
Tumor Neoepitope-Based Vaccines: A Scoping Review on Current Predictive Computational Strategies

Abstract
1. Introduction
2. Methods
- Scopus: article type, abstract, keywords; all open access; publication stage: final; article journal.
- Pubmed: free full text, full text.
- Web of science: open access; articles; English language.
- Science Direct: open access and open archive; research article (search in “title, abstract, keywords”).
- Dataset (next-generation sequencing data sequencing)
- Type of tumor
- Variant calling
- Neoepitope prediction program
- Alleles of MHC-I and II
- Statistics (the score used to classify epitopes)
- Evaluated immunological characteristics (biological and biochemical aspects affecting the immune response)
- Whether a structural model was evaluated
- Whether in vitro and in vivo validations were performed
- Positive/negative aspects
- Whether mutational data were evaluated
- Length of MHC-I and II epitopes
- Algorithm/matrix used
- Training data (tumor or microbiome)
- Steps of prioritization of neoepitopes
- Performance
3. Results and Discussion
3.1. Critical Steps in Identifying Immunogenic Neoepitopes
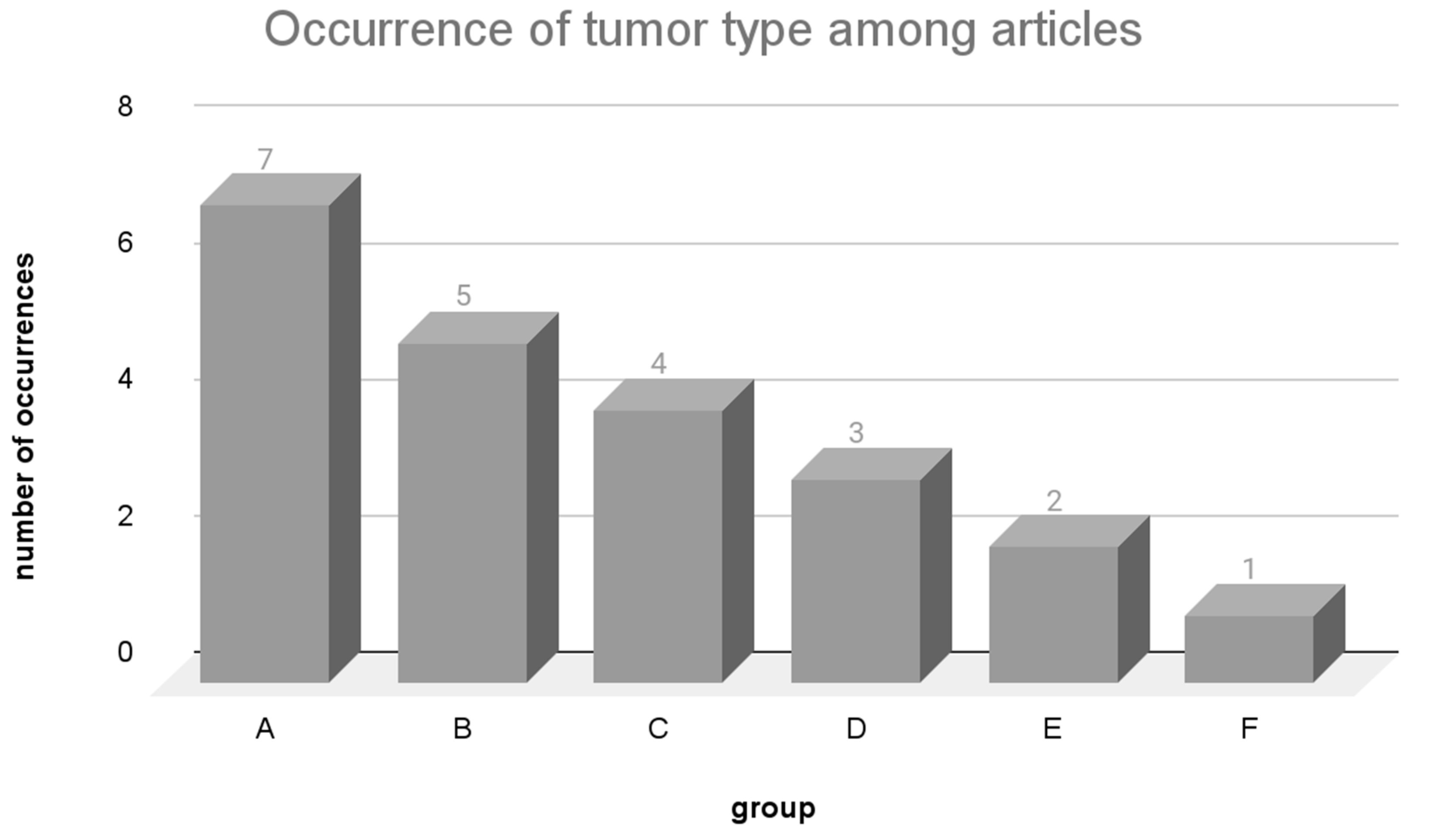
3.2. Main Types of Tumors Studied and Variant Calling
3.2.1. Mutation Frequency
3.2.2. Variant Calling
Limitations of Variant Calling
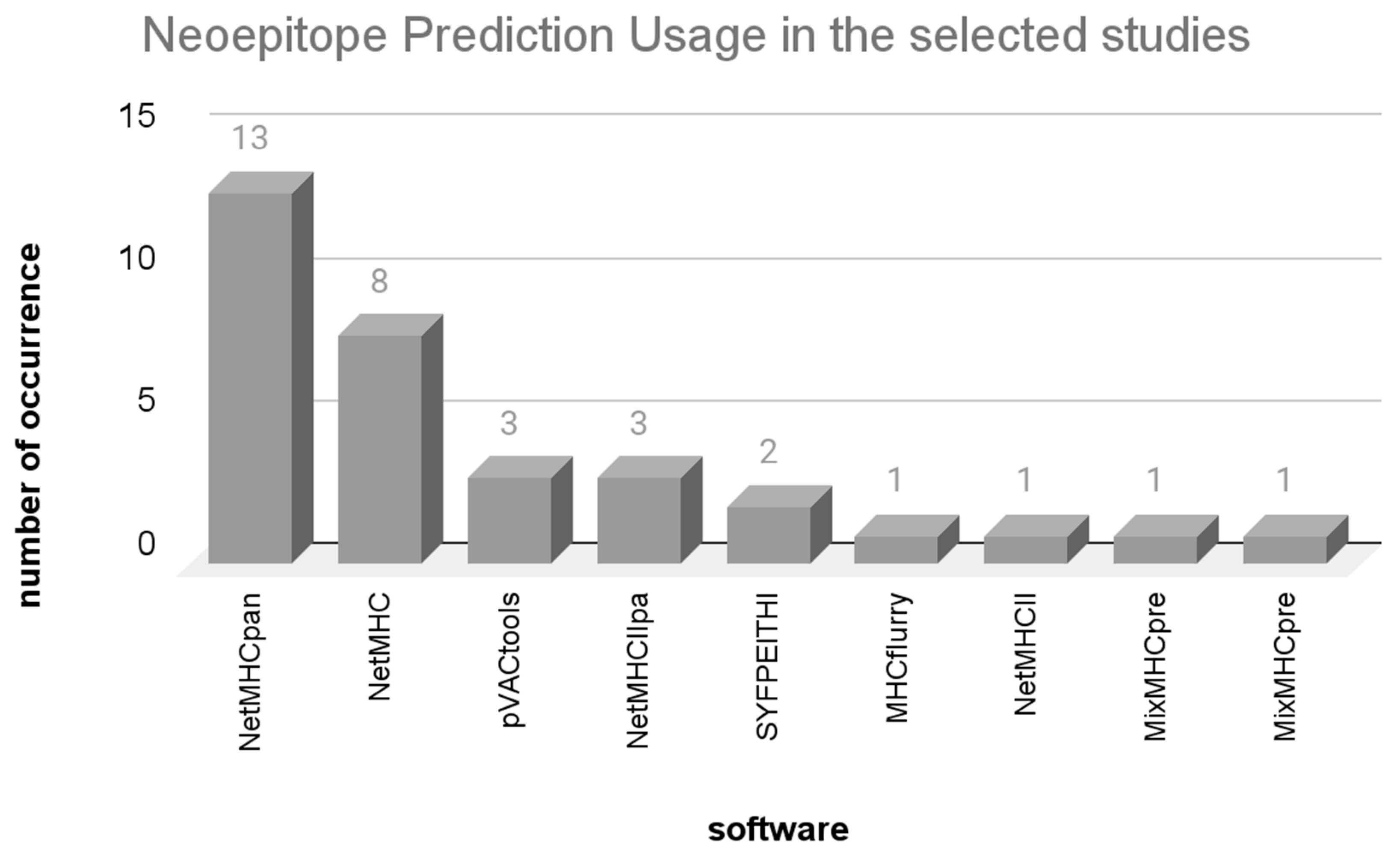
3.3. Algorithms Employed for Prediction and Prioritization of Neoepitopes
3.3.1. Neoepitope Prediction Programs
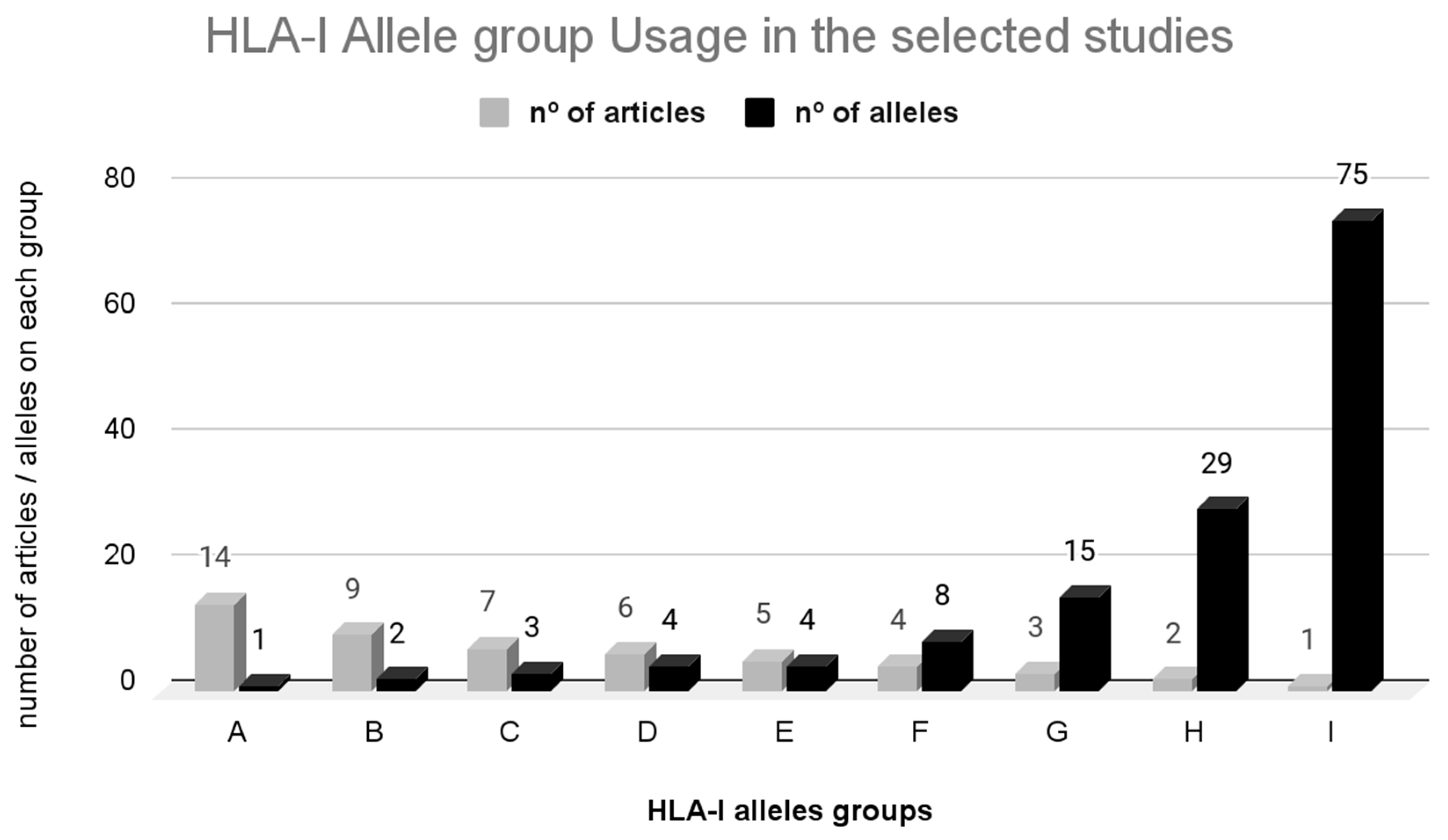
3.3.2. HLA-I Restriction
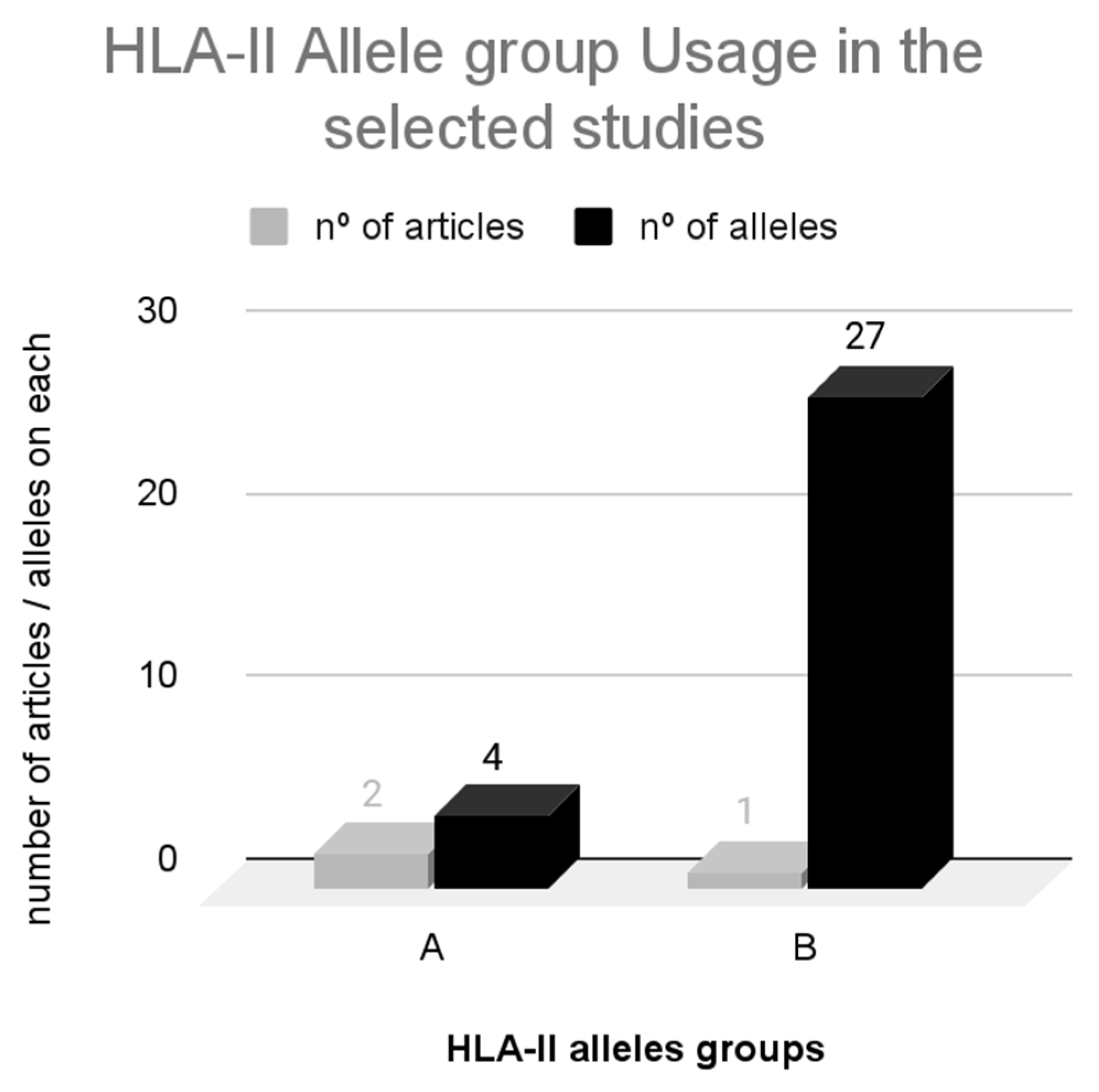
3.3.3. HLA-II Restriction
3.3.4. Prediction Algorithms for Neoepitopes
3.3.5. Limitations in MHC-I Neoepitope Prediction
3.3.6. Limitations in MHC-II Neoepitope Prediction
3.3.7. Prioritization Methods of Immunogenic Neoepitopes
3.4. Training Datasets Models
3.5. Performance and In Silico Validation
3.6. Can This Prediction Accurately Guide the Identification of Potential Targets for the Development of Therapeutic Vaccines?
