

Decoding NAD+ Metabolism in COVID-19: Implications for Immune Modulation and Therapy


Abstract
:1. Introduction
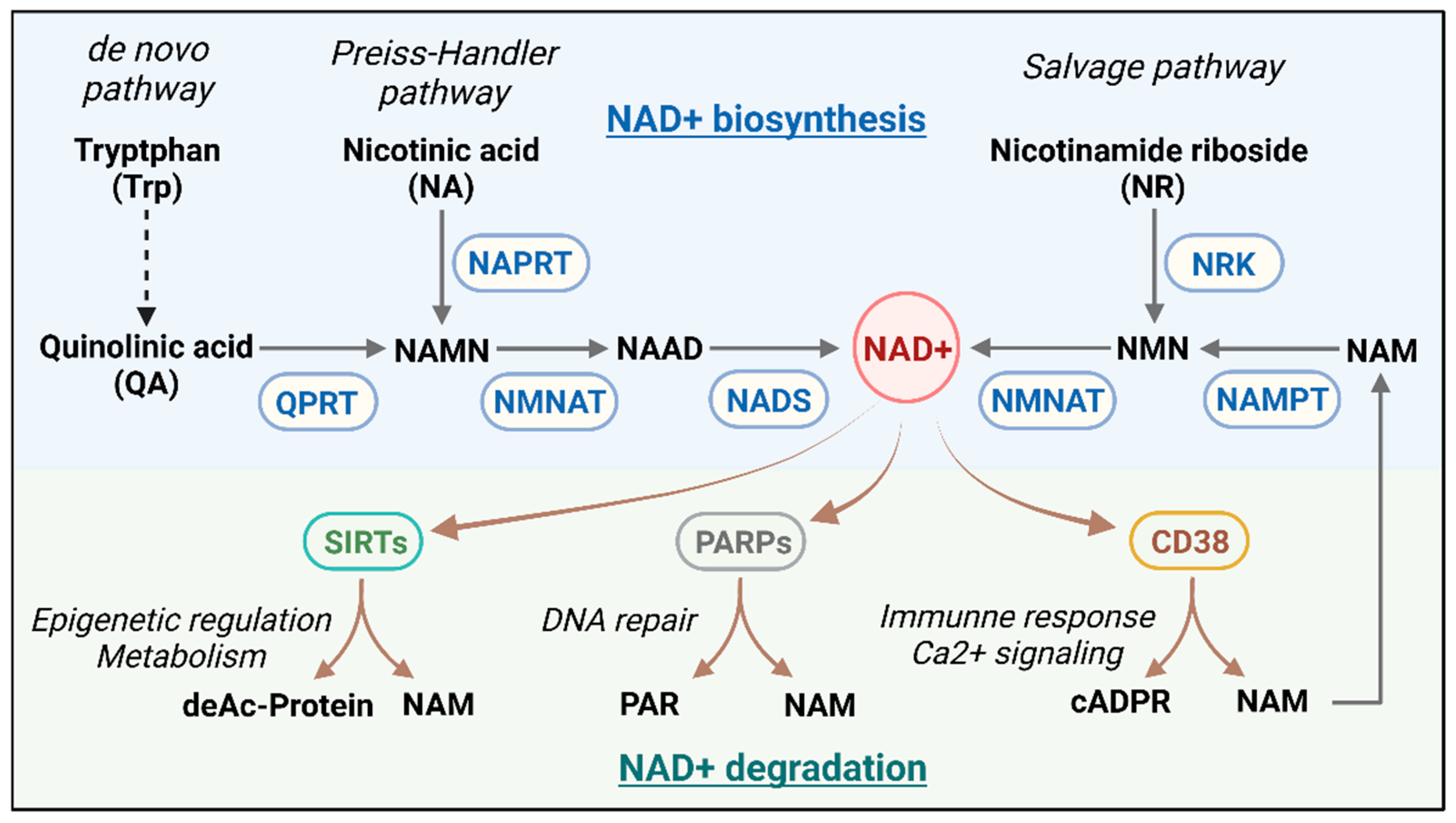
2. NAD+ Metabolism
2.1. The NAD+ Metabolic Axis and Its Regulation
2.2. The Importance of NAD+ in Cellular Energy Metabolism and Redox Reactions
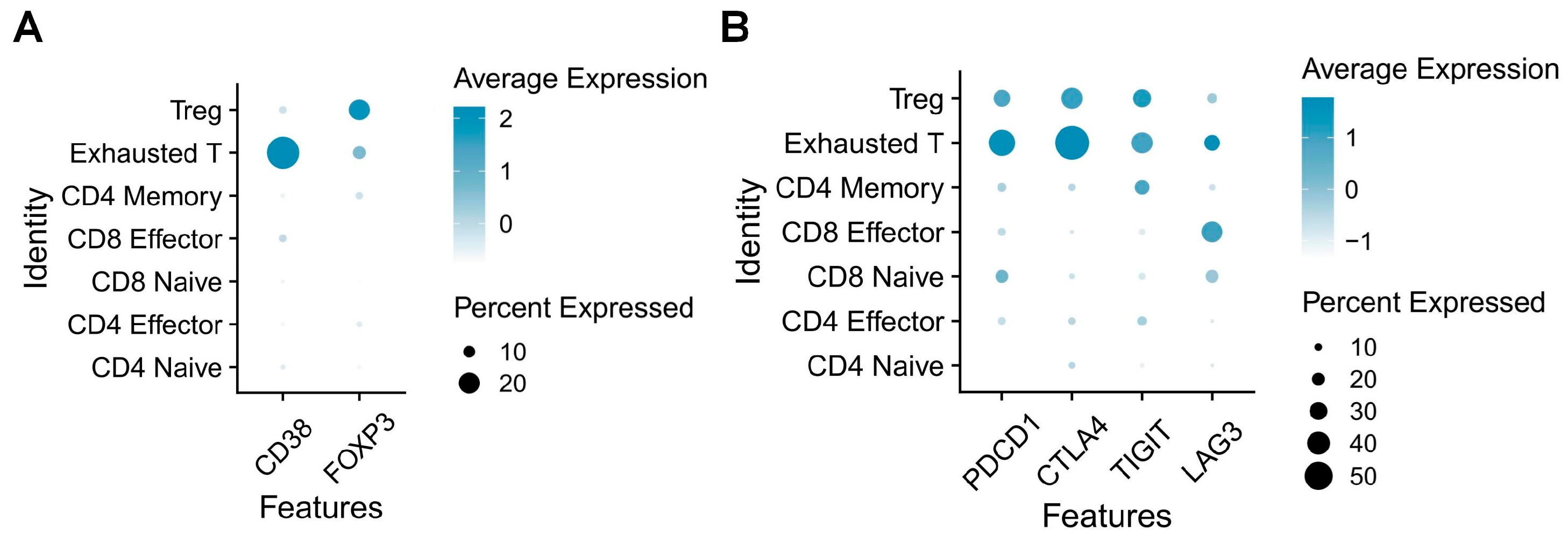
2.3. Role of CD38 in Immunoregulation and Cell Signaling
3. NAD+ Metabolism in Infectious and Non-Infectious Diseases
3.1. NAD+ Metabolism in Infectious Diseases
3.2. NAD+ Metabolism in COVID-19
3.3. NAD+ Metabolism in Non-Infectious Diseases
4. NAD+ Metabolism, Aging, and COVID-19
4.1. NAD+ and Aging
4.2. COVID-19, Cellular Senescence, and Aging
5. Potential of Modulating NAD+ Metabolism in COVID-19 Treatment
5.1. Clinical Studies of NAD+ and Its Precursors as Therapeutic Interventions in COVID-19
5.2. Targeting NAD+-Consuming Enzymes for COVID-19 Therapy
5.3. Restoration Strategies and Future Perspectives
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Lan, S.H.; Lai, C.C.; Huang, H.T.; Chang, S.P.; Lu, L.C.; Hsueh, P.R. Tocilizumab for severe COVID-19: A systematic review and meta-analysis. Int. J. Antimicrob. Agents 2020, 56, 106103. [Google Scholar] [CrossRef] [PubMed]
- Rezaei, S.; Fatemi, B.; Karimi Majd, Z.; Minaei, H.; Peikanpour, M.; Anjidani, N.; Taheri, A.; Dastan, F.; Mosaed, R. Efficacy and safety of Tocilizumab in severe and critical COVID-19: A Systematic Review and Meta-Analysis. Expert Rev. Clin. Immunol. 2021, 17, 499–511. [Google Scholar] [CrossRef]
- Ely, E.W.; Ramanan, A.V.; Kartman, C.E.; de Bono, S.; Liao, R.; Piruzeli, M.L.B.; Goldman, J.D.; Saraiva, J.F.K.; Chakladar, S.; Marconi, V.C. Efficacy and safety of baricitinib plus standard of care for the treatment of critically ill hospitalised adults with COVID-19 on invasive mechanical ventilation or extracorporeal membrane oxygenation: An exploratory, randomised, placebo-controlled trial. Lancet Respir. Med. 2022, 10, 327–336. [Google Scholar] [CrossRef] [PubMed]
- Marconi, V.C.; Ramanan, A.V.; de Bono, S.; Kartman, C.E.; Krishnan, V.; Liao, R.; Piruzeli, M.L.B.; Goldman, J.D.; Alatorre-Alexander, J.; de Cassia Pellegrini, R.; et al. Efficacy and safety of baricitinib for the treatment of hospitalised adults with COVID-19 (COV-BARRIER): A randomised, double-blind, parallel-group, placebo-controlled phase 3 trial. Lancet Respir. Med. 2021, 9, 1407–1418. [Google Scholar] [CrossRef]
