
Changes in Phenotypic and Molecular Features of Naïve and Central Memory T Helper Cell Subsets following SARS-CoV-2 Vaccination

 , , , , , ,
, , , , , ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Vaccinated Donor Samples and Measurements
2.2. Sample Collection and Processing for PBMC Isolation
2.3. CyTOF for Phenotyping
2.4. Cell Sorting of T Cell Populations
2.5. RNA Isolation and Sequencing
2.6. DNA Isolation and ATAC-Sequencing
2.7. TCR Repertoire Sequencing
3. Analysis
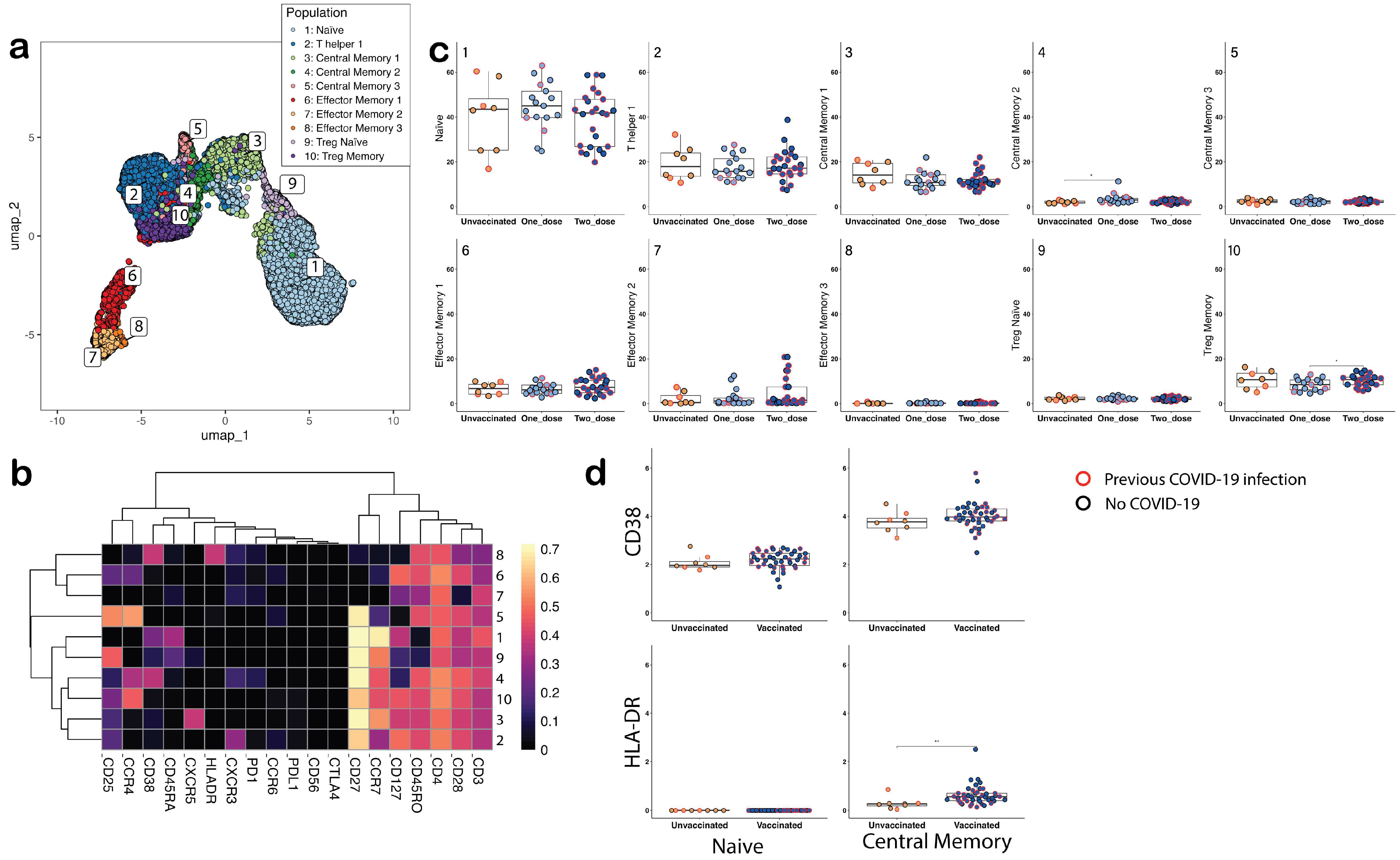
3.1. CyTOF Analysis
3.2. RNA-Sequencing Analysis
3.3. ATACseq Analysis
3.4. TCR Repertoire Analysis
4. Results
4.1. Study Cohort
4.2. Comparisons of Phenotypic Changes in CD4+ T Cell Subsets between Groups
4.3. Comparisons of Molecular Features in Naïve and Central Memory T Helper Cell Subsets
4.4. Within-Group Comparisons
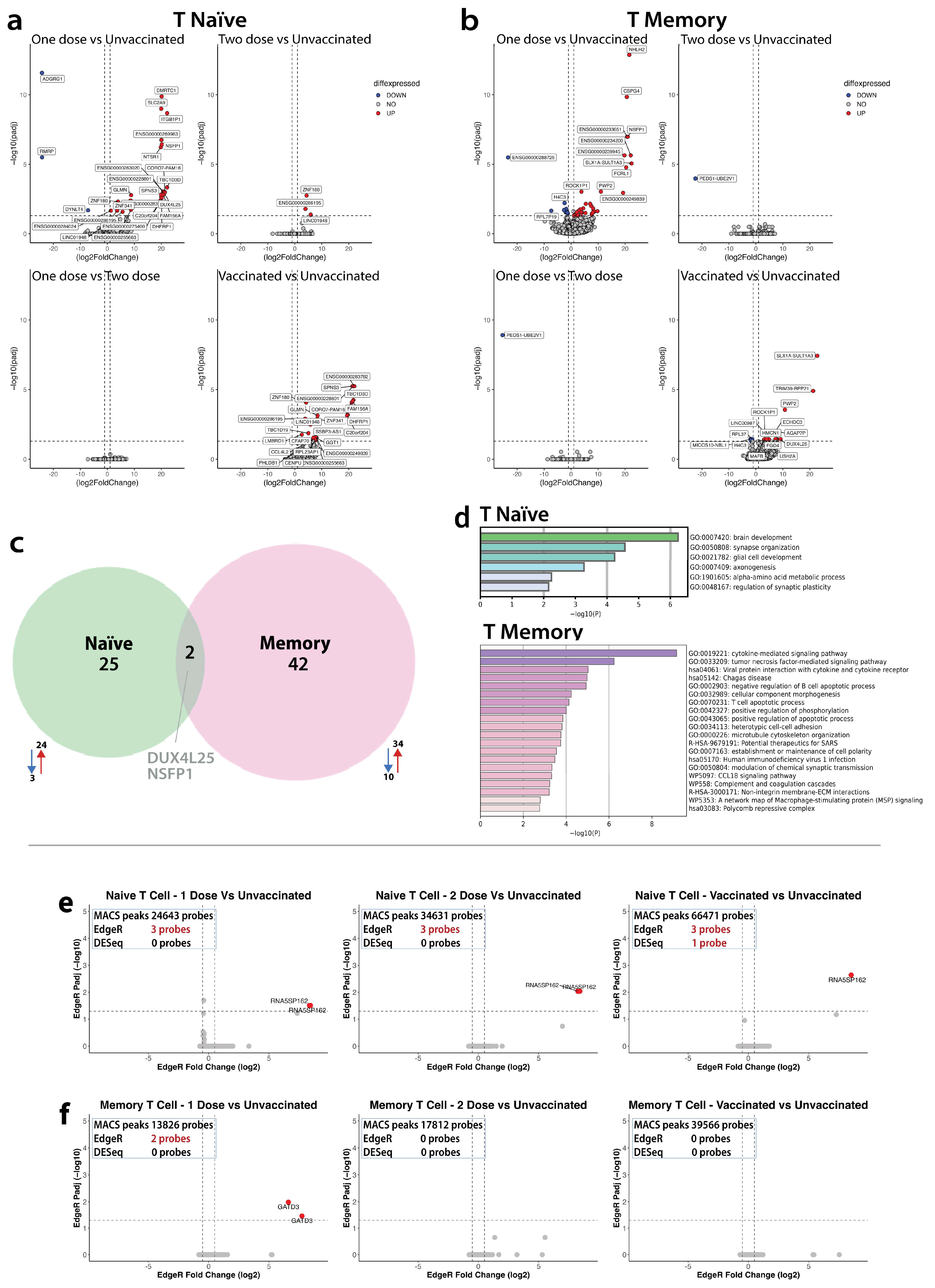
4.4.1. Transcriptome
4.4.2. Epigenome
4.5. Between Group Comparisons
4.5.1. Between Group Comparisons of Molecular Mechanisms in CD4+Naïve Subset
Transcriptome
Epigenome
4.5.2. Between Group Comparisons of Molecular Mechanisms in CD4+CM Subset
Transcriptome
Epigenome
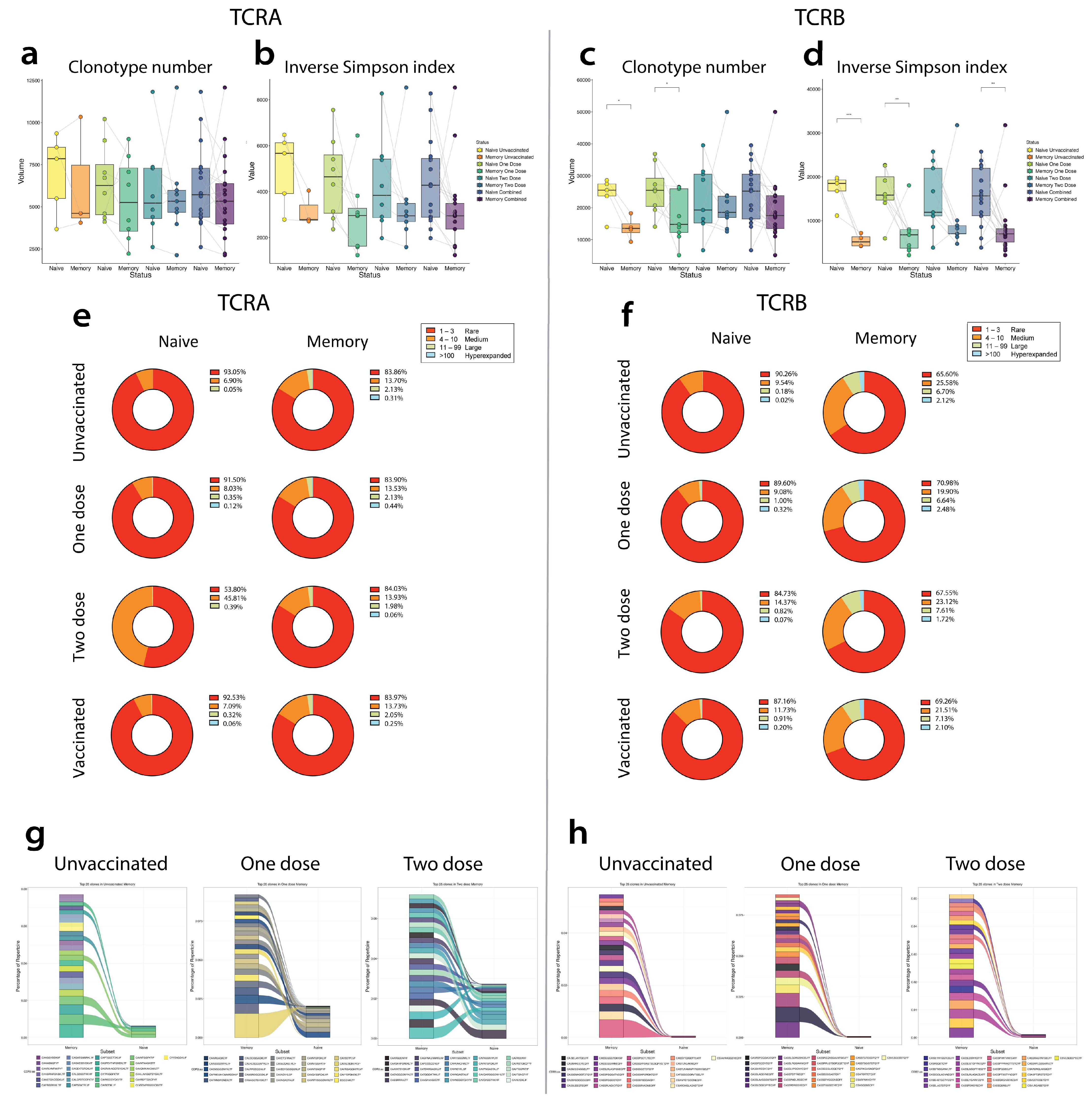
4.6. Comparisons of Repertoire Features between Unvaccinated, Single-Dose Vaccinated, Two-Dose Vaccinated, and Overall Vaccinated Groups
4.6.1. TCRA Comparisons
