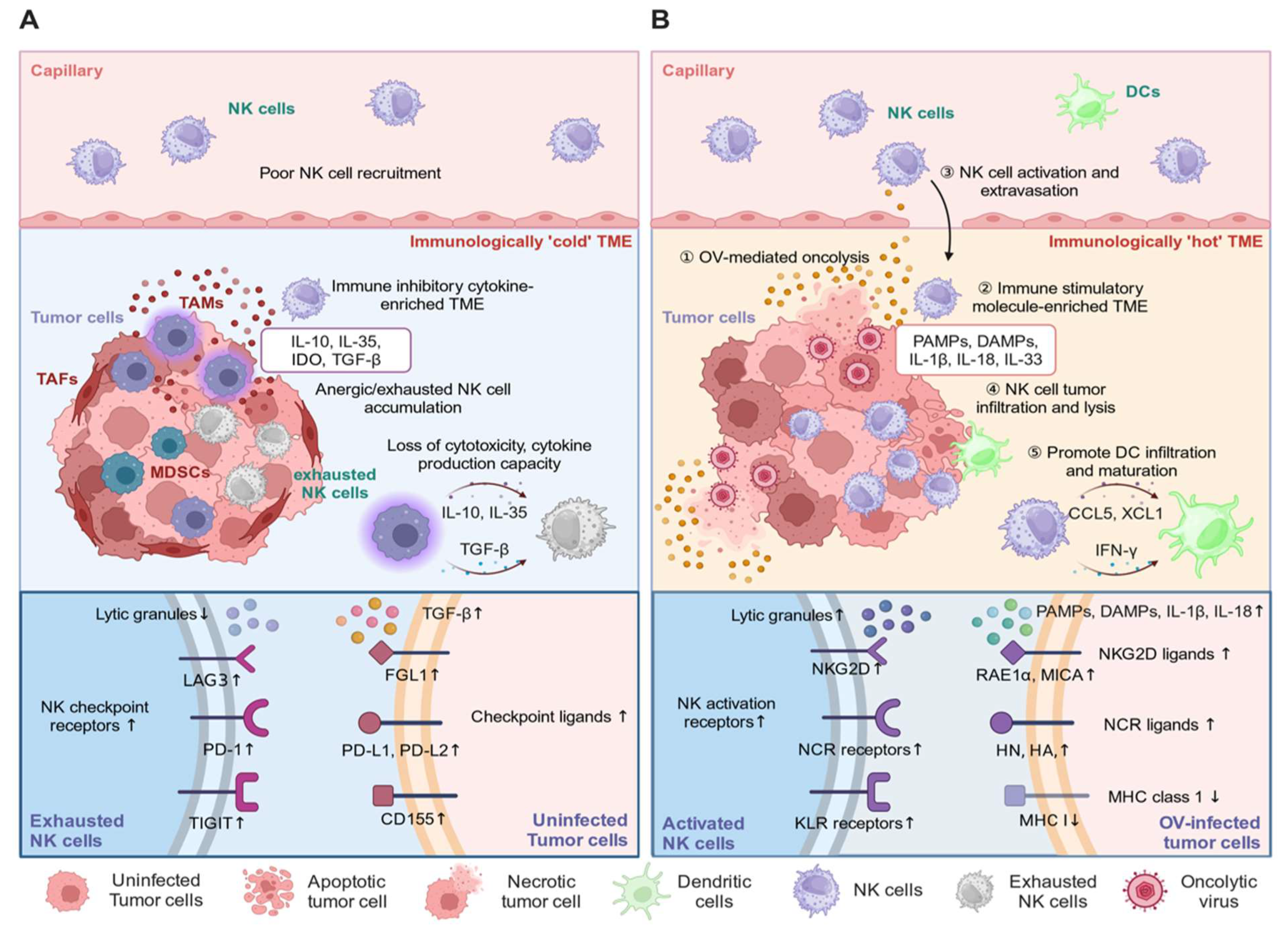
4.4. The Controversial Role of the Host Immune System in OVT
5. NK Cell Biology
5.1. NK Cell Antitumor Responses
5.2. NK Cell Antiviral Responses
5.3. NK Cell Memory Responses
6. NK Cell Biology in OVT
6.1. NK Cell Antitumor and Antiviral Responses in OVT
6.2. Enhancing NK Cell Antitumor Responses
6.3. Inhibiting NK Cell Antiviral Responses
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ADAM | a disintegrin and metalloprotease |
| ADCC | antibody-dependent cellular cytotoxicity |
| ADSCs | adipose-derived stem cells |
| AML | acute myeloid leukemia |
| APCs | antigen-presenting cells |
| BAG6 | BCL2-associated athanogene 6 |
| BiKE | bispecific killer engager |
| CAR | chimeric antigen receptor |
| CCL5 | C-C motif chemokine ligand 5 |
| CCR5 | C-C chemokine receptor 5 |
| CDH1 | cadherin 1 |
| CI cycle | cancer-immunity cycle |
| CIML NK cells | cytokine-induced memory-like NK cells |
| CTLA-4 | cytotoxic T lymphocyte-associated protein 4 |
| DAMPs | damage-associated molecular pattern molecules |
| DCs | dendritic cells |
| DRR | durable response rate |
| EBV | Epstein Barr virus |
| ECM | extracellular matrix |
| EGFR | epidermal growth factor receptor |
| Erp5 | endoplasmic reticulum protein 5 |
| GBM | glioblastoma multiforme |
| gGEHV-1 | equine herpes virus-1 glycoprotein G |
| GM-CSF | granulocyte–macrophage colony-stimulating factor |
| HCMV | human cytomegalovirus |
| HSP | heat shock protein |
| HN | hemagglutinin–neuraminidase |
| HPV | human papilloma virus |
| HSV | herpes simplex virus |
| ICB | immune checkpoint blockade |
| IDO | indoleamine 2:3-dioxygenase |
| IFN-γ | interferon-gamma |
| IL | interleukin |
| KIR | killer immunoglobulin-like receptors |
| KLRG1 | killer cell lectin-like receptor subfamily G member 1 |
| LILRB4 | leukocyte immunoglobulin-like receptor subfamily B member 4 |
| LCMV | lymphocytic choriomeningitis virus |
| MCMV | murine cytomegalovirus |
| MDSCs | myeloid-derived suppressor cells |
| MHC | major histocompatibility complex |
| MICA | MHC class I-related polypeptide sequence A |
| MMP14 | matrix metallopeptidase 14 |
| NCRs | natural cytotoxicity receptors |
| NCR3LG1 | natural killer cell cytotoxicity receptor 3 ligand 1 |
| NDV | Newcastle disease virus |
| NK cells | natural killer cells |
| oAds | oncolytic adenoviruses |
| oHSV | oncolytic herpes simplex virus |
| OVs | oncolytic viruses |
| OVT | oncolytic virotherapy |
| PAMPs | pathogen-associated molecular pattern molecules |
| PD-1 | programmed cell death protein 1 |
| PD-L1 | programmed cell death ligand 1 |
| PD-L2 | programmed cell death ligand 2 |
| PKR | protein kinase R |
| RAE1α | retinoic acid early transcript 1 alpha |
| scFv | single-chain variable fragment |
| TAAs | tumor-associated antigens |
| TAMs | tumor-associated macrophages |
| Tcf-1 | T-cell factor 1 |
| TGF | transforming growth factor |
| TIL | tumor-infiltrating lymphocytes |
| TLRs | toll-like receptors |
| TME | tumor microenvironment |
| Tregs | regulatory T-cells |
| TNF-α | tumor necrosis factor alpha |
| T-VEC | talimogene laherparepvec |
| VSV | vestibular stomatitis virus |
| VV | vaccinia virus |
| VZV | varicella zoster virus |
| XCL1 | X-C motif chemokine ligand 1 |
References
- Sharma, P.; Hu-Lieskovan, S.; Wargo, J.A.; Ribas, A. Primary, Adaptive, and Acquired Resistance to Cancer Immunotherapy. Cell 2017, 168, 707–723. [Google Scholar] [CrossRef] [PubMed]
- Ma, R.; Li, Z.; Chiocca, E.A.; Caligiuri, M.A.; Yu, J. The emerging field of oncolytic virus-based cancer immunotherapy. Trends Cancer 2023, 9, 122–139. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, H.M.; Saha, D. The Current State of Oncolytic Herpes Simplex Virus for Glioblastoma Treatment. Oncolytic Virother. 2021, 10, 1–27. [Google Scholar] [CrossRef] [PubMed]
- Yin, J.; Markert, J.M.; Leavenworth, J.W. Modulation of the Intratumoral Immune Landscape by Oncolytic Herpes Simplex Virus Virotherapy. Front. Oncol. 2017, 7, 136. [Google Scholar] [CrossRef] [PubMed]
- Huntington, N.D.; Cursons, J.; Rautela, J. The cancer–natural killer cell immunity cycle. Nat. Rev. Cancer 2020, 20, 437–454. [Google Scholar] [CrossRef] [PubMed]
- Mellman, I.; Chen, D.S.; Powles, T.; Turley, S.J. The cancer-immunity cycle: Indication, genotype, and immunotype. Immunity 2023, 56, 2188–2205. [Google Scholar] [CrossRef]
- Chen, D.S.; Mellman, I. Oncology Meets Immunology: The Cancer-Immunity Cycle. Immunity 2013, 39, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Spranger, S.; Bao, R.; Gajewski, T.F. Melanoma-intrinsic β-catenin signalling prevents anti-tumour immunity. Nature 2015, 523, 231–235. [Google Scholar] [CrossRef] [PubMed]
- Carvajal-Hausdorf, D.; Altan, M.; Velcheti, V.; Gettinger, S.N.; Herbst, R.S.; Rimm, D.L.; Schalper, K.A. Expression and clinical significance of PD-L1, B7-H3, B7-H4 and TILs in human small cell lung Cancer (SCLC). J. Immunother. Cancer 2019, 7, 65. [Google Scholar] [CrossRef]
- Hansen, A.R.; Massard, C.; Ott, P.A.; Haas, N.B.; Lopez, J.S.; Ejadi, S.; Wallmark, J.M.; Keam, B.; Delord, J.P.; Aggarwal, R.; et al. Pembrolizumab for advanced prostate adenocarcinoma: Findings of the KEYNOTE-028 study. Ann. Oncol. 2018, 29, 1807–1813. [Google Scholar] [CrossRef]
- Kim, O.-H.; Kang, G.-H.; Noh, H.; Cha, J.-Y.; Lee, H.-J.; Yoon, J.-H.; Mamura, M.; Nam, J.-S.; Lee, D.H.; Kim, Y.A.; et al. Proangiogenic TIE2+/CD31+ macrophages are the predominant population of tumor-associated macrophages infiltrating metastatic lymph nodes. Mol. Cells 2013, 36, 432–438. [Google Scholar] [CrossRef]
- Jarnicki, A.G.; Lysaght, J.; Todryk, S.; Mills, K.H.G. Suppression of Antitumor Immunity by IL-10 and TGF-β-Producing T Cells Infiltrating the Growing Tumor: Influence of Tumor Environment on the Induction of CD4+ and CD8+ Regulatory T Cells1. J. Immunol. 2006, 177, 896–904. [Google Scholar] [CrossRef]
- Collison, L.W.; Workman, C.J.; Kuo, T.T.; Boyd, K.; Wang, Y.; Vignali, K.M.; Cross, R.; Sehy, D.; Blumberg, R.S.; Vignali, D.A. The inhibitory cytokine IL-35 contributes to regulatory T-cell function. Nature 2007, 450, 566–569. [Google Scholar] [CrossRef]
- Yaseen, M.M.; Abuharfeil, N.M.; Darmani, H.; Daoud, A. Mechanisms of immune suppression by myeloid-derived suppressor cells: The role of interleukin-10 as a key immunoregulatory cytokine. Open Biol. 2020, 10, 200111. [Google Scholar] [CrossRef]
- Mariathasan, S.; Turley, S.J.; Nickles, D.; Castiglioni, A.; Yuen, K.; Wang, Y.; Kadel Iii, E.E.; Koeppen, H.; Astarita, J.L.; Cubas, R. TGFβ attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature 2018, 554, 544–548. [Google Scholar] [CrossRef]
- Shen, J.; Choi, Y.L.; Lee, T.; Kim, H.; Chae, Y.K.; Dulken, B.W.; Bogdan, S.; Huang, M.; Fisher, G.A.; Park, S.; et al. Inflamed immune phenotype predicts favorable clinical outcomes of immune checkpoint inhibitor therapy across multiple cancer types. J. Immunother. Cancer 2024, 12, e008339. [Google Scholar] [CrossRef] [PubMed]
- Rizvi, N.A.; Hellmann, M.D.; Snyder, A.; Kvistborg, P.; Makarov, V.; Havel, J.J.; Lee, W.; Yuan, J.; Wong, P.; Ho, T.S. Mutational landscape determines sensitivity to PD-1 blockade in non–small cell lung cancer. Science 2015, 348, 124–128. [Google Scholar] [CrossRef]
- Paijens, S.T.; Vledder, A.; de Bruyn, M.; Nijman, H.W. Tumor-infiltrating lymphocytes in the immunotherapy era. Cell Mol. Immunol. 2021, 18, 842–859. [Google Scholar] [CrossRef]
- Ayers, M.; Lunceford, J.; Nebozhyn, M.; Murphy, E.; Loboda, A.; Kaufman, D.R.; Albright, A.; Cheng, J.D.; Kang, S.P.; Shankaran, V.; et al. IFN-γ-related mRNA profile predicts clinical response to PD-1 blockade. J. Clin. Investig. 2017, 127, 2930–2940. [Google Scholar] [CrossRef]
- Kawase, K.; Kawashima, S.; Nagasaki, J.; Inozume, T.; Tanji, E.; Kawazu, M.; Hanazawa, T.; Togashi, Y. High Expression of MHC Class I Overcomes Cancer Immunotherapy Resistance Due to IFNγ Signaling Pathway Defects. Cancer Immunol. Res. 2023, 11, 895–908. [Google Scholar] [CrossRef]
- Bian, Y.; Walter, D.L.; Zhang, C. Efficiency of Interferon-γ in Activating Dendritic Cells and Its Potential Synergy with Toll-like Receptor Agonists. Viruses 2023, 15, 1198. [Google Scholar] [CrossRef] [PubMed]