- Liu, W.; Liu, T.; Zheng, Y.; Xia, Z. Metabolic Reprogramming and Its Regulatory Mechanism in Sepsis-Mediated Inflammation. J. Inflamm. Res. 2023, 16, 1195–1207. [Google Scholar] [CrossRef]
- Giamarellos-Bourboulis, E.J.; Aschenbrenner, A.C.; Bauer, M.; Bock, C.; Calandra, T.; Gat-Viks, I.; Kyriazopoulou, E.; Lupse, M.; Monneret, G.; Pickkers, P.; et al. The pathophysiology of sepsis and precision-medicine-based immunotherapy. Nat. Immunol. 2024, 25, 19–28. [Google Scholar] [CrossRef] [PubMed]
- Rudiansyah, M.; Jasim, S.A.; Mohammad Pour, Z.G.; Athar, S.S.; Jeda, A.S.; Doewes, R.I.; Jalil, A.T.; Bokov, D.O.; Mustafa, Y.F.; Noroozbeygi, M.; et al. Coronavirus disease 2019 (COVID-19) update: From metabolic reprogramming to immunometabolism. J. Med. Virol. 2022, 94, 4611–4627. [Google Scholar] [CrossRef]
- Cengiz, M.; Borku Uysal, B.; Ikitimur, H.; Ozcan, E.; Islamoğlu, M.S.; Aktepe, E.; Yavuzer, H.; Yavuzer, S. Effect of oral l-Glutamine supplementation on Covid-19 treatment. Clin. Nutr. Exp. 2020, 33, 24–31. [Google Scholar] [CrossRef]
- Gamarra-Morales, Y.; Herrera-Quintana, L.; Molina-López, J.; Vázquez-Lorente, H.; Machado-Casas, J.F.; Castaño-Pérez, J.; Pérez-Villares, J.M.; Planells, E. Response to Intravenous N-Acetylcysteine Supplementation in Critically Ill Patients with COVID-19. Nutrients 2023, 15, 2235. [Google Scholar] [CrossRef]
- Wang, S.; Tsun, Z.Y.; Wolfson, R.L.; Shen, K.; Wyant, G.A.; Plovanich, M.E.; Yuan, E.D.; Jones, T.D.; Chantranupong, L.; Comb, W.; et al. Metabolism. Lysosomal amino acid transporter SLC38A9 signals arginine sufficiency to mTORC1. Science 2015, 347, 188–194. [Google Scholar] [CrossRef]
- Tosato, M.; Calvani, R.; Picca, A.; Ciciarello, F.; Galluzzo, V.; Coelho-Júnior, H.J.; Di Giorgio, A.; Di Mario, C.; Gervasoni, J.; Gremese, E.; et al. Effects of l-Arginine Plus Vitamin C Supplementation on Physical Performance, Endothelial Function, and Persistent Fatigue in Adults with Long COVID: A Single-Blind Randomized Controlled Trial. Nutrients 2022, 14, 4984. [Google Scholar] [CrossRef] [PubMed]
- Slankamenac, J.; Ranisavljev, M.; Todorovic, N.; Ostojic, J.; Stajer, V.; Ostojic, S.M. Effects of six-month creatine supplementation on patient- and clinician-reported outcomes, and tissue creatine levels in patients with post-COVID-19 fatigue syndrome. Food Sci. Nutr. 2023, 11, 6899–6906. [Google Scholar] [CrossRef] [PubMed]