4.6.2. TCRB Comparisons
4.6.3. TCR-Matched Epitopes
5. Discussion
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Barbier, A.J.; Jiang, A.Y.; Zhang, P.; Wooster, R.; Anderson, D.G. The clinical progress of mRNA vaccines and immunotherapies. Nat. Biotechnol. 2022, 40, 840–854. [Google Scholar] [CrossRef] [PubMed]
- Chaudhary, N.; Weissman, D.; Whitehead, K.A. mRNA vaccines for infectious diseases: Principles, delivery and clinical translation. Nat. Rev. Drug Discov. 2021, 20, 817–838. [Google Scholar] [CrossRef] [PubMed]
- Hou, X.; Zaks, T.; Langer, R.; Dong, Y. Lipid nanoparticles for mRNA delivery. Nat. Rev. Mater. 2021, 6, 1078–1094. [Google Scholar] [CrossRef] [PubMed]
- Folegatti, P.M.; Ewer, K.J.; Aley, P.K.; Angus, B.; Becker, S.; Belij-Rammerstorfer, S.; Bellamy, D.; Bibi, S.; Bittaye, M.; Clutterbuck, E.A.; et al. Safety and immunogenicity of the ChAdOx1 nCoV-19 vaccine against SARS-CoV-2: A preliminary report of a phase 1/2, single-blind, randomised controlled trial. Lancet 2020, 396, 467–478. [Google Scholar] [CrossRef]
- Mascellino, M.T.; Di Timoteo, F.; De Angelis, M.; Oliva, A. Overview of the Main Anti-SARS-CoV-2 Vaccines: Mechanism of Action, Efficacy and Safety. Infect. Drug Resist. 2021, 14, 3459–3476. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Xiang, T.; Liang, B.; Deng, H.; Wang, H.; Feng, X.; Quan, X.; Wang, X.; Li, S.; Lu, S.; et al. Characterization of SARS-CoV-2-Specific Humoral and Cellular Immune Responses Induced by Inactivated COVID-19 Vaccines in a Real-World Setting. Front. Immunol. 2021, 12, 802858. [Google Scholar] [CrossRef] [PubMed]
- Guerrera, G.; Picozza, M.; D’Orso, S.; Placido, R.; Pirronello, M.; Verdiani, A.; Termine, A.; Fabrizio, C.; Giannessi, F.; Sambucci, M.; et al. BNT162b2 vaccination induces durable SARS-CoV-2–specific T cells with a stem cell memory phenotype. Sci. Immunol. 2021, 6, eabl5344. [Google Scholar] [CrossRef]
- Pang, A.P.S.; Higgins-Chen, A.T.; Comite, F.; Raica, I.; Arboleda, C.; Went, H.; Mendez, T.; Schotsaert, M.; Dwaraka, V.; Smith, R.; et al. Longitudinal Study of DNA Methylation and Epigenetic Clocks Prior to and Following Test-Confirmed COVID-19 and mRNA Vaccination. Front. Genet. 2022, 13, 819749. [Google Scholar] [CrossRef]
- Napoli, C.; Coscioni, E.; Trama, U.; Strozziero, M.G.; Benincasa, G. An evidence-based debate on epigenetics and immunosenescence in COVID-19. Curr. Res. Immunol. 2023, 4, 100069. [Google Scholar] [CrossRef]
- Maecker, H.T.; McCoy, J.P.; Nussenblatt, R. Standardizing immunophenotyping for the Human Immunology Project. Nat. Rev. Immunol. 2012, 12, 191–200. [Google Scholar] [CrossRef]