- Wijdeven, R.H.; van Luijn, M.M.; Wierenga-Wolf, A.F.; Akkermans, J.J.; van den Elsen, P.J.; Hintzen, R.Q.; Neefjes, J. Chemical and genetic control of IFNγ-induced MHCII expression. EMBO Rep. 2018, 19, e45553. [Google Scholar] [CrossRef] [PubMed]
- Fenton, S.E.; Saleiro, D.; Platanias, L.C. Type I and II Interferons in the Anti-Tumor Immune Response. Cancers 2021, 13, 1037. [Google Scholar] [CrossRef]
- Gao, Q.; Li, F.; Wang, S.; Shen, Z.; Cheng, S.; Ping, Y.; Qin, G.; Chen, X.; Yang, L.; Cao, L. A cycle involving HMGB1, IFN-γ and dendritic cells plays a putative role in anti-tumor immunity. Cell. Immunol. 2019, 343, 103850. [Google Scholar] [CrossRef]
- Lasek, W. Cancer immunoediting hypothesis: History, clinical implications and controversies. Cent. Eur. J. Immunol. 2022, 47, 168–174. [Google Scholar] [CrossRef]
- Zhou, F. Molecular mechanisms of IFN-gamma to up-regulate MHC class I antigen processing and presentation. Int. Rev. Immunol. 2009, 28, 239–260. [Google Scholar] [CrossRef]
- Dunn, G.P.; Bruce, A.T.; Ikeda, H.; Old, L.J.; Schreiber, R.D. Cancer immunoediting: From immunosurveillance to tumor escape. Nat. Immunol. 2002, 3, 991–998. [Google Scholar] [CrossRef]
- Zitvogel, L.; Tesniere, A.; Kroemer, G. Cancer despite immunosurveillance: Immunoselection and immunosubversion. Nat. Rev. Immunol. 2006, 6, 715–727. [Google Scholar] [CrossRef]
- Wu, Y.; Biswas, D.; Swanton, C. Impact of cancer evolution on immune surveillance and checkpoint inhibitor response. Semin. Cancer Biol. 2022, 84, 89–102. [Google Scholar] [CrossRef]
- Sulica, A.; Morel, P.; Metes, D.; Herberman, R.B. Ig-binding receptors on human NK cells as effector and regulatory surface molecules. Int. Rev. Immunol. 2001, 20, 371–414. [Google Scholar] [CrossRef]
- Steplewski, Z.; Lubeck, M.D.; Koprowski, H. Human macrophages armed with murine immunoglobulin G2a antibodies to tumors destroy human cancer cells. Science 1983, 221, 865–867. [Google Scholar] [CrossRef]
- Adams, G.P.; Weiner, L.M. Monoclonal antibody therapy of cancer. Nat. Biotechnol. 2005, 23, 1147–1157. [Google Scholar] [CrossRef] [PubMed]
- Gong, Y.; Klein Wolterink, R.G.J.; Wang, J.; Bos, G.M.J.; Germeraad, W.T.V. Chimeric antigen receptor natural killer (CAR-NK) cell design and engineering for cancer therapy. J. Hematol. Oncol. 2021, 14, 73. [Google Scholar] [CrossRef]
- Graham, C.; Hewitson, R.; Pagliuca, A.; Benjamin, R. Cancer immunotherapy with CAR-T cells—Behold the future. Clin. Med. 2018, 18, 324–328. [Google Scholar] [CrossRef]
- Lin, D.; Shen, Y.; Liang, T. Oncolytic virotherapy: Basic principles, recent advances and future directions. Signal Transduct. Target. Ther. 2023, 8, 156. [Google Scholar] [CrossRef] [PubMed]
- De Pace, N. Sulla scomparsa di un enorme cancro vegetante del callo dell’utero senza cura chirurgica. Ginecologia 1912, 9, 82–88. [Google Scholar]
- Russell, S.J.; Peng, K.W. Measles virus for cancer therapy. Curr. Top. Microbiol. Immunol. 2009, 330, 213–241. [Google Scholar] [CrossRef] [PubMed]
- Toyoda, H.; Yin, J.; Mueller, S.; Wimmer, E.; Cello, J. Oncolytic treatment and cure of neuroblastoma by a novel attenuated poliovirus in a novel poliovirus-susceptible animal model. Cancer Res. 2007, 67, 2857–2864. [Google Scholar] [CrossRef]
- Tsang, E.S.; Munster, P.N. Vaccinia (Smallpox) for the Treatment of Ovarian Cancer—Turning an Old Foe Into a Friend? JAMA Oncol. 2023, 9, 894–896. [Google Scholar] [CrossRef]
- Conry, R.M.; Westbrook, B.; McKee, S.; Norwood, T.G. Talimogene laherparepvec: First in class oncolytic virotherapy. Hum. Vaccin. Immunother. 2018, 14, 839–846. [Google Scholar] [CrossRef]
- Hastie, E.; Grdzelishvili, V.Z. Vesicular stomatitis virus as a flexible platform for oncolytic virotherapy against cancer. J. Gen. Virol. 2012, 93, 2529–2545. [Google Scholar] [CrossRef]
- Boozari, B.; Mundt, B.; Woller, N.; Strüver, N.; Gürlevik, E.; Schache, P.; Kloos, A.; Knocke, S.; Manns, M.P.; Wirth, T.C.; et al. Antitumoural immunity by virus-mediated immunogenic apoptosis inhibits metastatic growth of hepatocellular carcinoma. Gut 2010, 59, 1416–1426. [Google Scholar] [CrossRef]
- Cejalvo, J.M.; Falato, C.; Villanueva, L.; Tolosa, P.; González, X.; Pascal, M.; Canes, J.; Gavilá, J.; Manso, L.; Pascual, T.; et al. Oncolytic viruses: A new immunotherapeutic approach for breast cancer treatment? Cancer Treat. Rev. 2022, 106, 102392. [Google Scholar] [CrossRef]
- Matveeva, O.V.; Chumakov, P.M. Defects in interferon pathways as potential biomarkers of sensitivity to oncolytic viruses. Rev. Med. Virol. 2018, 28, e2008. [Google Scholar] [CrossRef]
- Stojdl, D.F.; Lichty, B.; Knowles, S.; Marius, R.; Atkins, H.; Sonenberg, N.; Bell, J.C. Exploiting tumor-specific defects in the interferon pathway with a previously unknown oncolytic virus. Nat. Med. 2000, 6, 821–825. [Google Scholar] [CrossRef]
- Krysko, D.V.; Garg, A.D.; Kaczmarek, A.; Krysko, O.; Agostinis, P.; Vandenabeele, P. Immunogenic cell death and DAMPs in cancer therapy. Nat. Rev. Cancer 2012, 12, 860–875. [Google Scholar] [CrossRef]
- Gujar, S.; Pol, J.G.; Kim, Y.; Lee, P.W.; Kroemer, G. Antitumor Benefits of Antiviral Immunity: An Underappreciated Aspect of Oncolytic Virotherapies. Trends Immunol. 2018, 39, 209–221. [Google Scholar] [CrossRef]
- Burke, S.; Shergold, A.; Elder, M.J.; Whitworth, J.; Cheng, X.; Jin, H.; Wilkinson, R.W.; Harper, J.; Carroll, D.K. Oncolytic Newcastle disease virus activation of the innate immune response and priming of antitumor adaptive responses in vitro. Cancer Immunol. Immunother. 2020, 69, 1015–1027. [Google Scholar] [CrossRef]
- Gujar, S.A.; Pan, D.A.; Marcato, P.; Garant, K.A.; Lee, P.W. Oncolytic virus-initiated protective immunity against prostate cancer. Mol. Ther. 2011, 19, 797–804. [Google Scholar] [CrossRef]
- Prestwich, R.J.; Errington, F.; Steele, L.P.; Ilett, E.J.; Morgan, R.S.; Harrington, K.J.; Pandha, H.S.; Selby, P.J.; Vile, R.G.; Melcher, A.A. Reciprocal human dendritic cell-natural killer cell interactions induce antitumor activity following tumor cell infection by oncolytic reovirus. J. Immunol. 2009, 183, 4312–4321. [Google Scholar] [CrossRef]
- van Vloten, J.P.; Matuszewska, K.; Minow, M.A.A.; Minott, J.A.; Santry, L.A.; Pereira, M.; Stegelmeier, A.A.; McAusland, T.M.; Klafuric, E.M.; Karimi, K.; et al. Oncolytic Orf virus licenses NK cells via cDC1 to activate innate and adaptive antitumor mechanisms and extends survival in a murine model of late-stage ovarian cancer. J. Immunother. Cancer 2022, 10, e004335. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Tai, L.H.; Ilkow, C.S.; Alkayyal, A.A.; Ananth, A.A.; de Souza, C.T.; Wang, J.; Sahi, S.; Ly, L.; Lefebvre, C.; et al. Maraba MG1 virus enhances natural killer cell function via conventional dendritic cells to reduce postoperative metastatic disease. Mol. Ther. 2014, 22, 1320–1332. [Google Scholar] [CrossRef]
- Kanerva, A.; Nokisalmi, P.; Diaconu, I.; Koski, A.; Cerullo, V.; Liikanen, I.; Tähtinen, S.; Oksanen, M.; Heiskanen, R.; Pesonen, S.; et al. Antiviral and antitumor T-cell immunity in patients treated with GM-CSF-coding oncolytic adenovirus. Clin. Cancer Res. 2013, 19, 2734–2744. [Google Scholar] [CrossRef]
- Lai, J.; Mardiana, S.; House, I.G.; Sek, K.; Henderson, M.A.; Giuffrida, L.; Chen, A.X.Y.; Todd, K.L.; Petley, E.V.; Chan, J.D.; et al. Adoptive cellular therapy with T cells expressing the dendritic cell growth factor Flt3L drives epitope spreading and antitumor immunity. Nat. Immunol. 2020, 21, 914–926. [Google Scholar] [CrossRef] [PubMed]
- Ricca, J.M.; Oseledchyk, A.; Walther, T.; Liu, C.; Mangarin, L.; Merghoub, T.; Wolchok, J.D.; Zamarin, D. Pre-existing Immunity to Oncolytic Virus Potentiates Its Immunotherapeutic Efficacy. Mol. Ther. 2018, 26, 1008–1019. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Zhao, J.; Yue, S.; Li, Z.; Duan, X.; Lin, Y.; Yang, Y.; He, J.; Gao, L.; Pan, Z.; et al. An oncolytic virus delivering tumor-irrelevant bystander T cell epitopes induces anti-tumor immunity and potentiates cancer immunotherapy. Nat. Cancer 2024, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Xie, D.; Tian, Y.; Hu, D.; Wang, Y.; Yang, Y.; Zhou, B.; Zhang, R.; Ren, Z.; Liu, M.; Xu, J.; et al. Oncolytic adenoviruses expressing checkpoint inhibitors for cancer therapy. Signal Transduct. Target. Ther. 2023, 8, 436. [Google Scholar] [CrossRef]