- Castro-Portuguez, R.; Sutphin, G.L. Kynurenine pathway, NAD(+) synthesis, and mitochondrial function: Targeting tryptophan metabolism to promote longevity and healthspan. Exp. Gerontol. 2020, 132, 110841. [Google Scholar] [CrossRef]
- Rajman, L.; Chwalek, K.; Sinclair, D.A. Therapeutic Potential of NAD-Boosting Molecules: The In Vivo Evidence. Cell Metab. 2018, 27, 529–547. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, K.N.; Peics, J.; Ma, T.; Karavaeva, I.; Dall, M.; Chubanava, S.; Basse, A.L.; Dmytriyeva, O.; Treebak, J.T.; Gerhart-Hines, Z. NAMPT-mediated NAD(+) biosynthesis is indispensable for adipose tissue plasticity and development of obesity. Mol. Metab. 2018, 11, 178–188. [Google Scholar] [CrossRef] [PubMed]
- Yoshino, J.; Mills, K.F.; Yoon, M.J.; Imai, S. Nicotinamide mononucleotide, a key NAD(+) intermediate, treats the pathophysiology of diet- and age-induced diabetes in mice. Cell Metab. 2011, 14, 528–536. [Google Scholar] [CrossRef] [PubMed]
- Carafa, V.; Rotili, D.; Forgione, M.; Cuomo, F.; Serretiello, E.; Hailu, G.S.; Jarho, E.; Lahtela-Kakkonen, M.; Mai, A.; Altucci, L. Sirtuin functions and modulation: From chemistry to the clinic. Clin. Epigenetics 2016, 8, 61. [Google Scholar] [CrossRef] [PubMed]
- Chalkiadaki, A.; Guarente, L. The multifaceted functions of sirtuins in cancer. Nat. Rev. Cancer 2015, 15, 608–624. [Google Scholar] [CrossRef]
- Schreiber, V.; Dantzer, F.; Ame, J.C.; de Murcia, G. Poly(ADP-ribose): Novel functions for an old molecule. Nat. Rev. Mol. Cell Biol. 2006, 7, 517–528. [Google Scholar] [CrossRef]
- Guse, A.H. Calcium mobilizing second messengers derived from NAD. Biochim. Biophys. Acta 2015, 1854, 1132–1137. [Google Scholar] [CrossRef]
- Chini, C.C.S.; Peclat, T.R.; Warner, G.M.; Kashyap, S.; Espindola-Netto, J.M.; de Oliveira, G.C.; Gomez, L.S.; Hogan, K.A.; Tarragó, M.G.; Puranik, A.S.; et al. CD38 ecto-enzyme in immune cells is induced during aging and regulates NAD(+) and NMN levels. Nat. Metab. 2020, 2, 1284–1304. [Google Scholar] [CrossRef] [PubMed]
- Katsyuba, E.; Romani, M.; Hofer, D.; Auwerx, J. NAD(+) homeostasis in health and disease. Nat. Metab. 2020, 2, 9–31. [Google Scholar] [CrossRef] [PubMed]
- Xie, N.; Zhang, L.; Gao, W.; Huang, C.; Huber, P.E.; Zhou, X.; Li, C.; Shen, G.; Zou, B. NAD(+) metabolism: Pathophysiologic mechanisms and therapeutic potential. Signal Transduct. Target. Ther. 2020, 5, 227. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef]