- Grimaldi, V.; Benincasa, G.; Moccia, G.; Sansone, A.; Signoriello, G.; Napoli, C. Evaluation of circulating leucocyte populations both in subjects with previous SARS-COV-2 infection and in healthy subjects after vaccination. J. Immunol. Methods 2022, 502, 113230. [Google Scholar] [CrossRef] [PubMed]
- Jameson, S.C.; Masopust, D. Understanding Subset Diversity in T Cell Memory. Immunity 2018, 48, 214–226. [Google Scholar] [CrossRef] [PubMed]
- Buenrostro, J.; Wu, B.; Chang, H.; Greenleaf, W. ATAC-seq: A Method for Assaying Chromatin Accessibility Genome-Wide. Curr. Protoc. Mol. Biol. 2015, 109, 21.29.1–21.29.9. [Google Scholar] [CrossRef] [PubMed]
- Corces, M.R.; Trevino, A.E.; Hamilton, E.G.; Greenside, P.G.; Sinnott-Armstrong, N.A.; Vesuna, S.; Satpathy, A.T.; Rubin, A.J.; Montine, K.S.; Wu, B.; et al. An improved ATAC-seq protocol reduces background and enables interrogation of frozen tissues. Nat. Methods 2017, 14, 959–962. [Google Scholar] [CrossRef]
- Finck, R.; Simonds, E.F.; Jager, A.; Krishnaswamy, S.; Sachs, K.; Fantl, W.; Pe’er, D.; Nolan, G.P.; Bendall, S.C. Normalization of mass cytometry data with bead standards. Cytom. Part J. Int. Soc. Anal. Cytol. 2013, 83, 483–494. [Google Scholar] [CrossRef]
- Crowell, H.L.; Zanotelli, V.R.T.; Chevrier, S.; Robinson, M.D. Bioconductor, CATALYST: Cytometry dATa anALYSis Tools. R Package Version 1.26.1. 2024. Available online: http://bioconductor.org/packages/CATALYST/ (accessed on 9 September 2021).