- Samson, A.; Bentham, M.J.; Scott, K.; Nuovo, G.; Bloy, A.; Appleton, E.; Adair, R.A.; Dave, R.; Peckham-Cooper, A.; Toogood, G.; et al. Oncolytic reovirus as a combined antiviral and anti-tumour agent for the treatment of liver cancer. Gut 2018, 67, 562–573. [Google Scholar] [CrossRef] [PubMed]
- Guse, K.; Cerullo, V.; Hemminki, A. Oncolytic vaccinia virus for the treatment of cancer. Expert. Opin. Biol. Ther. 2011, 11, 595–608. [Google Scholar] [CrossRef]
- Friedman, G.K.; Johnston, J.M.; Bag, A.K.; Bernstock, J.D.; Li, R.; Aban, I.; Kachurak, K.; Nan, L.; Kang, K.-D.; Totsch, S. Oncolytic HSV-1 G207 immunovirotherapy for pediatric high-grade gliomas. N. Engl. J. Med. 2021, 384, 1613–1622. [Google Scholar] [CrossRef]
- Peter, M.; Kühnel, F. Oncolytic Adenovirus in Cancer Immunotherapy. Cancers 2020, 12, 3354. [Google Scholar] [CrossRef] [PubMed]
- Ito, H.; Aoki, H.; Kühnel, F.; Kondo, Y.; Kubicka, S.; Wirth, T.; Iwado, E.; Iwamaru, A.; Fujiwara, K.; Hess, K.R.; et al. Autophagic cell death of malignant glioma cells induced by a conditionally replicating adenovirus. J. Natl. Cancer Inst. 2006, 98, 625–636. [Google Scholar] [CrossRef] [PubMed]
- Liikanen, I.; Ahtiainen, L.; Hirvinen, M.L.; Bramante, S.; Cerullo, V.; Nokisalmi, P.; Hemminki, O.; Diaconu, I.; Pesonen, S.; Koski, A.; et al. Oncolytic adenovirus with temozolomide induces autophagy and antitumor immune responses in cancer patients. Mol. Ther. 2013, 21, 1212–1223. [Google Scholar] [CrossRef] [PubMed]
- Di Somma, S.; Iannuzzi, C.A.; Passaro, C.; Forte, I.M.; Iannone, R.; Gigantino, V.; Indovina, P.; Botti, G.; Giordano, A.; Formisano, P.; et al. The Oncolytic Virus dl922-947 Triggers Immunogenic Cell Death in Mesothelioma and Reduces Xenograft Growth. Front. Oncol. 2019, 9, 564. [Google Scholar] [CrossRef] [PubMed]
- Marcato, P.; Shmulevitz, M.; Pan, D.; Stoltz, D.; Lee, P.W. Ras transformation mediates reovirus oncolysis by enhancing virus uncoating, particle infectivity, and apoptosis-dependent release. Mol. Ther. 2007, 15, 1522–1530. [Google Scholar] [CrossRef] [PubMed]
- Errington, F.; Steele, L.; Prestwich, R.; Harrington, K.J.; Pandha, H.S.; Vidal, L.; de Bono, J.; Selby, P.; Coffey, M.; Vile, R.; et al. Reovirus activates human dendritic cells to promote innate antitumor immunity. J. Immunol. 2008, 180, 6018–6026. [Google Scholar] [CrossRef] [PubMed]
- Lyles, D.; Kuzmin, I.; Rupprecht, C. Rhabdoviridae. Fields Virology; Wolters Kluwer: Philadelphia, PA, USA, 2013; pp. 885–2456. [Google Scholar]
- Naik, S.; Nace, R.; Federspiel, M.J.; Barber, G.N.; Peng, K.W.; Russell, S.J. Curative one-shot systemic virotherapy in murine myeloma. Leukemia 2012, 26, 1870–1878. [Google Scholar] [CrossRef] [PubMed]
- Nakashima, H.; Kaur, B.; Chiocca, E.A. Directing systemic oncolytic viral delivery to tumors via carrier cells. Cytokine Growth Factor. Rev. 2010, 21, 119–126. [Google Scholar] [CrossRef] [PubMed]
- Pipiya, T.; Sauthoff, H.; Huang, Y.Q.; Chang, B.; Cheng, J.; Heitner, S.; Chen, S.; Rom, W.N.; Hay, J.G. Hypoxia reduces adenoviral replication in cancer cells by downregulation of viral protein expression. Gene Ther. 2005, 12, 911–917. [Google Scholar] [CrossRef]
- Hiley, C.T.; Yuan, M.; Lemoine, N.R.; Wang, Y. Lister strain vaccinia virus, a potential therapeutic vector targeting hypoxic tumours. Gene Ther. 2010, 17, 281–287. [Google Scholar] [CrossRef]
- Pütz, M.M.; Midgley, C.M.; Law, M.; Smith, G.L. Quantification of antibody responses against multiple antigens of the two infectious forms of Vaccinia virus provides a benchmark for smallpox vaccination. Nat. Med. 2006, 12, 1310–1315. [Google Scholar] [CrossRef]
- Peters, C.; Rabkin, S.D. Designing Herpes Viruses as Oncolytics. Mol. Ther.-Oncolytics 2015, 2, 15010. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.-J.; Moon, D.; Kong, S.J.; Lee, Y.S.; Yoo, Y.; Kim, S.; Kim, C.; Chon, H.J.; Kim, J.-H.; Choi, K.-J. Antitumor effects of IL-12 and GM-CSF co-expressed in an engineered oncolytic HSV-1. Gene Ther. 2021, 28, 186–198. [Google Scholar] [CrossRef]
- Parker, J.N.; Gillespie, G.Y.; Love, C.E.; Randall, S.; Whitley, R.J.; Markert, J.M. Engineered herpes simplex virus expressing IL-12 in the treatment of experimental murine brain tumors. Proc. Natl. Acad. Sci. USA 2000, 97, 2208–2213. [Google Scholar] [CrossRef]
- Gaston, D.C.; Odom, C.I.; Li, L.; Markert, J.M.; Roth, J.C.; Cassady, K.A.; Whitley, R.J.; Parker, J.N. Production of bioactive soluble interleukin-15 in complex with interleukin-15 receptor alpha from a conditionally-replicating oncolytic HSV-1. PLoS ONE 2013, 8, e81768. [Google Scholar] [CrossRef]
- Ma, R.; Lu, T.; Li, Z.; Teng, K.-Y.; Mansour, A.G.; Yu, M.; Tian, L.; Xu, B.; Ma, S.; Zhang, J.; et al. An Oncolytic Virus Expressing IL15/IL15Rα Combined with Off-the-Shelf EGFR-CAR NK Cells Targets Glioblastoma. Cancer Res. 2021, 81, 3635–3648. [Google Scholar] [CrossRef]
- Mineta, T.; Rabkin, S.D.; Martuza, R.L. Treatment of malignant gliomas using ganciclovir-hypersensitive, ribonucleotide reductase-deficient herpes simplex viral mutant. Cancer Res. 1994, 54, 3963–3966. [Google Scholar] [PubMed]
- Ju, F.; Luo, Y.; Lin, C.; Jia, X.; Xu, Z.; Tian, R.; Lin, Y.; Zhao, M.; Chang, Y.; Huang, X.; et al. Oncolytic virus expressing PD-1 inhibitors activates a collaborative intratumoral immune response to control tumor and synergizes with CTLA-4 or TIM-3 blockade. J. Immunother. Cancer 2022, 10, e004762. [Google Scholar] [CrossRef]
- Wang, G.; Kang, X.; Chen, K.S.; Jehng, T.; Jones, L.; Chen, J.; Huang, X.F.; Chen, S.-Y. An engineered oncolytic virus expressing PD-L1 inhibitors activates tumor neoantigen-specific T cell responses. Nat. Commun. 2020, 11, 1395. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, J.R.; Vijayakumar, G.; Palese, P. A Recombinant Antibody-Expressing Influenza Virus Delays Tumor Growth in a Mouse Model. Cell Rep. 2018, 22, 1–7. [Google Scholar] [CrossRef]
- Wongthida, P.; Diaz, R.M.; Pulido, C.; Rommelfanger, D.; Galivo, F.; Kaluza, K.; Kottke, T.; Thompson, J.; Melcher, A.; Vile, R. Activating systemic T-cell immunity against self tumor antigens to support oncolytic virotherapy with vesicular stomatitis virus. Hum. Gene Ther. 2011, 22, 1343–1353. [Google Scholar] [CrossRef]
- Galanis, E.; Dooley, K.E.; Keith Anderson, S.; Kurokawa, C.B.; Carrero, X.W.; Uhm, J.H.; Federspiel, M.J.; Leontovich, A.A.; Aderca, I.; Viker, K.B.; et al. Carcinoembryonic antigen-expressing oncolytic measles virus derivative in recurrent glioblastoma: A phase 1 trial. Nat. Commun. 2024, 15, 493. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Zhang, M.; Ye, Q.; Liu, Y.; Qian, W. Preclinical and clinical trials of oncolytic vaccinia virus in cancer immunotherapy: A comprehensive review. Cancer Biol. Med. 2023, 20, 646–661. [Google Scholar] [CrossRef] [PubMed]
- Yu, F.; Wang, X.; Guo, Z.S.; Bartlett, D.L.; Gottschalk, S.M.; Song, X.T. T-cell engager-armed oncolytic vaccinia virus significantly enhances antitumor therapy. Mol. Ther. 2014, 22, 102–111. [Google Scholar] [CrossRef] [PubMed]
- Field, H.J.; Wildy, P. The pathogenicity of thymidine kinase-deficient mutants of herpes simplex virus in mice. J. Hyg. 1978, 81, 267–277. [Google Scholar] [CrossRef] [PubMed]
- Martuza, R.L.; Malick, A.; Markert, J.M.; Ruffner, K.L.; Coen, D.M. Experimental therapy of human glioma by means of a genetically engineered virus mutant. Science 1991, 252, 854–856. [Google Scholar] [CrossRef] [PubMed]
- Kanai, R.; Zaupa, C.; Sgubin, D.; Antoszczyk, S.J.; Martuza, R.L.; Wakimoto, H.; Rabkin, S.D. Effect of γ34.5 deletions on oncolytic herpes simplex virus activity in brain tumors. J. Virol. 2012, 86, 4420–4431. [Google Scholar] [CrossRef]
- Wilcox, D.R.; Longnecker, R. The Herpes Simplex Virus Neurovirulence Factor γ34.5: Revealing Virus-Host Interactions. PLoS Pathog. 2016, 12, e1005449. [Google Scholar] [CrossRef] [PubMed]
- Megison, M.L.; Gillory, L.A.; Stewart, J.E.; Nabers, H.C.; Mroczek-Musulman, E.; Waters, A.M.; Coleman, J.M.; Kelly, V.; Markert, J.M.; Gillespie, G.Y.; et al. Preclinical Evaluation of Engineered Oncolytic Herpes Simplex Virus for the Treatment of Pediatric Solid Tumors. PLoS ONE 2014, 9, e86843. [Google Scholar] [CrossRef]