- Blacker, T.S.; Mann, Z.F.; Gale, J.E.; Ziegler, M.; Bain, A.J.; Szabadkai, G.; Duchen, M.R. Separating NADH and NADPH fluorescence in live cells and tissues using FLIM. Nat. Commun. 2014, 5, 3936. [Google Scholar] [CrossRef] [PubMed]
- Ghisays, F.; Brace, C.S.; Yackly, S.M.; Kwon, H.J.; Mills, K.F.; Kashentseva, E.; Dmitriev, I.P.; Curiel, D.T.; Imai, S.I.; Ellenberger, T. The N-Terminal Domain of SIRT1 Is a Positive Regulator of Endogenous SIRT1-Dependent Deacetylation and Transcriptional Outputs. Cell Rep. 2015, 10, 1665–1673. [Google Scholar] [CrossRef] [PubMed]
- Hori, Y.S.; Kuno, A.; Hosoda, R.; Horio, Y. Regulation of FOXOs and p53 by SIRT1 modulators under oxidative stress. PLoS ONE 2013, 8, e73875. [Google Scholar] [CrossRef] [PubMed]
- Nemoto, S.; Fergusson, M.M.; Finkel, T. SIRT1 functionally interacts with the metabolic regulator and transcriptional coactivator PGC-1{alpha}. J. Biol. Chem. 2005, 280, 16456–16460. [Google Scholar] [CrossRef]
- Olmos, Y.; Sánchez-Gómez, F.J.; Wild, B.; García-Quintans, N.; Cabezudo, S.; Lamas, S.; Monsalve, M. SirT1 regulation of antioxidant genes is dependent on the formation of a FoxO3a/PGC-1α complex. Antioxid. Redox Signal 2013, 19, 1507–1521. [Google Scholar] [CrossRef]
- Bause, A.S.; Haigis, M.C. SIRT3 regulation of mitochondrial oxidative stress. Exp. Gerontol. 2013, 48, 634–639. [Google Scholar] [CrossRef] [PubMed]
- Qiu, X.; Brown, K.; Hirschey, M.D.; Verdin, E.; Chen, D. Calorie restriction reduces oxidative stress by SIRT3-mediated SOD2 activation. Cell Metab. 2010, 12, 662–667. [Google Scholar] [CrossRef] [PubMed]
- Bryan, S.; Baregzay, B.; Spicer, D.; Singal, P.K.; Khaper, N. Redox-inflammatory synergy in the metabolic syndrome. Can. J. Physiol. Pharmacol. 2013, 91, 22–30. [Google Scholar] [CrossRef]
- Fritze, C.E.; Verschueren, K.; Strich, R.; Easton Esposito, R. Direct evidence for SIR2 modulation of chromatin structure in yeast rDNA. EMBO J. 1997, 16, 6495–6509. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.C. Cyclic ADP-ribose and nicotinic acid adenine dinucleotide phosphate (NAADP) as messengers for calcium mobilization. J. Biol. Chem. 2012, 287, 31633–31640. [Google Scholar] [CrossRef] [PubMed]
- Wei, W.; Graeff, R.; Yue, J. Roles and mechanisms of the CD38/cyclic adenosine diphosphate ribose/Ca2+ signaling pathway. World J. Biol. Chem. 2014, 5, 58–67. [Google Scholar] [CrossRef]
- Skyline, G. Available online: http://rstats.immgen.org/Skyline_microarray/skyline.html (accessed on 20 September 2024).