- Kotecha, N.; Krutzik, P.O.; Irish, J.M. Web-Based Analysis and Publication of Flow Cytometry Experiments. Curr. Protoc. Cytom. 2010, 53, 10–17. [Google Scholar] [CrossRef]
- Van Gassen, S.; Callebaut, B.; Van Helden, M.J.; Lambrecht, B.N.; Demeester, P.; Dhaene, T.; Saeys, Y. FlowSOM: Using self-organizing maps for visualization and interpretation of cytometry data. Cytometry A 2015, 87, 636–645. [Google Scholar] [CrossRef]
- Chen, H.; Lau, M.C.; Wong, M.T.; Newell, E.W.; Poidinger, M.; Chen, J. Cytofkit: A Bioconductor Package for an Integrated Mass Cytometry Data Analysis Pipeline. PLoS Comput. Biol. 2016, 12, e1005112. [Google Scholar] [CrossRef]
- McInnes, L.; Healy, J.; Saul, N.; Großberger, L. UMAP: Uniform Manifold Approximation and Projection. J. Open Source Softw. 2018, 3, 861. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformaics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Andrews, S. Babraham Bioinformatics—FastQC A Quality Control Tool for High Throughput Sequence Data. 2010. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 25 October 2023).
- Patro, R.; Duggal, G.; Love, M.I.; Irizarry, R.A.; Kingsford, C. Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 2017, 14, 417–419. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Di Tommaso, P.; Chatzou, M.; Floden, E.W.; Barja, P.P.; Palumbo, E.; Notredame, C. Nextflow enables reproducible computational workflows. Nat. Biotechnol. 2017, 35, 316–319. [Google Scholar] [CrossRef] [PubMed]
- Ewels, P.A.; Peltzer, A.; Fillinger, S.; Patel, H.; Alneberg, J.; Wilm, A.; Garcia, M.U.; Di Tommaso, P.; Nahnsen, S. The nf-core framework for community-curated bioinformatics pipelines. Nat. Biotechnol. 2020, 38, 276–278. [Google Scholar] [CrossRef]
- Patel, H.; Espinosa-Carrasco, J.; Langer, B.; Ewels, P. Nf-core/atacseq: [2.1.2]—2022-08-07. Zenodo. 2023. Available online: https://zenodo.org/records/8222875 (accessed on 25 August 2023).
- Krueger, F. Babraham Bioinformatics—Trim Galore! 2012. Available online: https://www.bioinformatics.babraham.ac.uk/projects/trim_galore/ (accessed on 25 August 2023).
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef]
- Picard Tools—By Broad Institute. Available online: https://broadinstitute.github.io/picard/ (accessed on 25 August 2023).
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. 1000 Genome Project Data Processing Subgroup, 2009. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- Barnett, D.W.; Garrison, E.K.; Quinlan, A.R.; Strömberg, M.P.; Marth, G.T. BamTools: A C++ API and toolkit for analyzing and managing BAM files. Bioinformatics. 2011, 27, 1691–1692. [Google Scholar] [CrossRef] [PubMed]
- Ramírez, F.; Ryan, D.P.; Grüning, B.; Bhardwaj, V.; Kilpert, F.; Richter, A.S.; Heyne, S.; Dündar, F.; Manke, T. DeepTools2: A next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 2016, 44, W160–W165. [Google Scholar] [CrossRef]
- Zhang, Y.; Liu, T.; Meyer, C.A.; Eeckhoute, J.; Johnson, D.S.; Bernstein, B.E.; Nusbaum, C.; Myers, R.M.; Brown, M.; Li, W.; et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 2008, 9, R137. [Google Scholar] [CrossRef]
- Heinz, S.; Benner, C.; Spann, N.; Bertolino, E.; Lin, Y.C.; Laslo, P.; Cheng, J.X.; Murre, C.; Singh, H.; Glass, C.K. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell 2010, 38, 576–589. [Google Scholar] [CrossRef]
- R: The R Project for Statistical Computing. Available online: https://www.r-project.org/ (accessed on 8 September 2021).
- Andrews, S. Babraham Bioinformatics—SeqMonk Mapped Sequence Analysis Tool. 2007. Available online: https://www.bioinformatics.babraham.ac.uk/projects/seqmonk/ (accessed on 15 September 2023).