3.7. Restrictions Regarding the Article Selection Process
4. Conclusions and Perspectives
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Schmidt, J.; Smith, A.R.; Magnin, M.; Racle, J.; Devlin, J.R.; Bobisse, S.; Cesbron, J.; Bonnet, V.; Carmona, S.J.; Huber, F.; et al. Prediction of Neo-Epitope Immunogenicity Reveals TCR Recognition Determinants and Provides Insight into Immunoediting. Cell Rep. Med. 2021, 2, 100194. [Google Scholar] [CrossRef]
- Lundegaard, C.; Lamberth, K.; Harndahl, M.; Buus, S.; Lund, O.; Nielsen, M. NetMHC-3.0: Accurate web accessible predictions of human, mouse and monkey MHC class I affinities for peptides of length 8–11. Nucleic Acids Res. 2018, 36 (Suppl. 2), W509–W512. [Google Scholar] [CrossRef] [PubMed]
- Brennick, C.A.; George, M.M.; Corwin, W.L.; Srivastava, P.K.; Ebrahimi-Nik, H. Neoepitopes as cancer immunotherapy targets: Key challenges and opportunities. Immunotherapy 2017, 9, 361–371. [Google Scholar] [CrossRef] [PubMed]
- Nagpal, G.; Chaudhary, K.; Agrawal, P.; Raghava, G.P.S. Computer-aided prediction of antigen presenting cell modulators for designing peptide-based vaccine adjuvants. J. Transl. Med. 2018, 16, 181. [Google Scholar] [CrossRef] [PubMed]
- Tomar, N.; De, R.K. Immunoinformatics: An Integrated Scenario. Immunology 2010, 131, 153–168. [Google Scholar] [CrossRef]
- Reardon, B.; Koşaloğlu-Yalçın, Z.; Paul, S.; Peters, B.; Sette, A. Allele-specific thresholds of eluted ligands for T-cell epitope prediction. Mol.Cell. Proteom. 2021, 20, 100122. [Google Scholar] [CrossRef]
- Ghosh, M.; Di Marco, M.; Stevanović, S. Identification of MHC Ligands and Establishing MHC Class I Peptide Motifs. Met. Mol. Biol. 2019, 1988, 137–147. [Google Scholar] [CrossRef]
- Wang, P.; Sidney, J.; Kim, Y.; Sette, A.; Lund, O.; Nielsen, M.; Peters, B. Peptide Binding Predictions for HLA DR, DP and DQ Molecules. BMC Bioinform. 2010, 11, 568. [Google Scholar] [CrossRef] [PubMed]
- Lv, J.; Zhu, Y.; Ji, A.; Zhang, Q.; Liao, G. Mining TCGA Database for Tumor Mutation Burden and Their Clinical Significance in Bladder Cancer. Biosci. Rep. 2020, 40, BSR20194337. [Google Scholar] [CrossRef]
- Suda, K.; Tomizawa, K.; Mitsudomi, T. Biological and Clinical Significance of KRAS Mutations in Lung Cancer: An Oncogenic Driver That Contrasts with EGFR Mutation. Cancer Metastasis Rev. 2010, 29, 49–60. [Google Scholar] [CrossRef]
- Tomczak, K.; Czerwińska, P.; Wiznerowicz, M. Review The Cancer Genome Atlas (TCGA): An Immeasurable Source of Knowledge. Contemp. Oncol. 2015, 19, 68–77. [Google Scholar] [CrossRef] [PubMed]
- The UK10K Consortium; Writing Group; Walter, K.; Min, J.L.; Huang, J.; Crooks, L.; Memari, Y.; McCarthy, S.; Perry, J.R.B.; Xu, C.; et al. The UK10K Project Identifies Rare Variants in Health and Disease. Nature 2015, 526, 82–90. [Google Scholar] [CrossRef] [PubMed]
- Butterfield, L.H. Cancer Vaccines. BMJ 2015, 350, h988. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Trincado, J.L.; Gomez-Perosanz, M.; Reche, P.A. Fundamentals and methods for T- and B-cell epitope prediction. J. Immunol. Res. 2017, 2017, 2680160. [Google Scholar] [CrossRef] [PubMed]
- Moreira, A.L.; Eng, J. Personalized Therapy for Lung Cancer. Chest 2014, 146, 1649–1657. [Google Scholar] [CrossRef] [PubMed]
- Behjati, S.; Tarpey, P.S. What Is next Generation Sequencing? Arch. Dis. Child. Educ. Pract. Ed. 2013, 98, 236–238. [Google Scholar] [CrossRef] [PubMed]
- Blanc, E.; Holtgrewe, M.; Dhamodaran, A.; Messerschmidt, C.; Willimsky, G.; Blankenstein, T.; Beule, D. Identification and Ranking of Recurrent Neo-Epitopes in Cancer. BMC Med. Genom. 2019, 12, 171. [Google Scholar] [CrossRef] [PubMed]
- Tricco, A.C.; Lillie, E.; Zarin, W.; O’Brien, K.K.; Colquhoun, H.; Levac, D.; Moher, D.; Peters, M.D.; Horsley, T.; Weeks, L.; et al. PRISMA Extension for Scoping Reviews (PRISMAScR): Checklist and Explanation. Ann. Intern. Med. 2018, 169, 467–473. [Google Scholar] [CrossRef] [PubMed]
- Boegel, S.; Löwer, M.; Bukur, T.; Sahin, U.; Castle, J.C. A Catalog of HLA Type, HLA Expression, and Neo-Epitope Candidates in Human Cancer Cell Lines. Oncoimmunology 2014, 3, e954893. [Google Scholar] [CrossRef] [PubMed]
- Xiang, H.; Zhang, L.; Bu, F.; Guan, X.; Chen, L.; Zhang, H.; Zhao, Y.; Chen, H.; Zhang, W.; Li, Y.; et al. A Novel Proteogenomic Integration Strategy Expands the Breadth of Neo-Epitope Sources. Cancers 2022, 14, 3016. [Google Scholar] [CrossRef]
- de Mey, W.; De Schrijver, P.; Autaers, D.; Pfitzer, L.; Fant, B.; Locy, H.; Esprit, A.; Lybaert, L.; Bogaert, C.; Verdonck, M.; et al. A Synthetic DNA Template for Fast Manufacturing of Versatile Single Epitope MRNA. Mol. Ther. Nucleic Acids 2022, 29, 943–954. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Kim, S.; Hundal, J.; Herndon, J.; Li, S.; Petti, A.; Soysal, S.; Li, L.; McLellan, M.; Hoog, J.; et al. Breast Cancer Neoantigens Can Induce CD8+ T-Cell Responses and Antitumor Immunity. Cancer Immunol. Res. 2017, 5, 516–523. [Google Scholar] [CrossRef] [PubMed]
- Aparicio, B.; Repáraz, D.; Ruiz, M.; Llopiz, D.; Silva, L.; Vercher, E.; Theunissen, P.; Tamayo, I.; Smerdou, C.; Igea, A.; et al. Identification of HLA Class I-Restricted Immunogenic Neoantigens in Triple Negative Breast Cancer. Front. Immunol. 2022, 13, 985886. [Google Scholar] [CrossRef] [PubMed]
- Cafri, G.; Gartner, J.J.; Zaks, T.; Hopson, K.; Levin, N.; Paria, B.C.; Parkhurst, M.R.; Yossef, R.; Lowery, F.J.; Jafferji, M.S.; et al. MRNA Vaccine-Induced Neoantigen-Specific T Cell Immunity in Patients with Gastrointestinal Cancer. J. Clin. Investig. 2020, 130, 5976–5988. [Google Scholar] [CrossRef] [PubMed]
- Hartmaier, R.J.; Charo, J.; Fabrizio, D.; Goldberg, M.E.; Albacker, L.A.; Pao, W.; Chmielecki, J. Genomic Analysis of 63,220 Tumors Reveals Insights into Tumor Uniqueness and Targeted Cancer Immunotherapy Strategies. Genome Med. 2017, 9, 16. [Google Scholar] [CrossRef] [PubMed]
- Thakur, S.; Jain, M.; Zhang, C.; Major, C.; Bielamowicz, K.J.; Lacayo, N.J.; Vaske, O.; Lewis, V.; Murguia-Favela, L.; Narendran, A. Identification and in Vitro Validation of Neoantigens for Immune Activation against High-Risk Pediatric Leukemia Cells. Hum. Vaccin. Immunother. 2021, 17, 5558–5562. [Google Scholar] [CrossRef] [PubMed]
- Reimann, H.; Nguyen, A.; Sanborn, J.Z.; Vaske, C.J.; Benz, S.C.; Niazi, K.; Rabizadeh, S.; Spilman, P.; Mackensen, A.; Ruebner, M.; et al. Identification and Validation of Expressed HLA-Binding Breast Cancer Neoepitopes for Potential Use in Individualized Cancer Therapy. J. Immunother. Cancer 2021, 9, e002605. [Google Scholar] [CrossRef] [PubMed]
- Nonomura, C.; Otsuka, M.; Kondou, R.; Iizuka, A.; Miyata, H.; Ashizawa, T.; Sakura, N.; Yoshikawa, S.; Kiyohara, Y.; Ohshima, K.; et al. Identification of a Neoantigen Epitope in a Melanoma Patient with Good Response to Anti-PD-1 Antibody Therapy. Immunol. Lett. 2019, 208, 52–59. [Google Scholar] [CrossRef] [PubMed]
- James, C.A.; Ronning, P.; Cullinan, D.; Cotto, K.C.; Barnell, E.K.; Campbell, K.M.; Skidmore, Z.L.; Sanford, D.E.; Goedegebuure, S.P.; Gillanders, W.E.; et al. In Silico Epitope Prediction Analyses Highlight the Potential for Distracting Antigen Immunodominance with Allogeneic Cancer Vaccines. Cancer Res. Commun. 2021, 1, 115–126. [Google Scholar] [CrossRef]
- Martin, S.D.; Brown, S.D.; Wick, D.A.; Nielsen, J.S.; Kroeger, D.R.; Twumasi-Boateng, K.; Holt, R.A.; Nelson, B.H. Low Mutation Burden in Ovarian Cancer May Limit the Utility of Neoantigen-Targeted Vaccines. PLoS ONE 2016, 11, e0155189. [Google Scholar] [CrossRef]
- Sarivalasis, A.; Boudousquié, C.; Balint, K.; Stevenson, B.J.; Gannon, P.O.; Iancu, E.M.; Rossier, L.; Martin Lluesma, S.; Mathevet, P.; Sempoux, C.; et al. A Phase I/II Trial Comparing Autologous Dendritic Cell Vaccine Pulsed Either with Personalized Peptides (PEP-DC) or with Tumor Lysate (OC-DC) in Patients with Advanced High-Grade Ovarian Serous Carcinoma. J. Transl. Med. 2019, 17, 391. [Google Scholar] [CrossRef] [PubMed]