- Sandoval-Montes, C.; Santos-Argumedo, L. CD38 is expressed selectively during the activation of a subset of mature T cells with reduced proliferation but improved potential to produce cytokines. J. Leukoc. Biol. 2005, 77, 513–521. [Google Scholar] [CrossRef]
- Shubinsky, G.; Schlesinger, M. The CD38 lymphocyte differentiation marker: New insight into its ectoenzymatic activity and its role as a signal transducer. Immunity 1997, 7, 315–324. [Google Scholar] [CrossRef]
- Li, W.; Li, Y.; Jin, X.; Liao, Q.; Chen, Z.; Peng, H.; Zhou, Y. CD38: A Significant Regulator of Macrophage Function. Front. Oncol. 2022, 12, 775649. [Google Scholar] [CrossRef] [PubMed]
- Schneider, M.; Schumacher, V.; Lischke, T.; Lücke, K.; Meyer-Schwesinger, C.; Velden, J.; Koch-Nolte, F.; Mittrücker, H.W. CD38 is expressed on inflammatory cells of the intestine and promotes intestinal inflammation. PLoS ONE 2015, 10, e0126007. [Google Scholar] [CrossRef] [PubMed]
- Schiavoni, I.; Scagnolari, C.; Horenstein, A.L.; Leone, P.; Pierangeli, A.; Malavasi, F.; Ausiello, C.M.; Fedele, G. CD38 modulates respiratory syncytial virus-driven proinflammatory processes in human monocyte-derived dendritic cells. Immunology 2018, 154, 122–131. [Google Scholar] [CrossRef]
- Ben Baruch, B.; Blacher, E.; Mantsur, E.; Schwartz, H.; Vaknine, H.; Erez, N.; Stein, R. Stromal CD38 regulates outgrowth of primary melanoma and generation of spontaneous metastasis. Oncotarget 2018, 9, 31797–31811. [Google Scholar] [CrossRef]
- Levy, A.; Blacher, E.; Vaknine, H.; Lund, F.E.; Stein, R.; Mayo, L. CD38 deficiency in the tumor microenvironment attenuates glioma progression and modulates features of tumor-associated microglia/macrophages. Neuro Oncol. 2012, 14, 1037–1049. [Google Scholar] [CrossRef] [PubMed]
- van de Donk, N.W.; Janmaat, M.L.; Mutis, T.; Lammerts van Bueren, J.J.; Ahmadi, T.; Sasser, A.K.; Lokhorst, H.M.; Parren, P.W. Monoclonal antibodies targeting CD38 in hematological malignancies and beyond. Immunol. Rev. 2016, 270, 95–112. [Google Scholar] [CrossRef]
- Chen, L.; Diao, L.; Yang, Y.; Yi, X.; Rodriguez, B.L.; Li, Y.; Villalobos, P.A.; Cascone, T.; Liu, X.; Tan, L.; et al. CD38-Mediated Immunosuppression as a Mechanism of Tumor Cell Escape from PD-1/PD-L1 Blockade. Cancer Discov. 2018, 8, 1156–1175. [Google Scholar] [CrossRef] [PubMed]
- Navas, L.E.; Carnero, A. NAD(+) metabolism, stemness, the immune response, and cancer. Signal Transduct. Target. Ther. 2021, 6, 2. [Google Scholar] [CrossRef] [PubMed]
- Deaglio, S.; Aydin, S.; Grand, M.M.; Vaisitti, T.; Bergui, L.; D’Arena, G.; Chiorino, G.; Malavasi, F. CD38/CD31 interactions activate genetic pathways leading to proliferation and migration in chronic lymphocytic leukemia cells. Mol. Med. 2010, 16, 87–91. [Google Scholar] [CrossRef] [PubMed]
- Deaglio, S.; Dianzani, U.; Horenstein, A.L.; Fernández, J.E.; van Kooten, C.; Bragardo, M.; Funaro, A.; Garbarino, G.; Di Virgilio, F.; Banchereau, J.; et al. Human CD38 ligand. A 120-KDA protein predominantly expressed on endothelial cells. J. Immunol. 1996, 156, 727–734. [Google Scholar] [CrossRef]
- Horenstein, A.L.; Bracci, C.; Morandi, F.; Malavasi, F. CD38 in Adenosinergic Pathways and Metabolic Re-programming in Human Multiple Myeloma Cells: In-tandem Insights From Basic Science to Therapy. Front. Immunol. 2019, 10, 760. [Google Scholar] [CrossRef]
- Maluchenko, N.V.; Feofanov, A.V.; Studitsky, V.M. PARP-1-Associated Pathological Processes: Inhibition by Natural Polyphenols. Int. J. Mol. Sci. 2021, 22, 11441. [Google Scholar] [CrossRef] [PubMed]
- Pacher, P.; Szabo, C. Role of the peroxynitrite-poly(ADP-ribose) polymerase pathway in human disease. Am. J. Pathol. 2008, 173, 2–13. [Google Scholar] [CrossRef]
- Virág, L.; Robaszkiewicz, A.; Rodriguez-Vargas, J.M.; Oliver, F.J. Poly(ADP-ribose) signaling in cell death. Mol. Aspects Med. 2013, 34, 1153–1167. [Google Scholar] [CrossRef] [PubMed]