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed]
- Quinlan, A.R.; Hall, I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef] [PubMed]
- Bailey, T.L.; Johnson, J.; Grant, C.E.; Noble, W.S. The MEME Suite. Nucleic Acids Res. 2015, 43, W39–W49. [Google Scholar] [CrossRef]
- Machanick, P.; Bailey, T.L. MEME-ChIP: Motif analysis of large DNA datasets. Bioinformatics 2011, 27, 1696–1697. [Google Scholar] [CrossRef]
- Bolotin, D.A.; Poslavsky, S.; Mitrophanov, I.; Shugay, M.; Mamedov, I.Z.; Putintseva, E.V.; Chudakov, D.M. MiXCR: Software for comprehensive adaptive immunity profiling. Nat. Methods 2015, 12, 380–381. [Google Scholar] [CrossRef]
- Nazarov, V.I.; Tsvetkov, V.; Rumynskiy, E.; Popov, A.; Balashov, I.; Samokhina, M. Immunarch: Bioinformatics Analysis of T-Cell and B-Cell Immune Repertoires. 2023. Available online: https://github.com/immunomind/immunarch (accessed on 3 October 2023).
- Wickham, H. Ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2016. [Google Scholar]
- Gu, Y.; Gu, J.; Shen, K.; Zhou, H.; Hao, J.; Li, F.; Yu, H.; Chen, Y.; Li, J.; Li, Y.; et al. HOXA13 promotes colon cancer progression through β-catenin-dependent WNT pathway. Exp. Cell Res. 2020, 395, 112238. [Google Scholar] [CrossRef] [PubMed]
- Laity, J.H.; Lee, B.M.; Wright, P.E. Zinc finger proteins: New insights into structural and functional diversity. Curr. Opin. Struct. Biol. 2001, 11, 39–46. [Google Scholar] [CrossRef]
- Diana, P.; Carvalheira, G.M.G. NIBAN1, Exploring its Roles in Cell Survival Under Stress Context. Front. Cell Dev. Biol. 2022, 10, 867003. [Google Scholar] [CrossRef]
- Rathinam, V.A.K.; Jiang, Z.; Waggoner, S.N.; Sharma, S.; Cole, L.E.; Waggoner, L.; Vanaja, S.K.; Monks, B.G.; Ganesan, S.; Latz, E.; et al. The AIM2 inflammasome is essential for host defense against cytosolic bacteria and DNA viruses. Nat. Immunol. 2010, 11, 395–402. [Google Scholar] [CrossRef]
- Zhang, Y.; Liu, Q.; Yang, S.; Liao, Q. CD58 Immunobiology at a Glance. Front. Immunol. 2021, 12, 705260. [Google Scholar] [CrossRef]
- Nieto-Estevez, V.; Hsieh, J. CHD2: One Gene, Many Roles. Neuron 2018, 100, 1014–1016. [Google Scholar] [CrossRef] [PubMed]
- Hollenberg, M.D.; Compton, S.J. International Union of Pharmacology. XXVIII. Proteinase-Activated Receptors. Pharmacol. Rev. 2002, 54, 203–217. [Google Scholar] [CrossRef] [PubMed]
- Vazquez, S.E.; Mann, S.A.; Bodansky, A.; Kung, A.F.; Quandt, Z.; Ferré, E.M.; Landegren, N.; Eriksson, D.; Bastard, P.; Zhang, S.Y.; et al. Autoantibody discovery across monogenic, acquired, and COVID-19-associated autoimmunity with scalable PhIP-seq. eLife 2022, 11, e78550. [Google Scholar] [CrossRef] [PubMed]
- Dai, L.; Chai, C.M.; Shen, T.Y.; Tian, Y.; Shang, Z.Q.; Niu, Y.J. LncRNA ITGB1 promotes the development of bladder cancer through regulating microRNA-10a expression. Eur. Rev. Med. Pharmacol. Sci. 2019, 23, 6858–6867. [Google Scholar]
- Ehrnsperger, A.; Rehli, M.; Thu-Hang, P.; Kreutz, M. Epigenetic regulation of the dendritic cell-marker gene ADAM19. Biochem. Biophys. Res. Commun. 2005, 332, 456–464. [Google Scholar] [CrossRef]
- Szymanski, M.; Barciszewska, M.Z.; Erdmann, V.A.; Barciszewski, J. 5S Ribosomal RNA Database. Nucleic Acids Res. 2002, 30, 176–178. [Google Scholar] [CrossRef]
- Wan, Y.Y. GATA3: A master of many trades in immune regulation. Trends Immunol. 2014, 35, 233–242. [Google Scholar] [CrossRef]