- Gillory, L.A.; Megison, M.L.; Stewart, J.E.; Mroczek-Musulman, E.; Nabers, H.C.; Waters, A.M.; Kelly, V.; Coleman, J.M.; Markert, J.M.; Gillespie, G.Y.; et al. Preclinical Evaluation of Engineered Oncolytic Herpes Simplex Virus for the Treatment of Neuroblastoma. PLoS ONE 2013, 8, e77753. [Google Scholar] [CrossRef]
- Markert, J.M.; Cody, J.J.; Parker, J.N.; Coleman, J.M.; Price, K.H.; Kern, E.R.; Quenelle, D.C.; Lakeman, A.D.; Schoeb, T.R.; Palmer, C.A.; et al. Preclinical Evaluation of a Genetically Engineered Herpes Simplex Virus Expressing Interleukin-12. J. Virol. 2012, 86, 5304–5313. [Google Scholar] [CrossRef] [PubMed]
- Smyth, M.J.; Taniguchi, M.; Street, S.E.A. The Anti-Tumor Activity of IL-12: Mechanisms of Innate Immunity That Are Model and Dose Dependent1. J. Immunol. 2000, 165, 2665–2670. [Google Scholar] [CrossRef] [PubMed]
- Oka, N.; Markova, T.; Tsuzuki, K.; Li, W.; El-Darawish, Y.; Pencheva-Demireva, M.; Yamanishi, K.; Yamanishi, H.; Sakagami, M.; Tanaka, Y.; et al. IL-12 regulates the expansion, phenotype, and function of murine NK cells activated by IL-15 and IL-18. Cancer Immunol. Immunother. 2020, 69, 1699–1712. [Google Scholar] [CrossRef] [PubMed]
- Trinchieri, G. Interleukin-12 and the regulation of innate resistance and adaptive immunity. Nat. Rev. Immunol. 2003, 3, 133–146. [Google Scholar] [CrossRef]
- Roth, J.C.; Cassady, K.A.; Cody, J.J.; Parker, J.N.; Price, K.H.; Coleman, J.M.; Peggins, J.O.; Noker, P.E.; Powers, N.W.; Grimes, S.D.; et al. Evaluation of the Safety and Biodistribution of M032, an Attenuated Herpes Simplex Virus Type 1 Expressing hIL-12, After Intracerebral Administration to Aotus Nonhuman Primates. Hum. Gene Ther. Clin. Dev. 2014, 25, 16–27. [Google Scholar] [CrossRef] [PubMed]
- Saha, D.; Martuza, R.L.; Rabkin, S.D. Macrophage Polarization Contributes to Glioblastoma Eradication by Combination Immunovirotherapy and Immune Checkpoint Blockade. Cancer Cell 2017, 32, 253–267.e5. [Google Scholar] [CrossRef] [PubMed]
- Aisenbrey, C.; Sizun, C.; Koch, J.; Herget, M.; Abele, R.; Bechinger, B.; Tampé, R. Structure and dynamics of membrane-associated ICP47, a viral inhibitor of the MHC I antigen-processing machinery. J. Biol. Chem. 2006, 281, 30365–30372. [Google Scholar] [CrossRef]
- Kaufman, H.L.; Ruby, C.E.; Hughes, T.; Slingluff, C.L., Jr. Current status of granulocyte-macrophage colony-stimulating factor in the immunotherapy of melanoma. J. Immunother. Cancer 2014, 2, 11. [Google Scholar] [CrossRef]
- Kaufman, H.L.; Kim, D.W.; DeRaffele, G.; Mitcham, J.; Coffin, R.S.; Kim-Schulze, S. Local and Distant Immunity Induced by Intralesional Vaccination with an Oncolytic Herpes Virus Encoding GM-CSF in Patients with Stage IIIc and IV Melanoma. Ann. Surg. Oncol. 2010, 17, 718–730. [Google Scholar] [CrossRef]
- Andtbacka, R.H.; Agarwala, S.S.; Ollila, D.W.; Hallmeyer, S.; Milhem, M.; Amatruda, T.; Nemunaitis, J.J.; Harrington, K.J.; Chen, L.; Shilkrut, M.; et al. Cutaneous head and neck melanoma in OPTiM, a randomized phase 3 trial of talimogene laherparepvec versus granulocyte-macrophage colony-stimulating factor for the treatment of unresected stage IIIB/IIIC/IV melanoma. Head Neck 2016, 38, 1752–1758. [Google Scholar] [CrossRef]
- Andtbacka, R.H.; Kaufman, H.L.; Collichio, F.; Amatruda, T.; Senzer, N.; Chesney, J.; Delman, K.A.; Spitler, L.E.; Puzanov, I.; Agarwala, S.S.; et al. Talimogene Laherparepvec Improves Durable Response Rate in Patients With Advanced Melanoma. J. Clin. Oncol. 2015, 33, 2780–2788. [Google Scholar] [CrossRef] [PubMed]
- Ressler, J.M.; Karasek, M.; Koch, L.; Silmbrod, R.; Mangana, J.; Latifyan, S.; Aedo-Lopez, V.; Kehrer, H.; Weihsengruber, F.; Koelblinger, P.; et al. Real-life use of talimogene laherparepvec (T-VEC) in melanoma patients in centers in Austria, Switzerland and Germany. J. Immunother. Cancer 2021, 9, e001701. [Google Scholar] [CrossRef]
- Todo, T.; Martuza, R.L.; Rabkin, S.D.; Johnson, P.A. Oncolytic herpes simplex virus vector with enhanced MHC class I presentation and tumor cell killing. Proc. Natl. Acad. Sci. USA 2001, 98, 6396–6401. [Google Scholar] [CrossRef] [PubMed]
- Todo, T.; Ito, H.; Ino, Y.; Ohtsu, H.; Ota, Y.; Shibahara, J.; Tanaka, M. Intratumoral oncolytic herpes virus G47∆ for residual or recurrent glioblastoma: A phase 2 trial. Nat. Med. 2022, 28, 1630–1639. [Google Scholar] [CrossRef]
- Ballman, K.V.; Buckner, J.C.; Brown, P.D.; Giannini, C.; Flynn, P.J.; LaPlant, B.R.; Jaeckle, K.A. The relationship between six-month progression-free survival and 12-month overall survival end points for phase II trials in patients with glioblastoma multiforme. Neuro Oncol. 2007, 9, 29–38. [Google Scholar] [CrossRef]
- Miller, K.E.; Cassady, K.A.; Roth, J.C.; Clements, J.; Schieffer, K.M.; Leraas, K.; Miller, A.R.; Prasad, N.; Leavenworth, J.W.; Aban, I.B.; et al. Immune Activity and Response Differences of Oncolytic Viral Therapy in Recurrent Glioblastoma: Gene Expression Analyses of a Phase IB Study. Clin. Cancer Res. 2022, 28, 498–506. [Google Scholar] [CrossRef]
- Markert, J.M.; Razdan, S.N.; Kuo, H.C.; Cantor, A.; Knoll, A.; Karrasch, M.; Nabors, L.B.; Markiewicz, M.; Agee, B.S.; Coleman, J.M.; et al. A phase 1 trial of oncolytic HSV-1, G207, given in combination with radiation for recurrent GBM demonstrates safety and radiographic responses. Mol. Ther. 2014, 22, 1048–1055. [Google Scholar] [CrossRef]
- Ling, A.L.; Solomon, I.H.; Landivar, A.M.; Nakashima, H.; Woods, J.K.; Santos, A.; Masud, N.; Fell, G.; Mo, X.; Yilmaz, A.S.; et al. Clinical trial links oncolytic immunoactivation to survival in glioblastoma. Nature 2023, 623, 157–166. [Google Scholar] [CrossRef]
- Fujioka, N.; Akazawa, R.; Ohashi, K.; Fujii, M.; Ikeda, M.; Kurimoto, M. Interleukin-18 protects mice against acute herpes simplex virus type 1 infection. J. Virol. 1999, 73, 2401–2409. [Google Scholar] [CrossRef] [PubMed]
- Sergerie, Y.; Rivest, S.; Boivin, G. Tumor necrosis factor-alpha and interleukin-1 beta play a critical role in the resistance against lethal herpes simplex virus encephalitis. J. Infect. Dis. 2007, 196, 853–860. [Google Scholar] [CrossRef]
- Ma, Y.; He, B. Recognition of herpes simplex viruses: Toll-like receptors and beyond. J. Mol. Biol. 2014, 426, 1133–1147. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Fueyo, J. Healing after death: Antitumor immunity induced by oncolytic adenoviral therapy. Oncoimmunology 2014, 3, e947872. [Google Scholar] [CrossRef] [PubMed]
- Bridle, B.W.; Stephenson, K.B.; Boudreau, J.E.; Koshy, S.; Kazdhan, N.; Pullenayegum, E.; Brunellière, J.; Bramson, J.L.; Lichty, B.D.; Wan, Y. Potentiating cancer immunotherapy using an oncolytic virus. Mol. Ther. 2010, 18, 1430–1439. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Breckenridge, C.A.; Yu, J.; Price, R.; Wojton, J.; Pradarelli, J.; Mao, H.; Wei, M.; Wang, Y.; He, S.; Hardcastle, J.; et al. NK cells impede glioblastoma virotherapy through NKp30 and NKp46 natural cytotoxicity receptors. Nat. Med. 2012, 18, 1827–1834. [Google Scholar] [CrossRef] [PubMed]
- Allen, C.; Vongpunsawad, S.; Nakamura, T.; James, C.D.; Schroeder, M.; Cattaneo, R.; Giannini, C.; Krempski, J.; Peng, K.W.; Goble, J.M.; et al. Retargeted oncolytic measles strains entering via the EGFRvIII receptor maintain significant antitumor activity against gliomas with increased tumor specificity. Cancer Res. 2006, 66, 11840–11850. [Google Scholar] [CrossRef] [PubMed]
- Miller, C.G.; Fraser, N.W. Role of the immune response during neuro-attenuated herpes simplex virus-mediated tumor destruction in a murine intracranial melanoma model. Cancer Res. 2000, 60, 5714–5722. [Google Scholar] [PubMed]
- Heiber, J.F.; Barber, G.N. Vesicular stomatitis virus expressing tumor suppressor p53 is a highly attenuated, potent oncolytic agent. J. Virol. 2011, 85, 10440–10450. [Google Scholar] [CrossRef] [PubMed]
- Verron, Q.; Forslund, E.; Brandt, L.; Leino, M.; Frisk, T.W.; Olofsson, P.E.; Önfelt, B. NK cells integrate signals over large areas when building immune synapses but require local stimuli for degranulation. Sci. Signal. 2021, 14, eabe2740. [Google Scholar] [CrossRef] [PubMed]