- Michalak, T.I. The Initial Hepatitis B Virus-Hepatocyte Genomic Integrations and Their Role in Hepatocellular Oncogenesis. Int. J. Mol. Sci. 2023, 24, 14849. [Google Scholar] [CrossRef] [PubMed]
- Rivabene, R.; Straface, E.; Giammarioli, A.M.; Rainaldi, G.; Malorni, W. Combined effect of 3-aminobenzamide and N-acetylcysteine on HIV replication in chronically infected U937 cells. Redox Rep. 1997, 3, 145–151. [Google Scholar] [CrossRef]
- Rom, S.; Reichenbach, N.L.; Dykstra, H.; Persidsky, Y. The dual action of poly(ADP-ribose) polymerase -1 (PARP-1) inhibition in HIV-1 infection: HIV-1 LTR inhibition and diminution in Rho GTPase activity. Front. Microbiol. 2015, 6, 878. [Google Scholar] [CrossRef]
- Sharma, R.; Hartman, T.E.; Beites, T.; Kim, J.H.; Eoh, H.; Engelhart, C.A.; Zhu, L.; Wilson, D.J.; Aldrich, C.C.; Ehrt, S.; et al. Metabolically distinct roles of NAD synthetase and NAD kinase define the essentiality of NAD and NADP in Mycobacterium tuberculosis. mBio 2023, 14, e0034023. [Google Scholar] [CrossRef] [PubMed]
- Thirunavukkarasu, S.; Ahmed, M.; Rosa, B.A.; Boothby, M.; Cho, S.H.; Rangel-Moreno, J.; Mbandi, S.K.; Schreiber, V.; Gupta, A.; Zuniga, J.; et al. Poly(ADP-ribose) polymerase 9 mediates early protection against Mycobacterium tuberculosis infection by regulating type I IFN production. J. Clin. Investig. 2023, 133. [Google Scholar] [CrossRef] [PubMed]
- Alqarni, M.H.; Foudah, A.I.; Muharram, M.M.; Labrou, N.E. The Pleiotropic Function of Human Sirtuins as Modulators of Metabolic Pathways and Viral Infections. Cells 2021, 10, 460. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.W.; Sun, L.J.; Liu, W.; Zhao, Y.H.; Kang, P.; Yan, B.Z. Hepatitis C virus core protein induces hepatic metabolism disorders through down-regulation of the SIRT1-AMPK signaling pathway. Int. J. Infect. Dis. 2013, 17, e539–e545. [Google Scholar] [CrossRef]
- Ren, J.H.; Tao, Y.; Zhang, Z.Z.; Chen, W.X.; Cai, X.F.; Chen, K.; Ko, B.C.; Song, C.L.; Ran, L.K.; Li, W.Y.; et al. Sirtuin 1 regulates hepatitis B virus transcription and replication by targeting transcription factor AP-1. J. Virol. 2014, 88, 2442–2451. [Google Scholar] [CrossRef]
- He, M.; Gao, S.J. A novel role of SIRT1 in gammaherpesvirus latency and replication. Cell Cycle 2014, 13, 3328–3330. [Google Scholar] [CrossRef]
- Li, H.R.; Liu, Q.; Zhu, C.L.; Sun, X.Y.; Sun, C.Y.; Yu, C.M.; Li, P.; Deng, X.M.; Wang, J.F. β-Nicotinamide mononucleotide activates NAD+/SIRT1 pathway and attenuates inflammatory and oxidative responses in the hippocampus regions of septic mice. Redox Biol. 2023, 63, 102745. [Google Scholar] [CrossRef] [PubMed]
- Abdellatif, M.; Sedej, S.; Kroemer, G. NAD(+) Metabolism in Cardiac Health, Aging, and Disease. Circulation 2021, 144, 1795–1817. [Google Scholar] [CrossRef]
- Smulan, L.J.; Martinez, N.; Kiritsy, M.C.; Kativhu, C.; Cavallo, K.; Sassetti, C.M.; Singhal, A.; Remold, H.G.; Kornfeld, H. Sirtuin 3 Downregulation in Mycobacterium tuberculosis-Infected Macrophages Reprograms Mitochondrial Metabolism and Promotes Cell Death. mBio 2021, 12, e03140-20. [Google Scholar] [CrossRef]
- Piedra-Quintero, Z.L.; Wilson, Z.; Nava, P.; Guerau-de-Arellano, M. CD38: An Immunomodulatory Molecule in Inflammation and Autoimmunity. Front. Immunol. 2020, 11, 597959. [Google Scholar] [CrossRef] [PubMed]
- Montali, I.; Ceccatelli Berti, C.; Morselli, M.; Acerbi, G.; Barili, V.; Pedrazzi, G.; Montanini, B.; Boni, C.; Alfieri, A.; Pesci, M.; et al. Deregulated intracellular pathways define novel molecular targets for HBV-specific CD8 T cell reconstitution in chronic hepatitis B. J. Hepatol. 2023, 79, 50–60. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Alba, J.C.; Abrego-Peredo, A.; Gallardo-Hernández, C.; Pérez-Lara, J.; Santiago-Cruz, W.; Jiang, W.; Espinosa, E. HIV Disease Progression: Overexpression of the Ectoenzyme CD38 as a Contributory Factor? Bioessays 2019, 41, e1800128. [Google Scholar] [CrossRef] [PubMed]