- Löffler, M.; Mohr, C.; Bichmann, L.; Freudenmann, L.; Walzer, M.; Schroeder, C.; Trautwein, N.; Hilke, F.; Zinser, R.; Mühlenbruch, L.; et al. Multi-Omics Discovery of Exome-Derived Neoantigens in Hepatocellular Carcinoma. Genome Med. 2019, 11, 28. [Google Scholar] [CrossRef] [PubMed]
- Marcu, A.; Schlosser, A.; Keupp, A.; Trautwein, N.; Johann, P.; Wölfl, M.; Lager, J.; Monoranu, C.M.; Walz, J.S.; Henkel, L.M.; et al. Natural and Cryptic Peptides Dominate the Immunopeptidome of Atypical Teratoid Rhabdoid Tumors. J. ImmunoTherapy Cancer 2021, 9, e003404. [Google Scholar] [CrossRef]
- Chen, F.; Zou, Z.; Du, J.; Su, S.; Shao, J.; Meng, F.; Yang, J.; Xu, Q.; Ding, N.; Yang, Y.; et al. Neoantigen Identification Strategies Enable Personalized Immunotherapy in Refractory Solid Tumors. J. Clin. Investig. 2019, 129, 2056–2070. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, S.; Noguchi, E.; Bando, H.; Miyadera, H.; Morii, W.; Nakamura, T.; Hara, H. Neoantigen Prediction in Human Breast Cancer Using RNA Sequencing Data. Cancer Sci. 2021, 112, 465–475. [Google Scholar] [CrossRef] [PubMed]
- Keskin, D.; Anandappa, A.; Sun, J.; Tirosh, I.; Mathewson, N.; Li, S.; Oliveira, G.; Giobbie-Hurder, A.; Felt, K.; Gjini, E.; et al. Neoantigen Vaccine Generates Intratumoral T Cell Responses in Phase Ib Glioblastoma Trial. Nature 2019, 565, 234–239. [Google Scholar] [CrossRef] [PubMed]
- Repáraz, D.; Ruiz, M.; Llopiz, D.; Silva, L.; Vercher, E.; Aparicio, B.; Egea, J.; Tamayo-Uria, I.; Hervás-Stubbs, S.; García-Balduz, J.; et al. Neoantigens as Potential Vaccines in Hepatocellular Carcinoma. J. Immunother. Cancer 2022, 10, e003978. [Google Scholar] [CrossRef] [PubMed]
- Löffler, M.W.; Chandran, P.A.; Laske, K.; Schroeder, C.; Bonzheim, I.; Walzer, M.; Hilke, F.J.; Trautwein, N.; Kowalewski, D.J.; Schuster, H.; et al. Personalized Peptide Vaccine-Induced Immune Response Associated with Long-Term Survival of a Metastatic Cholangiocarcinoma Patient. J. Hepatol. 2016, 65, 849–855. [Google Scholar] [CrossRef]
- Han, K.-C.; Park, D.; Ju, S.; Lee, Y.E.; Heo, S.-H.; Kim, Y.-A.; Lee, J.E.; Lee, Y.; Park, K.H.; Park, S.-H.; et al. Streamlined Selection of Cancer Antigens for Vaccine Development through Integrative Multi-Omics and High-Content Cell Imaging. Sci. Rep. 2020, 10, 5885. [Google Scholar] [CrossRef]
- Rajasagi, M.; Shukla, S.A.; Fritsch, E.F.; Keskin, D.B.; DeLuca, D.; Carmona, E.; Zhang, W.; Sougnez, C.; Cibulskis, K.; Sidney, J.; et al. Systematic Identification of Personal Tumor-Specific Neoantigens in Chronic Lymphocytic Leukemia. Blood 2014, 124, 453–462. [Google Scholar] [CrossRef]
- McCann, K.; von Witzleben, A.; Thomas, J.; Wang, C.; Wood, O.; Singh, D.; Boukas, K.; Bendjama, K.; Silvestre, N.; Nielsen, F.C.; et al. Targeting the Tumor Mutanome for Personalized Vaccination in a TMB Low Non-Small Cell Lung Cancer. J. Immunother. Cancer 2022, 10, e003821. [Google Scholar] [CrossRef]
- Conev, A.; Devaurs, D.; Rigo, M.M.; Antunes, D.A.; Kavraki, L.E. 3pHLA-Score Improves Structure-Based Peptide-HLA Binding Affinity Prediction. Sci. Rep. 2022, 12, 10749. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Wang, X.; Fei, C. A Highly Effective System for Predicting MHC-II Epitopes with Immunogenicity. Front. Oncol. 2022, 12, 888556. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Wang, Z.; Hu, H.; Wan, F.; Chen, L.; Xiong, Y.; Wang, X.; Zhao, D.; Huang, W.; Zeng, J. ACME: Pan-Specific Peptide-MHC Class i Binding Prediction through Attention-Based Deep Neural Networks. Bioinformatics 2019, 35, 4946–4954. [Google Scholar] [CrossRef] [PubMed]
- Mettu, R.; Charles, T.; Landry, S. CD4+T-Cell Epitope Prediction Using Antigen Processing Constraints. J. Immunol. Methods 2016, 432, 72–81. [Google Scholar] [CrossRef]
- Li, G.; Iyer, B.; Prasath, V.B.S.; Ni, Y.; Salomonis, N. DeepImmuno: Deep Learning-Empowered Prediction and Generation of Immunogenic Peptides for T Cell Immunity. Brief. Bioinform. 2021, 22, bbab160. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Zhao, L.; Wei, F.; Li, J. DeepNetBim: Deep Learning Model for Predicting HLA-Epitope Interactions Based on Network Analysis by Harnessing Binding and Immunogenicity Information. BMC Bioinform. 2021, 22, 231. [Google Scholar] [CrossRef] [PubMed]
- Xin, K.; Wei, X.; Shao, J.; Chen, F.; Liu, Q.; Liu, B. Establishment of a Novel Tumor Neoantigen Prediction Tool for Personalized Vaccine Design. Hum. Vaccin. Immunother. 2024, 20, 2300881. [Google Scholar] [CrossRef]
- Hao, Q.; Wei, P.; Shu, Y.; Zhang, Y.-G.; Xu, H.; Zhao, J.-N. Improvement of Neoantigen Identification Through Convolution Neural Network. Front. Immunol. 2021, 12, 682103. [Google Scholar] [CrossRef]
- Wang, G.; Wan, H.; Jian, X.; Li, Y.; Ouyang, J.; Tan, X.; Zhao, Y.; Lin, Y.; Xie, L. INeo-Epp: A Novel T-Cell HLA Class-I Immunogenicity or Neoantigenic Epitope Prediction Method Based on Sequence-Related Amino Acid Features. Biomed. Res. Int. 2020, 2020, 5798356. [Google Scholar] [CrossRef]
- Moise, L.; Gutierrez, A.; Kibria, F.; Martin, R.; Tassone, R.; Liu, R.; Terry, F.; Martin, B.; De Groot, A.S. IVAX: An Integrated Toolkit for the Selection and Optimization of Antigens and the Design of Epitope-Driven Vaccines. Hum. Vaccin. Immunother. 2015, 11, 2312–2321. [Google Scholar] [CrossRef] [PubMed]
- O’Donnell, T.J.; Rubinsteyn, A.; Bonsack, M.; Riemer, A.B.; Laserson, U.; Hammerbacher, J. MHCflurry: Open-Source Class I MHC Binding Affinity Prediction. Cell Syst. 2018, 7, 129–132.e4. [Google Scholar] [CrossRef] [PubMed]
- Phloyphisut, P.; Pornputtapong, N.; Sriswasdi, S.; Chuangsuwanich, E. MHCSeqNet: A Deep Neural Network Model for Universal MHC Binding Prediction. BMC Bioinform. 2019, 20, 270. [Google Scholar] [CrossRef] [PubMed]
- Declercq, A.; Bouwmeester, R.; Hirschler, A.; Carapito, C.; Degroeve, S.; Martens, L.; Gabriels, R. MS2Rescore: Data-Driven Rescoring Dramatically Boosts Immunopeptide Identification Rates. Mol. Cell. Proteom. 2022, 21, 100266. [Google Scholar] [CrossRef] [PubMed]
- Richard, G.; De Groot, A.S.; Steinberg, G.D.; Garcia, T.I.; Kacew, A.; Ardito, M.; Martin, W.D.; Berdugo, G.; Princiotta, M.F.; Balar, A.V.; et al. Multi-Step Screening of Neoantigens’ HLA- and TCR-Interfaces Improves Prediction of Survival. Sci. Rep. 2021, 11, 9983. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Kim, H.S.; Kim, E.; Lee, M.G.; Shin, E.-C.; Paik, S.; Kim, S. Neopepsee: Accurate Genome-Level Prediction of Neoantigens by Harnessing Sequence and Amino Acid Immunogenicity Information. Ann. Oncol. 2018, 29, 1030–1036. [Google Scholar] [CrossRef] [PubMed]
- Jurtz, V.; Paul, S.; Andreatta, M.; Marcatili, P.; Peters, B.; Nielsen, M. NetMHCpan-4.0: Improved Peptide-MHC Class I Interaction Predictions Integrating Eluted Ligand and Peptide Binding Affinity Data. J. Immunol. 2017, 199, 3360–3368. [Google Scholar] [CrossRef] [PubMed]
- Saxena, S.; Animesh, S.; Fullwood, M.J.; Mu, Y. Onionmhc: A Deep Learning Model for Peptide—Hla-A*02:01 Binding Predictions Using Both Structure and Sequence Feature Sets. J. Micromechanics Mol. Phys. 2020, 5, 2050009. [Google Scholar] [CrossRef]
- Pyke, R.M.; Mellacheruvu, D.; Dea, S.; Abbott, C.W.; Zhang, S.V.; Phillips, N.A.; Harris, J.; Bartha, G.; Desai, S.; McClory, R.; et al. Precision Neoantigen Discovery Using Large-Scale Immunopeptidomes and Composite Modeling of MHC Peptide Presentation. Mol. Cell Proteom. 2021, 20, 100111. [Google Scholar] [CrossRef]
- Schneidman-Duhovny, D.; Khuri, N.; Dong, G.Q.; Winter, M.B.; Shifrut, E.; Friedman, N.; Craik, C.S.; Pratt, K.P.; Paz, P.; Aswad, F.; et al. Predicting CD4+ T-Cell Epitopes Based on Antigen Cleavage, MHCII Presentation, and TCR Recognition. PLoS ONE 2018, 13, e0206654. [Google Scholar] [CrossRef]