- Fenoglio, D.; Dentone, C.; Parodi, A.; Di Biagio, A.; Bozzano, F.; Vena, A.; Fabbi, M.; Ferrera, F.; Altosole, T.; Bruzzone, B.; et al. Characterization of T lymphocytes in severe COVID-19 patients. J. Med. Virol. 2021, 93, 5608–5613. [Google Scholar] [CrossRef]
- Khantakova, J.N.; Bulygin, A.S.; Sennikov, S.V. The Regulatory-T-Cell Memory Phenotype: What We Know. Cells 2022, 11, 1687. [Google Scholar] [CrossRef]
- Franco, A.; Song, J.; Chambers, C.; Sette, A.; Grifoni, A. SARS-CoV-2 spike-specific regulatory T cells (Treg) expand and develop memory in vaccine recipients suggesting a role for immune regulation in preventing severe symptoms in COVID-19. Autoimmunity 2023, 56, 2259133. [Google Scholar] [CrossRef]
- Giannotta, G.; Murrone, A.; Giannotta, N. COVID-19 mRNA Vaccines: The Molecular Basis of Some Adverse Events. Vaccines 2023, 11, 747. [Google Scholar] [CrossRef] [PubMed]
- Murata, K.; Nakao, N.; Ishiuchi, N.; Fukui, T.; Katsuya, N.; Fukumoto, W.; Oka, H.; Yoshikawa, N.; Nagao, T.; Namera, A.; et al. Four cases of cytokine storm after COVID-19 vaccination: Case report. Front. Immunol. 2022, 13, 967226. [Google Scholar] [CrossRef]
- Bradley, C.C.; Wang, C.; Gordon, A.J.; Wen, A.X.; Luna, P.N.; Cooke, M.B.; Kohrn, B.F.; Kennedy, S.R.; Avadhanula, V.; Piedra, P.A.; et al. Targeted accurate RNA consensus sequencing (tARC-seq) reveals mechanisms of replication error affecting SARS-CoV-2 divergence. Nat. Microbiol. 2024, 9, 1382–1392. [Google Scholar] [CrossRef] [PubMed]
- Kopp, R.; Krautloher, A.; Ramírez-Fernández, A.; Nicke, A. P2X7 Interactions and Signaling—Making Head or Tail of It. Front. Mol. Neurosci. 2019, 12, 183. [Google Scholar] [CrossRef]
- Pacheco, P.A.F.; Faria, R.X. The potential involvement of P2X7 receptor in COVID-19 pathogenesis: A new therapeutic target? Scand. J. Immunol. 2021, 93, e12960. [Google Scholar] [CrossRef]
- Pathinayake, P.S.; Awatade, N.T.; Wark, P.A.B. Type 2 Immunity and Its Impact on COVID-19 Infection in the Airways. Viruses 2023, 15, 402. [Google Scholar] [CrossRef]
- Shah, K.; Al-Haidari, A.; Sun, J.; Kazi, J.U. T cell receptor (TCR) signaling in health and disease. Signal Transduct. Target. Ther. 2021, 6, 412. [Google Scholar] [CrossRef] [PubMed]
- Sallusto, F.; Geginat, J.; Lanzavecchia, A. Central Memory and Effector Memory T Cell Subsets: Function, Generation, and Maintenance. Annu. Rev. Immunol. 2004, 22, 745–763. [Google Scholar] [CrossRef]
- Drury, R.E.; Camara, S.; Chelysheva, I.; Bibi, S.; Sanders, K.; Felle, S.; Emary, K.; Phillips, D.; Voysey, M.; Ferreira, D.M.; et al. Multi-omics analysis reveals COVID-19 vaccine induced attenuation of inflammatory responses during breakthrough disease. Nat. Commun. 2024, 15, 3402. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CyTOF Markers | ||
|---|---|---|
| CCR6 | CD25 | CCR7 |
| CD38 | CD127 | PD1 |
| CXCR5 | HLADR | CTLA4 |
| CD45RA | CCR4 | CD3 |
| CD45RO | CD28 | CD4 |
| CD27 | CD56 | |
| CXCR3 | PDL1 | |
| Marker | Fluorochrome | Manufacturer | Clone | Cat Number | Volume (µL) |
|---|---|---|---|---|---|
| CD3 | BUV395 | BD | SK7 | 564,001 | 2 |
| CD4 | BV786 | BD | SK3 | 563,877 | 2 |
| CD45RA | BV510 | BD | HI100 | 563,031 | 2 |
| CCR7 | PE-CF594 | BD | 150503 | 562,381 | 4 |
| CD19 | FITC | BD | 555,412 | 2 | |
| CD27 | BV711 | Biolegend | O323 | 302,834 | 2 |