- Dash, S.; Dash, C.; Pandhare, J. Therapeutic Significance of microRNA-Mediated Regulation of PARP-1 in SARS-CoV-2 Infection. Noncoding RNA 2021, 7, 60. [Google Scholar] [CrossRef] [PubMed]
- Zheng, M.; Schultz, M.B.; Sinclair, D.A. NAD(+) in COVID-19 and viral infections. Trends Immunol. 2022, 43, 283–295. [Google Scholar] [CrossRef]
- Horenstein, A.L.; Faini, A.C.; Malavasi, F. CD38 in the age of COVID-19: A medical perspective. Physiol. Rev. 2021, 101, 1457–1486. [Google Scholar] [CrossRef]
- Isman, A.; Nyquist, A.; Strecker, B.; Harinath, G.; Lee, V.; Zhang, X.; Zalzala, S. Low-dose naltrexone and NAD+ for the treatment of patients with persistent fatigue symptoms after COVID-19. Brain Behav. Immun. Health 2024, 36, 100733. [Google Scholar] [CrossRef] [PubMed]
- Legler, F.; Meyer-Arndt, L.; Mödl, L.; Kedor, C.; Freitag, H.; Stein, E.; Hoppmann, U.; Rust, R.; Wittke, K.; Siebert, N.; et al. Long-term symptom severity and clinical biomarkers in post-COVID-19/chronic fatigue syndrome: Results from a prospective observational cohort. EClinicalMedicine 2023, 63, 102146. [Google Scholar] [CrossRef]
- Xiao, N.; Nie, M.; Pang, H.; Wang, B.; Hu, J.; Meng, X.; Li, K.; Ran, X.; Long, Q.; Deng, H.; et al. Integrated cytokine and metabolite analysis reveals immunometabolic reprogramming in COVID-19 patients with therapeutic implications. Nat. Commun. 2021, 12, 1618. [Google Scholar] [CrossRef] [PubMed]
- Izadpanah, A.; Mudd, J.C.; Garcia, J.G.N.; Srivastav, S.; Abdel-Mohsen, M.; Palmer, C.; Goldman, A.R.; Kolls, J.K.; Qin, X.; Rappaport, J. SARS-CoV-2 infection dysregulates NAD metabolism. Front. Immunol. 2023, 14, 1158455. [Google Scholar] [CrossRef] [PubMed]
- Duan, T.; Xing, C.; Chu, J.; Deng, X.; Du, Y.; Liu, X.; Hu, Y.; Qian, C.; Yin, B.; Wang, H.Y.; et al. ACE2-dependent and -independent SARS-CoV-2 entries dictate viral replication and inflammatory response during infection. Nat. Cell Biol. 2024, 26, 628–644. [Google Scholar] [CrossRef] [PubMed]
- Ramasamy, S.; Subbian, S. Critical Determinants of Cytokine Storm and Type I Interferon Response in COVID-19 Pathogenesis. Clin. Microbiol. Rev. 2021, 34, e00299-20. [Google Scholar] [CrossRef] [PubMed]
- Omran, H.M.; Almaliki, M.S. Influence of NAD+ as an ageing-related immunomodulator on COVID 19 infection: A hypothesis. J. Infect. Public Health 2020, 13, 1196–1201. [Google Scholar] [CrossRef]
- Zhang, S.; Wang, J.; Wang, L.; Aliyari, S.; Cheng, G. SARS-CoV-2 virus NSP14 Impairs NRF2/HMOX1 activation by targeting Sirtuin 1. Cell. Mol. Immunol. 2022, 19, 872–882. [Google Scholar] [CrossRef]
- Walter, M.; Chen, I.P.; Vallejo-Gracia, A.; Kim, I.J.; Bielska, O.; Lam, V.L.; Hayashi, J.M.; Cruz, A.; Shah, S.; Soveg, F.W.; et al. SIRT5 is a proviral factor that interacts with SARS-CoV-2 Nsp14 protein. PLoS Pathog. 2022, 18, e1010811. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Wang, H.; Zhang, H.; Sui, L.; Li, L.; Xu, W.; Du, S.; Hao, P.; Jiang, Y.; Chen, J.; et al. The global succinylation of SARS-CoV-2-infected host cells reveals drug targets. Proc. Natl. Acad. Sci. USA 2022, 119, e2123065119. [Google Scholar] [CrossRef]
- Koutsakos, M.; Rowntree, L.C.; Hensen, L.; Chua, B.Y.; van de Sandt, C.E.; Habel, J.R.; Zhang, W.; Jia, X.; Kedzierski, L.; Ashhurst, T.M.; et al. Integrated immune dynamics define correlates of COVID-19 severity and antibody responses. Cell Rep. Med. 2021, 2, 100208. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, M.W.; Cheng, Y.; Zhang, J.; Jiang, X.M.; Wang, L.; Deng, J.; Wang, P.H. Increasing host cellular receptor-angiotensin-converting enzyme 2 expression by coronavirus may facilitate 2019-nCoV (or SARS-CoV-2) infection. J. Med. Virol. 2020, 92, 2693–2701. [Google Scholar] [CrossRef]