- Chen, B.; Khodadoust, M.S.; Olsson, N.; Wagar, L.E.; Fast, E.; Liu, C.L.; Muftuoglu, Y.; Sworder, B.J.; Diehn, M.; Levy, R.; et al. Predicting HLA Class II Antigen Presentation through Integrated Deep Learning. Nat. Biotechnol. 2019, 37, 1332–1343. [Google Scholar] [CrossRef]
- Rao, A.A.; Madejska, A.A.; Pfeil, J.; Paten, B.; Salama, S.R.; Haussler, D. ProTECT—Prediction of T-Cell Epitopes for Cancer Therapy. Front. Immunol. 2020, 11, 483296. [Google Scholar] [CrossRef]
- Zhou, C.; Wei, Z.; Zhang, Z.; Zhang, B.; Zhu, C.; Chen, K.; Chuai, G.; Qu, S.; Xie, L.; Gao, Y.; et al. PTuneos: Prioritizing Tumor Neoantigens from next-Generation Sequencing Data. Genome Med. 2019, 11, 67. [Google Scholar] [CrossRef]
- Hundal, J.; Kiwala, S.; McMichael, J.; Miller, C.A.; Xia, H.; Wollam, A.T.; Liu, C.J.; Zhao, S.; Feng, Y.-Y.; Graubert, A.P.; et al. PVACtools: A Computational Toolkit to Identify and Visualize Cancer Neoantigens. Cancer Immunol. Res. 2020, 8, 409–420. [Google Scholar] [CrossRef]
- Sherafat, E.; Force, J.; Măndoiu, I.I. Semi-Supervised Learning for Somatic Variant Calling and Peptide Identification in Personalized Cancer Immunotherapy. BMC Bioinform. 2020, 21, 498. [Google Scholar] [CrossRef] [PubMed]
- Diao, K.; Chen, J.; Wu, T.; Wang, X.; Wang, G.; Sun, X.; Zhao, X.; Wu, C.; Wang, J.; Yao, H.; et al. Seq2Neo: A Comprehensive Pipeline for Cancer Neoantigen Immunogenicity Prediction. Int. J. Mol. Sci. 2022, 23, 11624. [Google Scholar] [CrossRef] [PubMed]
- Ye, Y.; Shen, Y.; Wang, J.; Li, D.; Zhu, Y.; Zhao, Z.; Pan, Y.; Wang, Y.; Liu, X.; Wan, J. SIGANEO: Similarity Network with GAN Enhancement for Immunogenic Neoepitope Prediction. Comput. Struct. Biotechnol. J. 2023, 21, 5538–5543. [Google Scholar] [CrossRef]
- Australian Pancreatic Cancer Genome Initiative; ICGC Breast Cancer Consortium; ICGC MMML-Seq Consortium; ICGC PedBrain; Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.J.R.; Behjati, S.; Biankin, A.V.; et al. Signatures of Mutational Processes in Human Cancer. Nature 2013, 500, 415–421. [Google Scholar] [CrossRef] [PubMed]
- Hilf, N.; Kuttruff-Coqui, S.; Frenzel, K.; Bukur, V.; Stevanović, S.; Gouttefangeas, C.; Platten, M.; Tabatabai, G.; Dutoit, V.; Van Der Burg, S.H.; et al. Actively Personalized Vaccination Trial for Newly Diagnosed Glioblastoma. Nature 2019, 565, 240–245. [Google Scholar] [CrossRef]
- Martínez-Pérez, E.; Molina-Vila, M.A.; Marino-Buslje, C. Panels and Models for Accurate Prediction of Tumor Mutation Burden in Tumor Samples. NPJ Precis. Oncol. 2021, 5, 31. [Google Scholar] [CrossRef]
- Blaeschke, F.; Paul, M.C.; Schuhmann, M.U.; Rabsteyn, A.; Schroeder, C.; Casadei, N.; Matthes, J.; Mohr, C.; Lotfi, R.; Wagner, B.; et al. Low Mutational Load in Pediatric Medulloblastoma Still Translates into Neoantigens as Targets for Specific T-Cell Immunotherapy. Cytotherapy 2019, 21, 973–986. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. World Health Statistics 2023: Monitoring Health for the SDGs, Sustainable Development Goals; World Health Organization: Geneva, Switzerland, 2023. [Google Scholar]
- Vivekanandhan, S.; Bahr, D.; Kothari, A.; Ashary, M.A.; Baksh, M.; Gabriel, E. Immunotherapies in rare cancers. Mol. Cancer 2023, 22, 23. [Google Scholar] [CrossRef] [PubMed]
- Koboldt, D.C. Best Practices for Variant Calling in Clinical Sequencing. Genome Med. 2020, 12, 91. [Google Scholar] [CrossRef] [PubMed]
- do Valle, Í.F.; Giampieri, E.; Simonetti, G.; Padella, A.; Manfrini, M.; Ferrari, A.; Castellani, G. Optimized pipeline of MuTect and GATK tools to improve the detection of somatic single nucleotide polymorphisms in whole-exome sequencing data. BMC Bioinform. 2016, 17, 27–35. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Yuan, Y.; Chen, X.; Chen, J.; Lin, S.; Li, X.; Du, H. Systematic Comparison of Somatic Variant Calling Performance among Different Sequencing Depth and Mutation Frequency. Sci. Rep. 2020, 10, 3501. [Google Scholar] [CrossRef] [PubMed]
- Ghandi, M.; Huang, F.W.; Jané-Valbuena, J.; Kryukov, G.V.; Lo, C.C.; McDonald, E.R.; Barretina, J., 3rd; Gelfand, E.T.; Bielski, C.M.; Li, H.; et al. Next-generation characterization of the Cancer Cell Line Encyclopedia. Nature 2019, 569, 503–508. [Google Scholar] [CrossRef] [PubMed]
- Tate, J.G.; Bamford, S.; Jubb, H.C.; Sondka, Z.; Beare, D.M.; Bindal, N.; Boutselakis, H.; Cole, C.G.; Creatore, C.; Dawson, E.; et al. COSMIC: The Catalogue Of Somatic Mutations In Cancer. Nucleic Acids Res. 2019, 47, D941–D947. [Google Scholar] [CrossRef]
- Sherry, S.T.; Ward, M.H.; Kholodov, M.; Baker, J.; Phan, L.; Smigielski, E.M.; Sirotkin, K. dbSNP: The NCBI database of genetic variation. Nucleic Acids Res. 2001, 29, 308–311. [Google Scholar] [CrossRef]
- Broeckx, B.J.G.; Peelman, L.; Saunders, J.H.; Deforce, D.; Clement, L. Using variant databases for variant prioritization and to detect erroneous genotype-phenotype associations. BMC Bioinform. 2017, 18, 535. [Google Scholar] [CrossRef]
- Mullaney, J.M.; Mills, R.E.; Pittard, W.S.; Devine, S.E. Small Insertions and Deletions (INDELs) in Human Genomes. Hum. Mol. Genet. 2010, 19, R131–R136. [Google Scholar] [CrossRef]
- Weber, J.L.; David, D.; Heil, J.; Fan, Y.; Zhao, C.; Marth, G. Human diallelic insertion/deletion polymorphisms. Am. J. Hum. Genet. 2002, 71, 854–862. [Google Scholar] [CrossRef]
- Cibulskis, K.; Lawrence, M.S.; Carter, S.L.; Sivachenko, A.; Jaffe, D.; Sougnez, C.; Gabriel, S.; Meyerson, M.; Lander, E.S.; Getz, G. Sensitive Detection of Somatic Point Mutations in Impure and Heterogeneous Cancer Samples. Nat. Biotechnol. 2013, 31, 213–219. [Google Scholar] [CrossRef] [PubMed]
- Trevarton, A.J.; Chang, J.T.; Symmans, W.F. Simple combination of multiple somatic variant callers to increase accuracy. Sci. Rep. 2023, 13, 8463. [Google Scholar] [CrossRef] [PubMed]
- Fennemann, F.L.; de Vries, I.J.M.; Figdor, C.G.; Verdoes, M. Attacking Tumors From All Sides: Personalized Multiplex Vaccines to Tackle Intratumor Heterogeneity. Front. Immunol. 2019, 10, 824. [Google Scholar] [CrossRef] [PubMed]
- Van Dijk, E.L.; Jaszczyszyn, Y.; Naquin, D.; Thermes, C. The Third Revolution in Sequencing Technology. Trends Genet. 2018, 34, 666–681. [Google Scholar] [CrossRef]
- Wang, Z.; Liu, X.; Yang, B.-Z.; Gelernter, J. The Role and Challenges of Exome Sequencing in Studies of Human Diseases. Front. Genet. 2013, 4, 160. [Google Scholar] [CrossRef]
- Ding, S.; Chen, X.; Shen, K. Single-cell RNA sequencing in breast cancer: Understanding tumor heterogeneity and paving roads to individualized therapy. Cancer Commun. 2020, 40, 329–344. [Google Scholar] [CrossRef]
- Robinson, J.; Barker, D.J.; Georgiou, X.; Cooper, M.A.; Flicek, P.; Marsh, S.G.E. IPD-IMGT/HLA Database. Nucleic Acids Res. 2020, 48, D948–D955. [Google Scholar] [CrossRef]
- Hurley, C.K.; Kempenich, J.; Wadsworth, K.; Sauter, J.; Hofmann, J.A.; Schefzyk, D.; Schmidt, A.H.; Galarza, P.; Cardozo, M.B.R.; Dudkiewicz, M.; et al. Common, Intermediate and Well-documented HLA Alleles in World Populations: CIWD Version 3.0.0. HLA 2020, 95, 516–531. [Google Scholar] [CrossRef]
- Gonzalez-Galarza, F.F.; McCabe, A.; Santos, E.J.M.D.; Jones, J.; Takeshita, L.; Ortega-Rivera, N.D.; Cid-Pavon, G.M.D.; Ramsbottom, K.; Ghattaoraya, G.; Alfirevic, A.; et al. Allele Frequency Net Database (AFND) 2020 Update: Gold-Standard Data Classification, Open Access Genotype Data and New Query Tools. Nucleic Acids Res. 2020, 48, D783–D788. [Google Scholar] [CrossRef]
- Roider, J.; Meissner, T.; Kraut, F.; Vollbrecht, T.; Stirner, R.; Bogner, J.R.; Draenert, R. Comparison of Experimental Fine-mapping to in Silico Prediction Results of HIV-1 Epitopes Reveals Ongoing Need for Mapping Experiments. Immunology 2014, 143, 193–201. [Google Scholar] [CrossRef] [PubMed]
- Momburg, F.; Roelse, J.; Hämmerling, G.J.; Neefjes, J.J. Peptide size selection by the major histocompatibility complex-encoded peptide transporter. J. Exp. Med. 1994, 179, 1613–1623. [Google Scholar] [CrossRef]