| IgG | APC | BD | 550,931 | 5 | |
| IgM | PE | BD | 555,783 | 10 | |
| Live Dead | DAPI (1 mg) | BD | 564,907 | 2 µL of a 1:30 dilution |
| Characteristic | Unvaccinated | One Dose | Two Dose |
|---|---|---|---|
| (n = 8) | (n = 17) | (n = 26) | |
| Age, median (IQR) | 37 (23–53) | 35.5 (32.25–39.75) | 55.5 (48–60) |
| Male n (%) | 8 (100%) | 17 (100%) | 25 (96.2%) |
| Previous SARS-CoV-2 Infection, n (%) | |||
| Yes | 3 (37.5%) | 4 (23.5%) | 13 (50%) |
| No | 5 (62.5%) | 14 (82.3%) | 13 (50%) |
| Symptoms, n (%) | |||
| Loss of smell | 2 (66.6%) | 1 (25%) | 6 (46.2%) |
| Cough | 2 (66.6%) | 3 (75%) | 7 (53.8%) |
| Loss of appetite | 2 (66.6%) | 1 (25%) | 5 (38.5%) |
| Fatigue | 3 (100%) | 4 (100%) | 11 (84.6%) |
| Chest pain | 0 | 0 | 2 (15.4%) |
| Sore throat | 2 (66.6%) | 1 (25%) | 1 (7.7%) |
| Muscle pain | 3 (100%) | 3 (75%) | 8 (61.5%) |
| Hoarseness | 2 (66.6%) | 0 | 0 |
| Abdominal pain | 0 | 0 | 0 |
| Headache | 2 (66.6%) | 3 (75%) | 8 (61.5%) |
| Fever | 1 (33.3%) | 0 | 4 (30.8) |
| Shortness of breath | 1 (33.3%) | 0 | 5 (38.5%) |
| Diarrhoea | 0 | 0 | 1 (7.7%) |
| Confusion | 0 | 0 | 1 (7.7%) |
| Other | 0 | 1 (25%) | 1 (7.7%) |
| Brand of vaccine first dose, n (%) | n/a | 17 | 26 |
| Pfizer | 11 (64.7%) | 8 (30.8%) | |
| Astra Zeneca | 5 (29.4%) | 18 (69.2%) | |
| Moderna | 2 (11.7%) | 0 | |
| Brand of vaccine second dose, n (%) | n/a | n/a | 26 |
| Pfizer | 8 (30.8%) | ||
| Astra Zeneca | 17 (65.4%) | ||
| Moderna | 0 | ||
| NA | 1 (3.8%) | ||
| Time between vaccination and sampling, median (IQR) days | n/a | 31 (20.5–42.75) | 47.5 (28.25–78.5) |
| Within Group Comparisons of CD4+Naïve vs. CD4+CM | |
|---|---|
| Comparison within unvaccinated group |
|
| Comparisons within vaccinated group |
|
| Between Group Comparisons Vaccinated vs. Unvaccinated by the CD4+Naïve and CD4+CM Subset | |
| CD4+Naïve subset |
|
| CD4+CM subset |
|
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mosavie, M.; Rynne, J.; Fish, M.; Smith, P.; Jennings, A.; Singh, S.; Millar, J.; Harvala, H.; Mora, A.; Kaloyirou, F.; et al. Changes in Phenotypic and Molecular Features of Naïve and Central Memory T Helper Cell Subsets following SARS-CoV-2 Vaccination. Vaccines 2024, 12, 1040. https://doi.org/10.3390/vaccines12091040
Mosavie M, Rynne J, Fish M, Smith P, Jennings A, Singh S, Millar J, Harvala H, Mora A, Kaloyirou F, et al. Changes in Phenotypic and Molecular Features of Naïve and Central Memory T Helper Cell Subsets following SARS-CoV-2 Vaccination. Vaccines. 2024; 12(9):1040. https://doi.org/10.3390/vaccines12091040
Chicago/Turabian StyleMosavie, Mia, Jennifer Rynne, Matthew Fish, Peter Smith, Aislinn Jennings, Shivani Singh, Jonathan Millar, Heli Harvala, Ana Mora, Fotini Kaloyirou, and et al. 2024. "Changes in Phenotypic and Molecular Features of Naïve and Central Memory T Helper Cell Subsets following SARS-CoV-2 Vaccination" Vaccines 12, no. 9: 1040. https://doi.org/10.3390/vaccines12091040
APA StyleMosavie, M., Rynne, J., Fish, M., Smith, P., Jennings, A., Singh, S., Millar, J., Harvala, H., Mora, A., Kaloyirou, F., Griffiths, A., Hopkins, V., Washington, C., Estcourt, L. J., Roberts, D., & Shankar-Hari, M. (2024). Changes in Phenotypic and Molecular Features of Naïve and Central Memory T Helper Cell Subsets following SARS-CoV-2 Vaccination. Vaccines, 12(9), 1040. https://doi.org/10.3390/vaccines12091040