- Gazit, R.; Gruda, R.; Elboim, M.; Arnon, T.I.; Katz, G.; Achdout, H.; Hanna, J.; Qimron, U.; Landau, G.; Greenbaum, E.; et al. Lethal influenza infection in the absence of the natural killer cell receptor gene Ncr1. Nat. Immunol. 2006, 7, 517–523. [Google Scholar] [CrossRef] [PubMed]
- Werner, J.M.; Heller, T.; Gordon, A.M.; Sheets, A.; Sherker, A.H.; Kessler, E.; Bean, K.S.; Stevens, M.; Schmitt, J.; Rehermann, B. Innate immune responses in hepatitis C virus-exposed healthcare workers who do not develop acute infection. Hepatology 2013, 58, 1621–1631. [Google Scholar] [CrossRef]
- Draghi, M.; Pashine, A.; Sanjanwala, B.; Gendzekhadze, K.; Cantoni, C.; Cosman, D.; Moretta, A.; Valiante, N.M.; Parham, P. NKp46 and NKG2D Recognition of Infected Dendritic Cells Is Necessary for NK Cell Activation in the Human Response to Influenza Infection1. J. Immunol. 2007, 178, 2688–2698. [Google Scholar] [CrossRef] [PubMed]
- Vivier, E.; Raulet, D.H.; Moretta, A.; Caligiuri, M.A.; Zitvogel, L.; Lanier, L.L.; Yokoyama, W.M.; Ugolini, S. Innate or adaptive immunity? The example of natural killer cells. Science 2011, 331, 44–49. [Google Scholar] [CrossRef] [PubMed]
- Brandt, C.S.; Baratin, M.; Yi, E.C.; Kennedy, J.; Gao, Z.; Fox, B.; Haldeman, B.; Ostrander, C.D.; Kaifu, T.; Chabannon, C. The B7 family member B7-H6 is a tumor cell ligand for the activating natural killer cell receptor NKp30 in humans. J. Exp. Med. 2009, 206, 1495–1503. [Google Scholar] [CrossRef] [PubMed]
- Armeanu, S.; Bitzer, M.; Lauer, U.M.; Venturelli, S.; Pathil, A.; Krusch, M.; Kaiser, S.; Jobst, J.; Smirnow, I.; Wagner, A.; et al. Natural Killer Cell–Mediated Lysis of Hepatoma Cells via Specific Induction of NKG2D Ligands by the Histone Deacetylase Inhibitor Sodium Valproate. Cancer Res. 2005, 65, 6321. [Google Scholar] [CrossRef] [PubMed]
- Guerra, N.; Tan, Y.X.; Joncker, N.T.; Choy, A.; Gallardo, F.; Xiong, N.; Knoblaugh, S.; Cado, D.; Greenberg, N.R.; Raulet, D.H. NKG2D-Deficient Mice Are Defective in Tumor Surveillance in Models of Spontaneous Malignancy. Immunity 2008, 28, 571–580. [Google Scholar] [CrossRef] [PubMed]
- Vieillard, V.; Baychelier, F.; Debré, P. NKp44L: A new tool for fighting cancer. Oncoimmunology 2014, 3, e27988. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.-Y.; Fu, T.; Jiang, Y.-Z.; Shao, Z.-M. Natural killer cells in cancer biology and therapy. Mol. Cancer 2020, 19, 120. [Google Scholar] [CrossRef] [PubMed]
- Bauer, S.; Groh, V.; Wu, J.; Steinle, A.; Phillips, J.H.; Lanier, L.L.; Spies, T. Activation of NK cells and T cells by NKG2D, a receptor for stress-inducible MICA. Science 1999, 285, 727–729. [Google Scholar] [CrossRef]
- Textor, S.; Bossler, F.; Henrich, K.-O.; Gartlgruber, M.; Pollmann, J.; Fiegler, N.; Arnold, A.; Westermann, F.; Waldburger, N.; Breuhahn, K.; et al. The proto-oncogene Myc drives expression of the NK cell-activating NKp30 ligand B7-H6 in tumor cells. Oncoimmunology 2016, 5, e1116674. [Google Scholar] [CrossRef]
- Guyton, D.; Caobi, A.; Singh, S.K.; Peyser, M.; Tevfik Dorak, M. 14-OR: HLA REGION AND LUNG CANCER SUSCEPTIBILITY: CONFIRMATION OF BAT3/BAG6 ASSOCIATION AND FUNCTIONAL REPLICATION. Hum. Immunol. 2012, 73, 12. [Google Scholar] [CrossRef]
- Hu, C.; Yang, J.; Qi, Z.; Wu, H.; Wang, B.; Zou, F.; Mei, H.; Liu, J.; Wang, W.; Liu, Q. Heat shock proteins: Biological functions, pathological roles, and therapeutic opportunities. MedComm 2022, 3, e161. [Google Scholar] [CrossRef] [PubMed]
- Ichiyanagi, T.; Imai, T.; Kajiwara, C.; Mizukami, S.; Nakai, A.; Nakayama, T.; Udono, H. Essential role of endogenous heat shock protein 90 of dendritic cells in antigen cross-presentation. J. Immunol. 2010, 185, 2693–2700. [Google Scholar] [CrossRef] [PubMed]
- Udono, H.; Ichiyanagi, T.; Mizukami, S.; Imai, T. Heat shock proteins in antigen trafficking—Implications on antigen presentation to T cells. Int. J. Hyperth. 2009, 25, 617–625. [Google Scholar] [CrossRef] [PubMed]
- Albakova, Z.; Armeev, G.A.; Kanevskiy, L.M.; Kovalenko, E.I.; Sapozhnikov, A.M. HSP70 Multi-Functionality in Cancer. Cells 2020, 9, 587. [Google Scholar] [CrossRef] [PubMed]
- Guliy, O.I.; Staroverov, S.A.; Dykman, L.A. Heat Shock Proteins in Cancer Diagnostics. Appl. Biochem. Microbiol. 2023, 59, 395–407. [Google Scholar] [CrossRef]
- Linder, M.; Pogge von Strandmann, E. The Role of Extracellular HSP70 in the Function of Tumor-Associated Immune Cells. Cancers 2021, 13, 4721. [Google Scholar] [CrossRef] [PubMed]
- Multhoff, G. Activation of natural killer cells by heat shock protein 70. Int. J. Hyperth. 2002, 18, 576–585. [Google Scholar] [CrossRef] [PubMed]
- Specht, H.M.; Ahrens, N.; Blankenstein, C.; Duell, T.; Fietkau, R.; Gaipl, U.S.; Günther, C.; Gunther, S.; Habl, G.; Hautmann, H.; et al. Heat Shock Protein 70 (Hsp70) Peptide Activated Natural Killer (NK) Cells for the Treatment of Patients with Non-Small Cell Lung Cancer (NSCLC) after Radiochemotherapy (RCTx)—From Preclinical Studies to a Clinical Phase II Trial. Front. Immunol. 2015, 6, 162. [Google Scholar] [CrossRef] [PubMed]
- Elsner, L.; Muppala, V.; Gehrmann, M.; Lozano, J.; Malzahn, D.; Bickeböller, H.; Brunner, E.; Zientkowska, M.; Herrmann, T.; Walter, L.; et al. The heat shock protein HSP70 promotes mouse NK cell activity against tumors that express inducible NKG2D ligands. J. Immunol. 2007, 179, 5523–5533. [Google Scholar] [CrossRef]
- Dean, I.; Lee, C.Y.C.; Tuong, Z.K.; Li, Z.; Tibbitt, C.A.; Willis, C.; Gaspal, F.; Kennedy, B.C.; Matei-Rascu, V.; Fiancette, R.; et al. Rapid functional impairment of natural killer cells following tumor entry limits anti-tumor immunity. Nat. Commun. 2024, 15, 683. [Google Scholar] [CrossRef]
- Wu, Y.; Kuang, D.M.; Pan, W.D.; Wan, Y.L.; Lao, X.M.; Wang, D.; Li, X.F.; Zheng, L. Monocyte/macrophage-elicited natural killer cell dysfunction in hepatocellular carcinoma is mediated by CD48/2B4 interactions. Hepatology 2013, 57, 1107–1116. [Google Scholar] [CrossRef] [PubMed]
- Bunting, M.D.; Vyas, M.; Requesens, M.; Langenbucher, A.; Schiferle, E.B.; Manguso, R.T.; Lawrence, M.S.; Demehri, S. Extracellular matrix proteins regulate NK cell function in peripheral tissues. Sci. Adv. 2022, 8, eabk3327. [Google Scholar] [CrossRef] [PubMed]
- Rossi, G.R.; Trindade, E.S.; Souza-Fonseca-Guimaraes, F. Tumor Microenvironment-Associated Extracellular Matrix Components Regulate NK Cell Function. Front. Immunol. 2020, 11, 73. [Google Scholar] [CrossRef] [PubMed]
- Itagaki, F.; Nakatsuka, K.; Sakai, H.; Endo, S.; Su, M.-T.; Takai, T. Fibronectin on target cells attenuates natural cytotoxicity of NK cells via myeloid immune checkpoint ILT3/LILRB4/gp49B. Int. Immunol. 2023, 35, 339–348. [Google Scholar] [CrossRef] [PubMed]
- Mai, Z.; Lin, Y.; Lin, P.; Zhao, X.; Cui, L. Modulating extracellular matrix stiffness: A strategic approach to boost cancer immunotherapy. Cell Death Dis. 2024, 15, 307. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Li, Y.; Xu, C.; Dong, J.; Wei, J. An oncolytic vaccinia virus encoding hyaluronidase reshapes the extracellular matrix to enhance cancer chemotherapy and immunotherapy. J. Immunother. Cancer 2024, 12, e008431. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Han, X.; Hu, X.; Jin, F.; Gao, Z.; Yin, L.; Qin, J.; Yin, F.; Li, C.; Wang, Y. IDO1 impairs NK cell cytotoxicity by decreasing NKG2D/NKG2DLs via promoting miR-18a. Mol. Immunol. 2018, 103, 144–155. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Atteridge, C.L.; Wang, X.; Lundgren, A.D.; Wu, J.D. The membrane type matrix metalloproteinase MMP14 mediates constitutive shedding of MHC class I chain-related molecule A independent of A disintegrin and metalloproteinases. J. Immunol. 2010, 184, 3346–3350. [Google Scholar] [CrossRef] [PubMed]
- Zocchi, M.R.; Catellani, S.; Canevali, P.; Tavella, S.; Garuti, A.; Villaggio, B.; Zunino, A.; Gobbi, M.; Fraternali-Orcioni, G.; Kunkl, A.; et al. High ERp5/ADAM10 expression in lymph node microenvironment and impaired NKG2D ligands recognition in Hodgkin lymphomas. Blood 2012, 119, 1479–1489. [Google Scholar] [CrossRef] [PubMed]