- Sahin, U.; Derhovanessian, E.; Miller, M.; Kloke, B.-P.; Simon, P.; Löwer, M.; Bukur, V.; Tadmor, A.D.; Luxemburger, U.; Schrörs, B.; et al. Personalized RNA Mutanome Vaccines Mobilize Poly-Specific Therapeutic Immunity against Cancer. Nature 2017, 547, 222–226. [Google Scholar] [CrossRef]
- Falk, K.; Rötzschke, O.; Stevanovié, S.; Jung, G.; Rammensee, H.-G. Allele-Specific Motifs Revealed by Sequencing of Self-Peptides Eluted from MHC Molecules. Nature 1991, 351, 290–296. [Google Scholar] [CrossRef] [PubMed]
- Lundegaard, C.; Lund, O.; Nielsen, M. Prediction of Epitopes Using Neural Network Based Methods. J. Immunol. Methods 2011, 374, 26–34. [Google Scholar] [CrossRef] [PubMed]
- Luo, H.; Ye, H.; Ng, H.W.; Shi, L.; Tong, W.; Mendrick, D.L.; Hong, H. Machine Learning Methods for Predicting HLA-Peptide Binding Activity. Bioinform. Biol. Insights 2015, 9 (Suppl. 3), 21–29. [Google Scholar] [CrossRef]
- Jesdale, B.M.; Deocampo, G.; Meisell, J.; Beall, J.; Marinello, M.J.; Chicz, R.M.; De Groot, A.S. Matrix-based prediction of MHC-binding peptides: The EpiMatrix algorithm, reagent for HIV research. Vaccines 1997, 57–64. [Google Scholar]
- Chen, T.; Guestrin, C. XGBoost: A Scalable Tree Boosting System. In Proceedings of the 22nd ACM SIGKDD International Conference on Knowledge Discovery and Data Mining (KDD ‘16), San Francisco, CA, USA, 13–17 August 2016; Association for Computing Machinery: New York, NY, USA, 2016; pp. 785–794. [Google Scholar] [CrossRef]
- Ghansah, B.; Wu, S.; Ghansah, N. Rankboost-based result merging. In Proceedings of the International Conference on Computer and Information Technology; Ubiquitous Computing and Communications; Dependable, Autonomic and Secure Computing; Pervasive Intelligence and Computing, Liverpool, UK, 26–28 October 2015; pp. 907–914. [Google Scholar]
- Song, D.; Chen, J.; Chen, G.; Li, N.; Li, J.; Fan, J.; Li, S.C. Parameterized BLOSUM matrices for protein alignment. Trans. Comput. Biol. Bioinform. 2014, 12, 686–694. [Google Scholar] [CrossRef] [PubMed]
- Rigatti, S.J. Random Forest. J. Insur. Med. 2017, 47, 31–39. [Google Scholar] [CrossRef]
- Hesterberg, T. Bootstrap. Wiley Interdiscip. Rev. Comput. Stat. 2011, 3, 497–526. [Google Scholar] [CrossRef]
- Freitas, P.; Silva, F.; Sousa, J.V.; Ferreira, R.M.; Figueiredo, C.; Pereira, T.; Oliveira, H.P. Machine learning-based approaches for cancer prediction using microbiome data. Sci. Rep. 2023, 13, 11821. [Google Scholar] [CrossRef] [PubMed]
- Bassani-Sternberg, M.; Pletscher-Frankild, S.; Jensen, L.J.; Mann, M. Mass Spectrometry of Human Leukocyte Antigen Class I Peptidomes Reveals Strong Effects of Protein Abundance and Turnover on Antigen Presentation. Mol. Cell. Proteom. 2015, 14, 658–673. [Google Scholar] [CrossRef] [PubMed]
- Blum, J.S.; Wearsch, P.A.; Cresswell, P. Pathways of Antigen Processing. Annu. Rev. Immunol. 2013, 31, 443–473. [Google Scholar] [CrossRef] [PubMed]
- Alspach, E.; Lussier, D.M.; Miceli, A.P.; Kizhvatov, I.; DuPage, M.; Luoma, A.M.; Meng, W.; Lichti, C.F.; Esaulova, E.; Vomund, A.N.; et al. MHC-II Neoantigens Shape Tumour Immunity and Response to Immunotherapy. Nature 2019, 574, 696–701. [Google Scholar] [CrossRef] [PubMed]
- Reynisson, B.; Alvarez, B.; Paul, S.; Peters, B.; Nielsen, M. NetMHCpan-4.1 and NetMHCIIpan-4.0: Improved Predictions of MHC Antigen Presentation by Concurrent Motif Deconvolution and Integration of MS MHC Eluted Ligand Data. Nucleic Acids Res. 2020, 48, W449–W454. [Google Scholar] [CrossRef] [PubMed]
- Fritsch, E.F.; Rajasagi, M.; Ott, P.A.; Brusic, V.; Hacohen, N.; Wu, C.J. HLA-binding properties of tumor neoepitopes in humans. Cancer Immunol. Res. 2014, 2, 522–529. [Google Scholar] [CrossRef]
- Bulik-Sullivan, B.; Busby, J.; Palmer, C.D.; Davis, M.J.; Murphy, T.; Clark, A.; Busby, M.; Duke, F.; Yang, A.; Young, L.; et al. Deep learning using tumor HLA peptide mass spectrometry datasets improves neoantigen identification. Nat. Biotechnol. 2018, 37, 55–63. [Google Scholar] [CrossRef]
- O’Donnell, T.J.; Rubinsteyn, A.; Laserson, U. MHCflurry 2.0: Improved Pan-Allele Prediction of MHC Class I-Presented Peptides by Incorporating Antigen Processing. Cell Syst. 2020, 11, 42–48.e7. [Google Scholar] [CrossRef]
- Vitiello, A.; Zanetti, M. Neoantigen prediction and the need for validation. Nat. Biotechnol. 2017, 35, 815–817. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ref. | NGS Dataset Used | Type of Tumor | Variant Calling Program/DATABASE | Neoepitope Prediction Programs | MHC Class Used for Predictions | Prediction Selection Scores | Immunological Characteristics Evaluated | In Vivo Validation | In Vitro Validation | In Silico Validation | Clinical Trial | Positive Aspects | Limitations |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| [19] | NGS RNA-seq of 177 lineages | Melanoma, squamous cell carcinoma, lung carcinoma, gastric adenocarcinoma, Burkitt’s lymphoma, breast carcinoma, colorectal adenocarcinoma, ovarian carcinoma, B-cell lymphocytic leukemia, colorectal carcinoma, cervical adenocarcinoma, liver carcinoma, chronic myeloid leukemia, gastric adenocarcinoma, prostate carcinoma, neuroblastoma, glioblastoma, osteosarcoma | CCLE e COSMIC | NetMHCpan | MHC-I | ic50 ≤ 500 nM | Affinity to MHC-I expression | - | - | HLA confirmation compared to other methods | - | Allows for the selection of cell lines based on HLA expression | The literature: limited immunological characterization |
| [20] | NGS (WES) | Colorectal carcinoma | Mutect2: snp | NetMHCpan pNovo3 | MHC-I | ic50 ≤ 500 nM | MHC-I affinity cleavage processing | - | - | - | - | It is also possible to identify non-linear epitopes and those arising from changes in other stages of antigen processing | The new peptides have not yet been validated—other mechanisms of peptide alteration are not well understood |
| [21] | NGS (WGS RNA-seq) | Melanoma | myNEO’s (snp and indel) Immuno Engine | MHCflurry neoIM (T cell activation) | MHC-I | ic50 ≤ 500 nM | MHC-I binding expression T cell activation | - | IFN-γ detection | - | - | Presented a cheaper and faster method of constructing mRNA | T cell prediction and activation steps are not described |
| [22] | NGS (WES) | Breast carcinoma | SAMtools Somatic Sniper VarScan Somatic Strelka (snp) | NetMHC pVACseq | MHC-I | ic50 ≤ 500 nM | MHC-I binding variant frequency expression | Measurement of tumor size and detection of interferon-gamma and TNF-alpha | Activation of CD8+ T cells (detection of interferon-gamma and TNF-alpha) | - | - | It was possible to identify some antigenic neoepitopes for each patient | Does not evaluate other biological characteristics for prediction resulting in a high false negative rate |
| [23] | NGS (WES e RNA-seq) | Breast carcinoma | Mutect Varscan Somatic Sniper Strelka | NetMHCpan | MHC-I | ic50 ≤ 500 nM | Binding to MHC-I expression | Activation of CD8+ T cells (IFN-γ detection) | - | - | - | These patients harbor a sufficient number of immunogenic neoAgs suitable for vaccine development setting the basis for future combinatorial therapies containing vaccines | Only one allele and mutation type were considered |
| [24] | NGS (WES e RNA-seq) | Gastric adenocarcinoma | Strelka Somatic Sniper Varscan2 Mutect (snp and indel) | NetMHCpan, NetMHCpan II | MHC-I MHC-II | ic50 ≤ 500 nM | Binding to MHC-I expression | Activation of CD8+ T cells (IFN-γ detection | Activation of CD8+ T cells (IFN-γ detection | - | mRNA vaccine encoding defined neoantigen mutations in driver genes and HLA-I–predicted epitopes in patients with metastatic gastrointestinal cancer | mRNA neoantigen vaccine could also possibly be used to improve adoptive T cell therapy with neoantigen-specific cells by restimulating T cells in vivo | Lack of tumor shrinkage in the present pilot trial |