- Chitadze, G.; Lettau, M.; Bhat, J.; Wesch, D.; Steinle, A.; Fürst, D.; Mytilineos, J.; Kalthoff, H.; Janssen, O.; Oberg, H.H.; et al. Shedding of endogenous MHC class I-related chain molecules A and B from different human tumor entities: Heterogeneous involvement of the “a disintegrin and metalloproteases” 10 and 17. Int. J. Cancer 2013, 133, 1557–1566. [Google Scholar] [CrossRef]
- Lee, J.-C.; Lee, K.-M.; Kim, D.-W.; Heo, D.S. Elevated TGF-β1 secretion and down-modulation of NKG2D underlies impaired NK cytotoxicity in cancer patients. J. Immunol. 2004, 172, 7335–7340. [Google Scholar] [CrossRef] [PubMed]
- Groh, V.; Wu, J.; Yee, C.; Spies, T. Tumour-derived soluble MIC ligands impair expression of NKG2D and T-cell activation. Nature 2002, 419, 734–738. [Google Scholar] [CrossRef] [PubMed]
- Baragaño Raneros, A.; Suarez Álvarez, B.; López Larrea, C. Secretory pathways generating immunosuppressive NKG2D ligands: New targets for therapeutic intervention. Oncoimmunology 2014, 3, e28497. [Google Scholar] [CrossRef] [PubMed]
- Vyas, M.; Reinartz, S.; Hoffmann, N.; Reiners, K.S.; Lieber, S.; Jansen, J.M.; Wagner, U.; Müller, R.; von Strandmann, E.P. Soluble NKG2D ligands in the ovarian cancer microenvironment are associated with an adverse clinical outcome and decreased memory effector T cells independent of NKG2D downregulation. Oncoimmunology 2017, 6, e1339854. [Google Scholar] [CrossRef] [PubMed]
- Orange, J.S. Natural killer cell deficiency. J. Allergy Clin. Immunol. 2013, 132, 515–525. [Google Scholar] [CrossRef] [PubMed]
- Gineau, L.; Cognet, C.; Kara, N.; Lach, F.P.; Dunne, J.; Veturi, U.; Picard, C.; Trouillet, C.; Eidenschenk, C.; Aoufouchi, S.; et al. Partial MCM4 deficiency in patients with growth retardation, adrenal insufficiency, and natural killer cell deficiency. J. Clin. Investig. 2012, 122, 821–832. [Google Scholar] [CrossRef] [PubMed]
- Shaw, R.K.; Issekutz, A.C.; Fraser, R.; Schmit, P.; Morash, B.; Monaco-Shawver, L.; Orange, J.S.; Fernandez, C.V. Bilateral adrenal EBV-associated smooth muscle tumors in a child with a natural killer cell deficiency. Blood 2012, 119, 4009–4012. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, K.N.; Leung, B.; Kwong, M.; Zarember, K.A.; Satyal, S.; Navas, T.A.; Wang, F.; Godowski, P.J. APC-independent activation of NK cells by the Toll-like receptor 3 agonist double-stranded RNA. J. Immunol. 2004, 172, 138–143. [Google Scholar] [CrossRef] [PubMed]
- Hart, O.M.; Athie-Morales, V.; O’Connor, G.M.; Gardiner, C.M. TLR7/8-mediated activation of human NK cells results in accessory cell-dependent IFN-gamma production. J. Immunol. 2005, 175, 1636–1642. [Google Scholar] [CrossRef]
- Nguyen, K.B.; Salazar-Mather, T.P.; Dalod, M.Y.; Van Deusen, J.B.; Wei, X.Q.; Liew, F.Y.; Caligiuri, M.A.; Durbin, J.E.; Biron, C.A. Coordinated and distinct roles for IFN-alpha beta, IL-12, and IL-15 regulation of NK cell responses to viral infection. J. Immunol. 2002, 169, 4279–4287. [Google Scholar] [CrossRef]
- Gumá, M.; Angulo, A.; López-Botet, M. NK cell receptors involved in the response to human cytomegalovirus infection. Curr. Top. Microbiol. Immunol. 2006, 298, 207–223. [Google Scholar] [CrossRef]
- Petersen, J.L.; Morris, C.R.; Solheim, J.C. Virus Evasion of MHC Class I Molecule Presentation1. J. Immunol. 2003, 171, 4473–4478. [Google Scholar] [CrossRef]
- Sun, J.C.; Beilke, J.N.; Lanier, L.L. Adaptive immune features of natural killer cells. Nature 2009, 457, 557–561. [Google Scholar] [CrossRef]
- Nabekura, T.; Kanaya, M.; Shibuya, A.; Fu, G.; Gascoigne, N.R.J.; Lanier, L.L. Costimulatory Molecule DNAM-1 Is Essential for Optimal Differentiation of Memory Natural Killer Cells during Mouse Cytomegalovirus Infection. Immunity 2014, 40, 225–234. [Google Scholar] [CrossRef]
- Romee, R.; Schneider, S.E.; Leong, J.W.; Chase, J.M.; Keppel, C.R.; Sullivan, R.P.; Cooper, M.A.; Fehniger, T.A. Cytokine activation induces human memory-like NK cells. Blood 2012, 120, 4751–4760. [Google Scholar] [CrossRef]
- Cooper, M.A.; Elliott, J.M.; Keyel, P.A.; Yang, L.; Carrero, J.A.; Yokoyama, W.M. Cytokine-induced memory-like natural killer cells. Proc. Natl. Acad. Sci. USA 2009, 106, 1915–1919. [Google Scholar] [CrossRef]
- Min-Oo, G.; Bezman, N.A.; Madera, S.; Sun, J.C.; Lanier, L.L. Proapoptotic Bim regulates antigen-specific NK cell contraction and the generation of the memory NK cell pool after cytomegalovirus infection. J. Exp. Med. 2014, 211, 1289–1296. [Google Scholar] [CrossRef]
- Sun, J.C.; Beilke, J.N.; Bezman, N.A.; Lanier, L.L. Homeostatic proliferation generates long-lived natural killer cells that respond against viral infection. J. Exp. Med. 2011, 208, 357–368. [Google Scholar] [CrossRef]
- Lee, J.; Zhang, T.; Hwang, I.; Kim, A.; Nitschke, L.; Kim, M.; Scott, J.M.; Kamimura, Y.; Lanier, L.L.; Kim, S. Epigenetic modification and antibody-dependent expansion of memory-like NK cells in human cytomegalovirus-infected individuals. Immunity 2015, 42, 431–442. [Google Scholar] [CrossRef]
- Liu, L.L.; Landskron, J.; Ask, E.H.; Enqvist, M.; Sohlberg, E.; Traherne, J.A.; Hammer, Q.; Goodridge, J.P.; Larsson, S.; Jayaraman, J.; et al. Critical Role of CD2 Co-stimulation in Adaptive Natural Killer Cell Responses Revealed in NKG2C-Deficient Humans. Cell Rep. 2016, 15, 1088–1099. [Google Scholar] [CrossRef]
- Rückert, T.; Lareau, C.A.; Mashreghi, M.F.; Ludwig, L.S.; Romagnani, C. Clonal expansion and epigenetic inheritance of long-lasting NK cell memory. Nat. Immunol. 2022, 23, 1551–1563. [Google Scholar] [CrossRef]
- Hammer, Q.; Rückert, T.; Borst, E.M.; Dunst, J.; Haubner, A.; Durek, P.; Heinrich, F.; Gasparoni, G.; Babic, M.; Tomic, A.; et al. Peptide-specific recognition of human cytomegalovirus strains controls adaptive natural killer cells. Nat. Immunol. 2018, 19, 453–463. [Google Scholar] [CrossRef]
- Jost, S.; Lucar, O.; Lee, E.; Yoder, T.; Kroll, K.; Sugawara, S.; Smith, S.; Jones, R.; Tweet, G.; Werner, A.; et al. Antigen-specific memory NK cell responses against HIV and influenza use the NKG2/HLA-E axis. Sci. Immunol. 2023, 8, eadi3974. [Google Scholar] [CrossRef]
- Escobar, G.; Mangani, D.; Anderson, A.C. T cell factor 1: A master regulator of the T cell response in disease. Sci. Immunol. 2020, 5, eabb9726. [Google Scholar] [CrossRef]
- Raghu, D.; Xue, H.H.; Mielke, L.A. Control of Lymphocyte Fate, Infection, and Tumor Immunity by TCF-1. Trends Immunol. 2019, 40, 1149–1162. [Google Scholar] [CrossRef]
- Weber, B.N.; Chi, A.W.; Chavez, A.; Yashiro-Ohtani, Y.; Yang, Q.; Shestova, O.; Bhandoola, A. A critical role for TCF-1 in T-lineage specification and differentiation. Nature 2011, 476, 63–68. [Google Scholar] [CrossRef]
- Zhou, X.; Yu, S.; Zhao, D.M.; Harty, J.T.; Badovinac, V.P.; Xue, H.H. Differentiation and persistence of memory CD8(+) T cells depend on T cell factor 1. Immunity 2010, 33, 229–240. [Google Scholar] [CrossRef]
- Jeannet, G.; Boudousquié, C.; Gardiol, N.; Kang, J.; Huelsken, J.; Held, W. Essential role of the Wnt pathway effector Tcf-1 for the establishment of functional CD8 T cell memory. Proc. Natl. Acad. Sci. USA 2010, 107, 9777–9782. [Google Scholar] [CrossRef]
- Connolly, K.A.; Kuchroo, M.; Venkat, A.; Khatun, A.; Wang, J.; William, I.; Hornick, N.I.; Fitzgerald, B.L.; Damo, M.; Kasmani, M.Y.; et al. A reservoir of stem-like CD8(+) T cells in the tumor-draining lymph node preserves the ongoing antitumor immune response. Sci. Immunol. 2021, 6, eabg7836. [Google Scholar] [CrossRef]
- Im, S.J.; Hashimoto, M.; Gerner, M.Y.; Lee, J.; Kissick, H.T.; Burger, M.C.; Shan, Q.; Hale, J.S.; Lee, J.; Nasti, T.H.; et al. Defining CD8+ T cells that provide the proliferative burst after PD-1 therapy. Nature 2016, 537, 417–421. [Google Scholar] [CrossRef]
- Jeevan-Raj, B.; Gehrig, J.; Charmoy, M.; Chennupati, V.; Grandclément, C.; Angelino, P.; Delorenzi, M.; Held, W. The Transcription Factor Tcf1 Contributes to Normal NK Cell Development and Function by Limiting the Expression of Granzymes. Cell Rep. 2017, 20, 613–626. [Google Scholar] [CrossRef]
- Torcellan, T.; Friedrich, C.; Doucet-Ladevèze, R.; Ossner, T.; Solé, V.V.; Riedmann, S.; Ugur, M.; Imdahl, F.; Rosshart, S.P.; Arnold, S.J.; et al. Circulating NK cells establish tissue residency upon acute infection of skin and mediate accelerated effector responses to secondary infection. Immunity 2024, 57, 124–140.e127. [Google Scholar] [CrossRef]
- Kujur, W.; Murillo, O.; Adduri, R.S.R.; Vankayalapati, R.; Konduru, N.V.; Mulik, S. Memory like NK cells display stem cell like properties after Zika virus infection. PLoS Pathog. 2021, 16, e1009132. [Google Scholar] [CrossRef]
- Wang, Y.; Lifshitz, L.; Gellatly, K.; Vinton, C.L.; Busman-Sahay, K.; McCauley, S.; Vangala, P.; Kim, K.; Derr, A.; Jaiswal, S.; et al. HIV-1-induced cytokines deplete homeostatic innate lymphoid cells and expand TCF7-dependent memory NK cells. Nat. Immunol. 2020, 21, 274–286. [Google Scholar] [CrossRef]
- Keppel, M.P.; Yang, L.; Cooper, M.A. Murine NK cell intrinsic cytokine-induced memory-like responses are maintained following homeostatic proliferation. J. Immunol. 2013, 190, 4754–4762. [Google Scholar] [CrossRef]
- Bednarski, J.J.; Zimmerman, C.; Berrien-Elliott, M.M.; Foltz, J.A.; Becker-Hapak, M.; Neal, C.C.; Foster, M.; Schappe, T.; McClain, E.; Pence, P.P.; et al. Donor memory-like NK cells persist and induce remissions in pediatric patients with relapsed AML after transplant. Blood 2022, 139, 1670–1683. [Google Scholar] [CrossRef]
- Fehniger, T.A.; Cooper, M.A. Harnessing NK Cell Memory for Cancer Immunotherapy. Trends Immunol. 2016, 37, 877–888. [Google Scholar] [CrossRef]
- Parihar, R. Memory NK cells to forget relapsed AML. Blood 2022, 139, 1607–1608. [Google Scholar] [CrossRef]
- Romee, R.; Rosario, M.; Berrien-Elliott, M.M.; Wagner, J.A.; Jewell, B.A.; Schappe, T.; Leong, J.W.; Abdel-Latif, S.; Schneider, S.E.; Willey, S.; et al. Cytokine-induced memory-like natural killer cells exhibit enhanced responses against myeloid leukemia. Sci. Transl. Med. 2016, 8, 357ra123. [Google Scholar] [CrossRef]
- Hanna, G.J.; Coleman, K.; Birch, G.; Redd, R.A.; Alonso, A.; Bednarz, S.; Daley, H.; Hernandez Rodriguez, D.E.; Shaw, K.L.; Haddad, R.I.; et al. Abstract CT540: A phase 1 trial of cytokine-induced memory-like (CIML) natural killer (NK) cell therapy with IL-15 superagonist in advanced head and neck cancer: Part 1 results. Cancer Res. 2022, 82, CT540. [Google Scholar] [CrossRef]
- Uppendahl, L.D.; Felices, M.; Bendzick, L.; Ryan, C.; Kodal, B.; Hinderlie, P.; Boylan, K.L.M.; Skubitz, A.P.N.; Miller, J.S.; Geller, M.A. Cytokine-induced memory-like natural killer cells have enhanced function, proliferation, and in vivo expansion against ovarian cancer cells. Gynecol. Oncol. 2019, 153, 149–157. [Google Scholar] [CrossRef]
- Pal, M.; Schwab, L.; Yermakova, A.; Mace, E.M.; Claus, R.; Krahl, A.C.; Woiterski, J.; Hartwig, U.F.; Orange, J.S.; Handgretinger, R.; et al. Tumor-priming converts NK cells to memory-like NK cells. Oncoimmunology 2017, 6, e1317411. [Google Scholar] [CrossRef]
- El-Sherbiny, Y.M.; Holmes, T.D.; Wetherill, L.F.; Black, E.V.; Wilson, E.B.; Phillips, S.L.; Scott, G.B.; Adair, R.A.; Dave, R.; Scott, K.J.; et al. Controlled infection with a therapeutic virus defines the activation kinetics of human natural killer cells in vivo. Clin. Exp. Immunol. 2015, 180, 98–107. [Google Scholar] [CrossRef]
- Miyamoto, S.; Inoue, H.; Nakamura, T.; Yamada, M.; Sakamoto, C.; Urata, Y.; Okazaki, T.; Marumoto, T.; Takahashi, A.; Takayama, K.; et al. Coxsackievirus B3 Is an Oncolytic Virus with Immunostimulatory Properties That Is Active against Lung Adenocarcinoma. Cancer Res. 2012, 72, 2609–2621. [Google Scholar] [CrossRef]
- Ahmed, J.; Chard, L.S.; Yuan, M.; Wang, J.; Howells, A.; Li, Y.; Li, H.; Zhang, Z.; Lu, S.; Gao, D.; et al. A new oncolytic Vacciniavirus augments antitumor immune responses to prevent tumor recurrence and metastasis after surgery. J. Immunother. Cancer 2020, 8, e000415. [Google Scholar] [CrossRef]
- Leung, E.Y.L.; Ennis, D.P.; Kennedy, P.R.; Hansell, C.; Dowson, S.; Farquharson, M.; Spiliopoulou, P.; Nautiyal, J.; McNamara, S.; Carlin, L.M.; et al. NK Cells Augment Oncolytic Adenovirus Cytotoxicity in Ovarian Cancer. Mol. Ther.-Oncolytics 2020, 16, 289–301. [Google Scholar] [CrossRef]
- Kottke, T.; Thompson, J.; Diaz, R.M.; Pulido, J.; Willmon, C.; Coffey, M.; Selby, P.; Melcher, A.; Harrington, K.; Vile, R.G. Improved systemic delivery of oncolytic reovirus to established tumors using preconditioning with cyclophosphamide-mediated Treg modulation and interleukin-2. Clin. Cancer Res. 2009, 15, 561–569. [Google Scholar] [CrossRef]
- Kottke, T.; Galivo, F.; Wongthida, P.; Maria Diaz, R.; Thompson, J.; Jevremovic, D.; Barber, G.N.; Hall, G.; Chester, J.; Selby, P.; et al. Treg Depletion–enhanced IL-2 Treatment Facilitates Therapy of Established Tumors Using Systemically Delivered Oncolytic Virus. Mol. Ther. 2008, 16, 1217–1226. [Google Scholar] [CrossRef]
- Kim, Y.; Yoo, J.Y.; Lee, T.J.; Liu, J.; Yu, J.; Caligiuri, M.A.; Kaur, B.; Friedman, A. Complex role of NK cells in regulation of oncolytic virus-bortezomib therapy. Proc. Natl. Acad. Sci. USA 2018, 115, 4927–4932. [Google Scholar] [CrossRef] [PubMed]
- Aurelian, L.; Bollino, D.; Colunga, A. The oncolytic virus ÃÂPK has multimodal anti-tumor activity. Pathog. Dis. 2016, 74, ftw050. [Google Scholar] [CrossRef] [PubMed]
- Wongthida, P.; Diaz, R.M.; Galivo, F.; Kottke, T.; Thompson, J.; Pulido, J.; Pavelko, K.; Pease, L.; Melcher, A.; Vile, R. Type III IFN interleukin-28 mediates the antitumor efficacy of oncolytic virus VSV in immune-competent mouse models of cancer. Cancer Res. 2010, 70, 4539–4549. [Google Scholar] [CrossRef] [PubMed]
- Jarahian, M.; Watzl, C.; Fournier, P.; Arnold, A.; Djandji, D.; Zahedi, S.; Cerwenka, A.; Paschen, A.; Schirrmacher, V.; Momburg, F. Activation of natural killer cells by newcastle disease virus hemagglutinin-neuraminidase. J. Virol. 2009, 83, 8108–8121. [Google Scholar] [CrossRef] [PubMed]
- Jarahian, M.; Fiedler, M.; Cohnen, A.; Djandji, D.; Hämmerling, G.J.; Gati, C.; Cerwenka, A.; Turner, P.C.; Moyer, R.W.; Watzl, C. Modulation of NKp30-and NKp46-mediated natural killer cell responses by poxviral hemagglutinin. PLoS Pathog. 2011, 7, e1002195. [Google Scholar] [CrossRef] [PubMed]
- Altomonte, J.; Wu, L.; Chen, L.; Meseck, M.; Ebert, O.; García-Sastre, A.; Fallon, J.; Woo, S.L.C. Exponential enhancement of oncolytic vesicular stomatitis virus potency by vector-mediated suppression of inflammatory responses in vivo. Mol. Ther. 2008, 16, 146–153. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Chen, X.; Chu, J.; Xu, B.; Meisen, W.H.; Chen, L.; Zhang, L.; Zhang, J.; He, X.; Wang, Q.E.; et al. TGFβ Treatment Enhances Glioblastoma Virotherapy by Inhibiting the Innate Immune Response. Cancer Res. 2015, 75, 5273–5282. [Google Scholar] [CrossRef] [PubMed]
- Fulci, G.; Breymann, L.; Gianni, D.; Kurozomi, K.; Rhee, S.S.; Yu, J.; Kaur, B.; Louis, D.N.; Weissleder, R.; Caligiuri, M.A.; et al. Cyclophosphamide enhances glioma virotherapy by inhibiting innate immune responses. Proc. Natl. Acad. Sci. USA 2006, 103, 12873–12878. [Google Scholar] [CrossRef] [PubMed]
- Altomonte, J.; Wu, L.; Meseck, M.; Chen, L.; Ebert, O.; Garcia-Sastre, A.; Fallon, J.; Mandeli, J.; Woo, S.L.C. Enhanced oncolytic potency of vesicular stomatitis virus through vector-mediated inhibition of NK and NKT cells. Cancer Gene Ther. 2009, 16, 266–278. [Google Scholar] [CrossRef]
- Westwood, J.A.; Kelly, J.M.; Tanner, J.E.; Kershaw, M.H.; Smyth, M.J.; Hayakawa, Y. Cutting Edge: Novel Priming of Tumor-Specific Immunity by NKG2D-Triggered NK Cell-Mediated Tumor Rejection and Th1-Independent CD4+ T Cell Pathway1. J. Immunol. 2004, 172, 757–761. [Google Scholar] [CrossRef] [PubMed]
- Kelly, J.M.; Darcy, P.K.; Markby, J.L.; Godfrey, D.I.; Takeda, K.; Yagita, H.; Smyth, M.J. Induction of tumor-specific T cell memory by NK cell–mediated tumor rejection. Nat. Immunol. 2002, 3, 83–90. [Google Scholar] [CrossRef]
- Mocikat, R.; Braumüller, H.; Gumy, A.; Egeter, O.; Ziegler, H.; Reusch, U.; Bubeck, A.; Louis, J.; Mailhammer, R.; Riethmüller, G. Natural killer cells activated by MHC class Ilow targets prime dendritic cells to induce protective CD8 T cell responses. Immunity 2003, 19, 561–569. [Google Scholar] [CrossRef]
- Böttcher, J.P.; Bonavita, E.; Chakravarty, P.; Blees, H.; Cabeza-Cabrerizo, M.; Sammicheli, S.; Rogers, N.C.; Sahai, E.; Zelenay, S.; Reis e Sousa, C. NK Cells Stimulate Recruitment of cDC1 into the Tumor Microenvironment Promoting Cancer Immune Control. Cell 2018, 172, 1022–1037.e14. [Google Scholar] [CrossRef] [PubMed]