| [25] | NGS | Lung carcinoma, breast carcinoma, colorectal carcinoma, glioma, ovarian carcinoma, pancreatic carcinoma, gastric adenocarcinoma, melanoma, liver carcinoma | Filtered germlines from dbSNP; COSMIC | NetMHCpan | MHC-I | ic50 ≤ 500 nM | Proteasome TAP binding to MHC-I and II | - | - | - | - | It is possible to find neoepitopes in common between specific groups of patients | - Some regions of the exome were left out of the analyses; - Only a small portion of the population can benefit from the technique when searching for universal neoantigens |
| [26] | NGS (WES RNA-seq) | B-cell lymphocytic leukemia | Strelka 2 (snp and indel) | NetMHCpan NetChop | MHC-I | ic50 ≤ 500 nM | MHC-I binding expression transport | - | IFN-γ detection | - | - | Allowed for the identification of disease-specific neoepitopes | Exclusion of many variables associated with the immune response |
| [27] | NGS (WGS e RNA-seq) | Lung carcinoma | TCGA (snp and indel) | NetMHC | MHC-I | ic50 ≤ 500 nM | Binding to MHC-I expression | - | Peptide expression; confirmation of T cell reactivity | - | - | A new algorithm that evaluates the possible factors that characterize the immunogenicity of a neoepitope | Does not perform a specific variant calling for the samples used |
| [28] | NGS (WES) | Melanoma | Germline filtered with dbSNP; (snp) | NetMHC | MHC-I | ic50 ≤ 500 nM | Binding to MHC-I expression | - | Activation of CD8+ T cells (interferon- gamma detection) | Docking | - | A single-cell RNA sequencing platform combined with microfluidics and NGS technology will allow every researcher to analyze biomarkers in terms of their genetic phenotypic and even functional properties | Validation for just one HLA allele |
| [29] | NGS | Melanoma, prostate carcinoma, pancreatic carcinoma, lung carcinoma, colorectal carcinoma, neuroblastoma, glioblastoma, renal carcinoma, myeloblastoma, mesothelioma | Mutect2: snp | pVACtools | MHC-I | ic50 ≤ 500 nM | Binding to MHC-I | - | - | - | - | Confirmation that there is great difficulty in constructing allogeneic vaccines for cancer | - Analysis is limited to a few epitope sizes; - There is no analysis of other biological factors for prioritizing neoepitopes |
| [30] | NGS (WGS e RNA-seq) | Ovarian carcinoma, lung carcinoma | Samtools (snp and indel) | NetMHCpan | MHC-I | ic50 ≤ 500 nM | Binding to MHC-I | There was activation of T CD8+ and T CD4+, but there was no tumor reduction | Activation of CD8+ T cells (detection of interferon-gamma and TNF-alpha) | - | - | Highlights of the limitation involved in the formulation of neoepitope vaccines in tumors with low mutational load | - Analysis is limited to a few epitope sizes; - There is no analysis of other biological factors for prioritizing neoepitopes |
| [31] | NGS | Ovarian carcinoma | GATK Haplotype Caller; MuTect; VarScan (snp and indel) | MixMHCpred MixMHC2pred | MHC-I MHC-II | ic50 ≤ 500 nM | Binding to MHC-I and II expression | - | IFN-γ detection | - | PEP-DC | NeoAgs are more likely to elicit strong T cell responses because T cell tolerance does not hamper their immunogenicity and consequently epitope spreading and a broad anti-tumor immune response | The process of sequencing immunopeptidomics analysis peptide manufacturing and good manufacturing practices for the manufacturing of NeoAg vaccines is long and expensive although costs may decrease as a result of technological improvements |
| [32] | NGS (WES e RNA-seq) | Liver carcinoma | Strelka 2 (snps and indels) | SYFPEITHI netMHC netMHCpan | MHC-I | ic50 ≤ 500 nM | Binding to MHC-I expression | - | Activation of CD8+ T cells (IFN-γ detection) | Identification of neoepitopes identified in TCGA | - | Evidence of results obtained depending on the type of tumor and mutational load | Validation is limited to what exists in the literature, hepatocellular carcinoma genomic data are limited |
| [33] | NGS (WGS) | Atypical teratoid rhabdoid tumor | Samtools Platypus | VaxiJen NetMHC | MHC-I | ic50 ≤ 500 nM | Binding to MHC-I, 3 physicochemical characteristics | - | Activation of CD8+ T cells (detection of IFN-γ and TNF-alpha) | - | - | It is possible to find immunogenic neoepitopes even in tumors with low mutational load | - Analyses carried out on low-quality samples - Lack of data on the type of tumor studied |
| [34] | NGS | Embryonal, liver carcinoma, colorectal carcinoma, gastric adenocarcinoma, endometrial, pancreatic carcinoma, ovarian carcinoma, glioma | GATK VarScan2 (snp and indel) FACTERA (fusion) | NetMHC NetMHCpan IEDB e NetMHCII | MHC-I MHC-II | ic50 ≤ 500 nM | Binding to MHC-I and MHC-II | - | Activation of CD8+ T cells (IFN-γ detection) | - | T cells activated for the A*11:01 allele | The neoepitopes selected for immunotherapy generated tumor regression | Difficulty controlling heterogeneous tumor |
| [35] | NGS (WES RNA-seq) | Breast carcinoma | Mutect 2 (WGS—WES) VarScan2 (WES—RNA-seq) | pVACtools | MHC-I | ic50 ≤ 500 nM | Binding to MHC-I expression | - | - | Intersection of 3 variant calling methods | - | The combination method has potential clinical applications, including vaccine target detection and prediction of therapeutic effects of immune checkpoint inhibitors | No experimental validation |
| [36] | NGS (WES RNA-seq) | Glioblastoma | Mutect 2 (snp) Indelocator e Strelka (indel) | NetMHCpan | MHC-I | ic50 ≤ 500 nM | Binding to MHC-I expression physicochemical characteristics | - | Activation of CD8+ T cells (detection of IFN-γ and TNF-alpha) | - | Testing for MHC-I allele epitopes included in MHC-II peptides | It was possible to identify epitopes and immune response even though it was a tumor with a low mutational load | - Clinical tests with patients receiving dexamethasone may have affected the T cell response against neoepitope - Few immunological characteristics evaluated |
| [37] | NGS (WES RNA-seq) | Liver carcinoma | Mutect2 (snp) | NetMHCpan NetMHCpan II | MHC-I MHC-II | ic50 ≤ 500 nM | Binding to MHC-I and II cleavage expression | Activation of CD8+ T cells (detection of interferon-gamma and TNF-alpha) | - | Comparison of mutated and wild-type neoepitopes | - | Generated responses in MHC-I and MHC-II | Test performed on only 2 alleles |
| [38] | NGS (WES RNA-seq) | Cholangiocarcinoma | Strelka (snp) | SYFPEITHI NetMHC | MHC-I | ic50 ≤ 500 nM | Binding to MHC-I expression | - | Activation of CD8+ T and CD4+ T cells (detection of IFN-γ and TNF-alpha) | - | 7 peptides tested 3 generated an immune response and showed no evidence of tumor recurrence | The immune response was efficient and prevented the emergence of new tumors during the period | None of the epitopes were validated by mass spectrometry |
| [39] | NGS (WES RNA-seq) | Lung carcinoma | GATK | NetMHC | MHC-I | ic50 ≤ 500 nM | Binding to MHC-I expression | - | IFN-γ detection high-throughput imaging | - | - | High-throughput imaging allows for the detection of expressed neoepitopes even at low concentrations; the combined method is highly selective | Few variables were explored |
| [40] | NGS (WES) | Chronic lymphocytic leukemia | Mutect | NetMHCpan | MHC-I | ic50 ≤ 500 nM | Binding to MHC-I expression | - | MHC binding IFN-γ detection | - | Combined treatment with transplant for 2 patients and vaccine led to tumor remission | Neoepitopes generated response and tumor remission | The number of identified neoepitopes is underestimated due to the cell therapy applied and the lack of sensitivity of the clonal expansion method |
| [41] | NGS (WES RNA-seq) | Lung carcinoma | TCGA (snp and indel) | NetMHCpan NetMHCIIpan | MHC-I MHC-II | ic50 ≤ 500 nM | Binding to MHC-I expression | - | IFN-γ detection | - | MyVac vaccine in preclinical testing against 18 mutated genes generated a response detected by IFN-γ ELISpot | The in silico approach allowed for the formulation of a vaccine even in a type of tumor with low mutational load | A relatively simple approach may be difficult in rarer alleles |
| Ref. | Program Training | Variant Calling Data | MHC-I Neoepitope Size/Alleles | MHC-II Neoepitope Size/Allele | Algorithm/Matrix for Neoepitope Prediction and Prioritization | Training Dataset for Neoepitope Prediction | Neoepitope Prioritization Criteria | Presented Performance |
|---|---|---|---|---|---|---|---|---|
| [42] | 3pHLA | None | 9–10 AA, 28 alleles | None | CART algorithm, random forest | Tumor microbiome | Structural analysis | Increase from 0.82 to 0.99 |
| [43] | FIONA | None | None | 9–25 AA | Convolutional neural networks | Tumor microbiome | None | AUC: 0.94 |
| [44] | ACME | None | 9–11 AA | None | Neural networks, BLOSUM50 | Tumor microbiome | Transport/cleavage; T cell activation | AUC: 0.9 (HLA-A), 0.88 (HLA-B) |
| [45] | (Unnamed) | None | None | 15–20 AA | NetMHCII, z-score (stability) | Tumor microbiome | None | AUC: 0.76 |
| [46] | DeepImmuno | None | 9–10 AA | None | Convolutional neural networks | Tumor microbiome | None | AUC: 0.85 |
| [47] | DeepNetBim | None | 9 AA, 104 alleles | None | Convolutional neural networks | Tumor microbiome | None | AUC: 0.93 |
| [48] | NUCC | None | 8–11 AA | None | Generalized linear model (GLM), random forest (RF), extreme gradient Boosting (Xgboost), gradient learning machine (GBM), fully connected neural network (FCNN) | Tumor microbiome | Binding affinity score, binding stability, and probability of presentation | AUC: 0.85 with XGboot and GMB |
| [49] | APPM | None | 8–11 AA | None | Convolutional neural networks | Tumor microbiome (larger proportion of mass spectrometry data) | None | PPV: 0.4 |
| [50] | INeo-Epp | None | 8–11 AA (does not predict neoepitopes) | None | Random forest, Boruta | Tumor microbiome | Physicochemical features | AUC: 0.78 |
| [51] | Ivax | None | 9 AA/6 alleles | 8 alleles/15–30 AA | Epitope (EpiMatrix, ClustiMer), homology (Conservatrix, JanusMatrix), HLA (EpiAssembler), epitope concatenation (VaccineCAD) | Microbiome | Transport/cleavage; T cell activation; homology | None |
| [52] | MHCflurry | None | 8–15 AA | None | neural networks | Tumor microbiome/mass spectrometry | None | AUC: 0.8 |
| [53] | MHCSeqNet | None | 9–11 AA | None | neural networks | Tumor microbiome | Transport/cleavage | AUC: 0.99 (common alleles), AUC: 0.79 (indistinct alleles) |
| [54] | MS2Rescore | None | None | None | XGBoost | Tumor microbiome | None | PCC: 0.94 |
| [55] | Ancer | None | 9–10 AA | 15–30 AA | EpiMatrix (neoepitope prediction), JanusMatrix (homology) | Tumor microbiome | Homology | AUC: 0.65 |
| [56] | Neopepsee | Snp/indel | None | None | NetCTL (nnalign), Gaussian naive Bayes (GNB), locally weighted naive Bayes (LNB), random forest (RF), and support vector machine (SVM) | Tumor microbiome | Transport/cleavage; hydrophobicity, polarity, and charge at positions 2, 3, 5, and 6; molecular size, entropy, differential antigenic index (DAI), and amino acid pairwise contact potentials (AAPPs); gene expression; homology | AUC: >0.9 |
| [57] | NetMHCpan | None | 8–11 AA | None | Neural networks (nnalign) | Tumor microbiome/mass spectrometry | None | AUC: 0.83—0.97 |
| [58] | OnionMHC | None | 9 AA/1 alleles | None | Convolutional neural networks, BLOSUM62 | Tumor microbiome | Modeling | AUC: 0.83 |
| [59] | SHERPA | None | 8–11 AA, 167 alleles | None | BLOSUM62 | Tumor | T cell activation; gene expression | 1.44-fold more accurate |
| [60] | ITCell | None | None | None | Neural networks | Microbiome | Transport/cleavage; modeling | None |
| [61] | MARIA | None | None | 8–26 AA | Neural networks | Tumor microbiome | Gene expression | AUC: 0.87 |
| [62] | ProTECT | Snp/indel/fusion | 9–10 AA | 15 AA | NetMHC * | Tumor microbiome | RankBoost; homology | None |
| [63] | PTuneos | Snp/indel | 9–11 AA | None | NetMHCpan (neural networks), random forest | Microbiome | T cell activation; gene expression; homology | AUC: 0.65, AUC: 0.83 |
| [64] | PVACtools | Snp/indel/fusion | None | None | NetMHCpan, NetMHC, NetMHCcons, PickPocket, SMM, SMMPMBEC, MHCflurry, MHCnuggets, NetMHCIIpan, SMMalign, NNalign, MHCnuggets | Tumor microbiome | Transport/cleavage; gene expression; homology | None |
| [65] | PLATO | Snp/indel | None | None | Bootstrap, random forest | Tumor microbiome/mass spectrometry | Transport/cleavage; F1 score; gene expression | AUC: 0.71–0.86 |
| [66] | Seq2Neo | Snp/indel/fusion | 8–11 AA | None | Convolutional neural networks | Tumor microbiome | Transport/cleavage; T cell activation; gene expression | AUC: 0.8 |
| [67] | SIGANEO | None | 8–11 AA | None | NetMHCpan, NetMHCstabpan, generative adversarial network | Tumor | Gene expression; stability; homology | AUROC: 0.94; AUPRC: 0.338 |
| Program | Advantages | Disadvantages |
|---|---|---|
| 3pHLA (https://github.com/KavrakiLab/3pHLA-score (accessed on 10 July 2024)) |
|
|
| ACME (https://github.com/HYsxe/ACME (accessed on 10 July 2024)) |
|
|
| Ancer (version not provided) |
|
|
| APPM (https://github.com/haoqing12/APPM.git (accessed on 10 July 2024)) |
|
|
| DeepImmuno (Version 1.0) |
|
|
| DeepNetBim (https://github.com/Li-Lab-Proteomics/DeepNetBim (accessed on 10 July 2024)) |
|
|
| FIONA (http://therarna.cn/fiona.html (accessed on 10 July 2024)) |
|
|
| INeo-Epp (http://www.biostatistics.online/ineo-epp/neoantigen.php (accessed on 10 July 2024)) |
|
|
| ITCell (http://salilab.org/itcell (accessed on 10 July 2024)) |
|
|
| Ivax (version not provided) |
|
|
| MARIA (https://maria.stanford.edu/ (accessed on 10 July 2024)) |
|
|
| MHCflurry (version 1.2.0) |
|
|
| MHCSeqNet https://github.com/cmbcu/MHCSeqNet (accessed on 10 July 2024)) |
|
|
| MixMHCpred (version 2.0.2) |
|
|
| MixMHC2pred (version 1.1) |
|
|
| MS2Rescore (version 2.1.2) |
|
|
| Neopepsee (http://sourceforge.net/projects/neopepsee/ (accessed on 10 July 2024)) |
|
|
| NetMHC (version 3.0 or higher) |
|
|
| NetMHCII (version 2.3) |
|
|
| NetMHCIIpan (version 4.3) |
|
|
| NetMHCpan (version 4.1) |
|
|
| NUCC (https://github.com/roubaokai/NUCC.git (accessed on 10 July 2024)) |
|
|
| OnionMHC (https://github.com/shikhar249/OnionMHC (accessed on 10 July 2024)) |
|
|
| PLATO (version not provided) |
|
|
| ProTECT (version 2.5.0) |
|
|
| PTuneos (version 1.0.2) |
|
|
| PVACtools (http://pvactools.org/ (accessed on 10 July 2024)) |
|
|
| Seq2Neo (prior to 2.1) |
|
|
| SHERPA (version not provided) |
|
|
| SIGANEO (https://github.com/NCTool/SIGANEO-GAN (accessed on 10 July 2024)) |
|
|
| SYFPEITHI (version 1.0) |
|
|
| Algorithm | Advantages | Disadvantages |
|---|---|---|
| Artificial Neural Network |
|
|
| Convolutional Neural Network |
|
|
| Random Forest |
|
|
| EpiMatrix |
|
|
| JanusMatrix |
|
|
| XGBoost |
|
|
| CART Algorithm |
|
|
| BLOSUM |
|
|
| Boruta |
|
|
| ClustiMer |
|
|
| Conservatrix |
|
|
| EpiAssembler |
|
|
| VaccineCAD |
|
|
| Gaussian Naive Bayes |
|
|
| Locally Weighted Naive Bayes |
|
|
| Support Vector Machine |
|
|
| Bootstrap |
|
|
| Generalized Linear Model |
|
|
| Gradient Learning Machine |
|
|
| Fully Connected Neural Network |
|
|
| Generative Adversarial Network |
|
|
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rocha, L.G.d.N.; Guimarães, P.A.S.; Carvalho, M.G.R.; Ruiz, J.C. Tumor Neoepitope-Based Vaccines: A Scoping Review on Current Predictive Computational Strategies. Vaccines 2024, 12, 836. https://doi.org/10.3390/vaccines12080836
Rocha LGdN, Guimarães PAS, Carvalho MGR, Ruiz JC. Tumor Neoepitope-Based Vaccines: A Scoping Review on Current Predictive Computational Strategies. Vaccines. 2024; 12(8):836. https://doi.org/10.3390/vaccines12080836
Chicago/Turabian StyleRocha, Luiz Gustavo do Nascimento, Paul Anderson Souza Guimarães, Maria Gabriela Reis Carvalho, and Jeronimo Conceição Ruiz. 2024. "Tumor Neoepitope-Based Vaccines: A Scoping Review on Current Predictive Computational Strategies" Vaccines 12, no. 8: 836. https://doi.org/10.3390/vaccines12080836
APA StyleRocha, L. G. d. N., Guimarães, P. A. S., Carvalho, M. G. R., & Ruiz, J. C. (2024). Tumor Neoepitope-Based Vaccines: A Scoping Review on Current Predictive Computational Strategies. Vaccines, 12(8), 836. https://doi.org/10.3390/vaccines12080836