- Moretta, A.; Marcenaro, E.; Sivori, S.; Chiesa, M.D.; Vitale, M.; Moretta, L. Early liaisons between cells of the innate immune system in inflamed peripheral tissues. Trends Immunol. 2005, 26, 668–675. [Google Scholar] [CrossRef]
- Frasca, L.; Nasso, M.; Spensieri, F.; Fedele, G.; Palazzo, R.; Malavasi, F.; Ausiello, C.M. IFN-γ Arms Human Dendritic Cells to Perform Multiple Effector Functions1. J. Immunol. 2008, 180, 1471–1481. [Google Scholar] [CrossRef] [PubMed]
- Rock, K.L.; Reits, E.; Neefjes, J. Present Yourself! By MHC Class I and MHC Class II Molecules. Trends Immunol. 2016, 37, 724–737. [Google Scholar] [CrossRef] [PubMed]
- Schoenborn, J.R.; Wilson, C.B. Regulation of interferon-gamma during innate and adaptive immune responses. Adv. Immunol. 2007, 96, 41–101. [Google Scholar] [CrossRef]
- Théry, C.; Amigorena, S. The cell biology of antigen presentation in dendritic cells. Curr. Opin. Immunol. 2001, 13, 45–51. [Google Scholar] [CrossRef]
- Colbert, J.D.; Cruz, F.M.; Rock, K.L. Cross-presentation of exogenous antigens on MHC I molecules. Curr. Opin. Immunol. 2020, 64, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Schuster, P.; Lindner, G.; Thomann, S.; Haferkamp, S.; Schmidt, B. Prospect of Plasmacytoid Dendritic Cells in Enhancing Anti-Tumor Immunity of Oncolytic Herpes Viruses. Cancers 2019, 11, 651. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Shin, D.-H.; Sung, C.K. The Optimal Balance between Oncolytic Viruses and Natural Killer Cells: A Mathematical Approach. Mathematics 2022, 10, 3370. [Google Scholar] [CrossRef]
- Senekal, N.S.; Mahasa, K.J.; Eladdadi, A.; de Pillis, L.; Ouifki, R. Natural Killer Cells Recruitment in Oncolytic Virotherapy: A Mathematical Model. Bull. Math. Biol. 2021, 83, 75. [Google Scholar] [CrossRef]
- Mgrditchian, T.; Arakelian, T.; Paggetti, J.; Noman, M.Z.; Viry, E.; Moussay, E.; Van Moer, K.; Kreis, S.; Guerin, C.; Buart, S.; et al. Targeting autophagy inhibits melanoma growth by enhancing NK cells infiltration in a CCL5-dependent manner. Proc. Natl. Acad. Sci. USA 2017, 114, e9271–e9279. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Sheng, Y.; Hou, W.; Sampath, P.; Byrd, D.; Thorne, S.; Zhang, Y. CCL5-armed oncolytic virus augments CCR5-engineered NK cell infiltration and antitumor efficiency. J. Immunother. Cancer 2020, 8, e000131. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Han, J.; Chu, J.; Zhang, L.; Zhang, J.; Chen, C.; Chen, L.; Wang, Y.; Wang, H.; Yi, L.; et al. A combinational therapy of EGFR-CAR NK cells and oncolytic herpes simplex virus 1 for breast cancer brain metastases. Oncotarget 2016, 7, 27764–27777. [Google Scholar] [CrossRef] [PubMed]
- Lee, G.A.; Liou, Y.-H.; Wang, S.-W.; Ko, K.-L.; Jiang, S.-T.; Liao, N.-S. Different NK Cell Developmental Events Require Different Levels of IL-15 Trans-Presentation. J. Immunol. 2011, 187, 1212–1221. [Google Scholar] [CrossRef] [PubMed]
- Lucas, M.; Schachterle, W.; Oberle, K.; Aichele, P.; Diefenbach, A. Dendritic cells prime natural killer cells by trans-presenting interleukin 15. Immunity 2007, 26, 503–517. [Google Scholar] [CrossRef] [PubMed]
- Mortier, E.; Woo, T.; Advincula, R.; Gozalo, S.; Ma, A. IL-15Ralpha chaperones IL-15 to stable dendritic cell membrane complexes that activate NK cells via trans presentation. J. Exp. Med. 2008, 205, 1213–1225. [Google Scholar] [CrossRef] [PubMed]
- Stoklasek, T.A.; Schluns, K.S.; Lefrançois, L. Combined IL-15/IL-15Ralpha immunotherapy maximizes IL-15 activity in vivo. J. Immunol. 2006, 177, 6072–6080. [Google Scholar] [CrossRef] [PubMed]
- Huntington, N.D.; Legrand, N.; Alves, N.L.; Jaron, B.; Weijer, K.; Plet, A.; Corcuff, E.; Mortier, E.; Jacques, Y.; Spits, H.; et al. IL-15 trans-presentation promotes human NK cell development and differentiation in vivo. J. Exp. Med. 2008, 206, 25–34. [Google Scholar] [CrossRef] [PubMed]
- Huntington, N.D.; Puthalakath, H.; Gunn, P.; Naik, E.; Michalak, E.M.; Smyth, M.J.; Tabarias, H.; Degli-Esposti, M.A.; Dewson, G.; Willis, S.N.; et al. Interleukin 15-mediated survival of natural killer cells is determined by interactions among Bim, Noxa and Mcl-1. Nat. Immunol. 2007, 8, 856–863. [Google Scholar] [CrossRef]
- Xu, B.; Ma, R.; Russell, L.; Yoo, J.Y.; Han, J.; Cui, H.; Yi, P.; Zhang, J.; Nakashima, H.; Dai, H.; et al. An oncolytic herpesvirus expressing E-cadherin improves survival in mouse models of glioblastoma. Nat. Biotechnol. 2019, 37, 45–54. [Google Scholar] [CrossRef]
- Ito, M.; Maruyama, T.; Saito, N.; Koganei, S.; Yamamoto, K.; Matsumoto, N. Killer cell lectin-like receptor G1 binds three members of the classical cadherin family to inhibit NK cell cytotoxicity. J. Exp. Med. 2006, 203, 289–295. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Hofmann, M.; Wang, Q.; Teng, L.; Chlewicki, L.K.; Pircher, H.; Mariuzza, R.A. Structure of natural killer cell receptor KLRG1 bound to E-cadherin reveals basis for MHC-independent missing self recognition. Immunity 2009, 31, 35–46. [Google Scholar] [CrossRef] [PubMed]
- Draganov, D.D.; Santidrian, A.F.; Minev, I.; Nguyen, D.; Kilinc, M.O.; Petrov, I.; Vyalkova, A.; Lander, E.; Berman, M.; Minev, B.; et al. Delivery of oncolytic vaccinia virus by matched allogeneic stem cells overcomes critical innate and adaptive immune barriers. J. Transl. Med. 2019, 17, 100. [Google Scholar] [CrossRef] [PubMed]
- Floerchinger, A.; Klein, J.E.; Finkbeiner, M.S.C.; Schäfer, T.E.; Fuchs, G.; Doerner, J.; Zirngibl, H.; Ackermann, M.; Kvasnicka, H.M.; Chester, K.A.; et al. A vector-encoded bispecific killer engager to harness virus-activated NK cells as anti-tumor effectors. Cell Death Dis. 2023, 14, 104. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
| OV | Tumor Model | Combination Therapy | Study/References |
|---|---|---|---|
| (Measles virus) MV-GFP-HSNS-EGFRvIII | Glioma (GBM39) | Not applicable (N/A) | [116] |
| (Herpes simplex virus-1) gamma34.5 mutant | Melanoma lines (Syngeneic DBA/2) | N/A | [117] |
| (Vesicular stomatitis virus) VSV-ΔM-mp53 | Melanoma lines (TS/A and B16) | N/A | [118] |
| (Reovirus) Reolysin | Leukemia cell line (K562) | N/A | [194] |
| Coxsackievirus B3 | Non-small-cell lung carcinoma line (A549) | N/A | [195] |
| (Vaccinia virus) VVΔTKΔN1L-IL12 | Lewis lung carcinoma | N/A | [196] |
| (Adenovirus) dl922-947, Ad3/Ad11p | Ovarian cancer (TOV21G and OVCAR4) | N/A | [197] |
| (Reovirus) WT type 3 Dearing strain | Melanoma line (B16) | Treg depletion and IL-2 treatment | [198] |
| (Vesicular stomatitis virus) VSV-GFP | Melanoma lines (B16 and YAC-1) | Treg depletion and IL-2 treatment | [199] |
| OV | Tumor Model | Combination Therapy | Study/References |
|---|---|---|---|
| (Herpes simplex virus-1) rQNestin34.5 | Human glioma (U87dEGFR) Syngeneic mouse glioma (KR158dEGFR) | N/A | [115] |
| (Vesicular stomatitis virus) rVSV-gG and rVSV-f | Hepatocellular carcinoma lines (BHK-21, McA-RH7777) | N/A | [205] |
| (Herpes simplex virus-1) rQNestin34.5 | Human GBM xenograft (GB30-FFL) Syngeneic mouse GBM (4C8) | TGF-β | [206] |
| (Herpes simplex virus-1) OV hrR3 | Rat glioma line (D74/HveC) | cyclophosphamide (CPA) | [207] |
| (Vesicular stomatitis virus) rVSV-UL141 | Hepatocellular carcinoma line (McA-RH7777) | N/A | [208] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Franks, M.L.; An, J.-H.; Leavenworth, J.W. The Role of Natural Killer Cells in Oncolytic Virotherapy: Friends or Foes? Vaccines 2024, 12, 721. https://doi.org/10.3390/vaccines12070721
Franks ML, An J-H, Leavenworth JW. The Role of Natural Killer Cells in Oncolytic Virotherapy: Friends or Foes? Vaccines. 2024; 12(7):721. https://doi.org/10.3390/vaccines12070721
Chicago/Turabian StyleFranks, Michael L., Ju-Hyun An, and Jianmei W. Leavenworth. 2024. "The Role of Natural Killer Cells in Oncolytic Virotherapy: Friends or Foes?" Vaccines 12, no. 7: 721. https://doi.org/10.3390/vaccines12070721
APA StyleFranks, M. L., An, J.-H., & Leavenworth, J. W. (2024). The Role of Natural Killer Cells in Oncolytic Virotherapy: Friends or Foes? Vaccines, 12(7), 721. https://doi.org/10.3390/vaccines12070721