- Du, J.; Wei, L.; Li, G.; Hua, M.; Sun, Y.; Wang, D.; Han, K.; Yan, Y.; Song, C.; Song, R.; et al. Persistent High Percentage of HLA-DR(+)CD38(high) CD8(+) T Cells Associated With Immune Disorder and Disease Severity of COVID-19. Front. Immunol. 2021, 12, 735125. [Google Scholar] [CrossRef] [PubMed]
- Hogan, K.A.; Chini, C.C.S.; Chini, E.N. The Multi-faceted Ecto-enzyme CD38: Roles in Immunomodulation, Cancer, Aging, and Metabolic Diseases. Front. Immunol. 2019, 10, 1187. [Google Scholar] [CrossRef]
- Linden, J.; Koch-Nolte, F.; Dahl, G. Purine Release, Metabolism, and Signaling in the Inflammatory Response. Annu. Rev. Immunol. 2019, 37, 325–347. [Google Scholar] [CrossRef]
- Nascimento, D.C.; Viacava, P.R.; Ferreira, R.G.; Damaceno, M.A.; Piñeros, A.R.; Melo, P.H.; Donate, P.B.; Toller-Kawahisa, J.E.; Zoppi, D.; Veras, F.P.; et al. Sepsis expands a CD39(+) plasmablast population that promotes immunosuppression via adenosine-mediated inhibition of macrophage antimicrobial activity. Immunity 2021, 54, 2024–2041.e8. [Google Scholar] [CrossRef] [PubMed]
- Yakymiv, Y.; Marchisio, S.; Ortolan, E.; Bracci, C.; Senetta, R.; Rumore, M.R.; Tampieri, C.; Fia, M.; Ribero, S.; Funaro, A.; et al. CD39/CD73 dysregulation and adenosine metabolism contribute to T-cell immunosuppression in patients with Sézary syndrome. Blood 2023, 141, 111–116. [Google Scholar] [CrossRef] [PubMed]
- Sica, A.; Colombo, M.P.; Trama, A.; Horn, L.; Garassino, M.C.; Torri, V. Immunometabolic Status of COVID-19 Cancer Patients. Physiol. Rev. 2020, 100, 1839–1850. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Fedele, G.; Celardo, I.; Loh, S.H.Y.; Martins, L.M. Parp mutations protect from mitochondrial toxicity in Alzheimer’s disease. Cell Death Dis. 2021, 12, 651. [Google Scholar] [CrossRef]
- Hou, Y.; Wei, Y.; Lautrup, S.; Yang, B.; Wang, Y.; Cordonnier, S.; Mattson, M.P.; Croteau, D.L.; Bohr, V.A. NAD(+) supplementation reduces neuroinflammation and cell senescence in a transgenic mouse model of Alzheimer’s disease via cGAS-STING. Proc. Natl. Acad. Sci. USA 2021, 118, e2011226118. [Google Scholar] [CrossRef]
- Acharya, G.; Mani, C.; Sah, N.; Saamarthy, K.; Young, R.; Reedy, M.B.; Sobol, R.W.; Palle, K. CHK1 inhibitor induced PARylation by targeting PARG causes excessive replication and metabolic stress and overcomes chemoresistance in ovarian cancer. Cell Death Discov. 2024, 10, 278. [Google Scholar] [CrossRef] [PubMed]
- Lau, C.H.; Seow, K.M.; Chen, K.H. The Molecular Mechanisms of Actions, Effects, and Clinical Implications of PARP Inhibitors in Epithelial Ovarian Cancers: A Systematic Review. Int. J. Mol. Sci. 2022, 23, 8125. [Google Scholar] [CrossRef]
- Kim, C.; Chen, C.; Yu, Y. Avoid the trap: Targeting PARP1 beyond human malignancy. Cell Chem. Biol. 2021, 28, 456–462. [Google Scholar] [CrossRef]
- Novak, E.A.; Crawford, E.C.; Mentrup, H.L.; Griffith, B.D.; Fletcher, D.M.; Flanagan, M.R.; Schneider, C.; Firek, B.; Rogers, M.B.; Morowitz, M.J.; et al. Epithelial NAD(+) depletion drives mitochondrial dysfunction and contributes to intestinal inflammation. Front. Immunol. 2023, 14, 1231700. [Google Scholar] [CrossRef]
- Cantó, C.; Houtkooper, R.H.; Pirinen, E.; Youn, D.Y.; Oosterveer, M.H.; Cen, Y.; Fernandez-Marcos, P.J.; Yamamoto, H.; Andreux, P.A.; Cettour-Rose, P.; et al. The NAD(+) precursor nicotinamide riboside enhances oxidative metabolism and protects against high-fat diet-induced obesity. Cell Metab. 2012, 15, 838–847. [Google Scholar] [CrossRef]
- Li, D.J.; Sun, S.J.; Fu, J.T.; Ouyang, S.X.; Zhao, Q.J.; Su, L.; Ji, Q.X.; Sun, D.Y.; Zhu, J.H.; Zhang, G.Y.; et al. NAD(+)-boosting therapy alleviates nonalcoholic fatty liver disease via stimulating a novel exerkine Fndc5/irisin. Theranostics 2021, 11, 4381–4402. [Google Scholar] [CrossRef]
- Katsuyama, E.; Suarez-Fueyo, A.; Bradley, S.J.; Mizui, M.; Marin, A.V.; Mulki, L.; Krishfield, S.; Malavasi, F.; Yoon, J.; Sui, S.J.H.; et al. The CD38/NAD/SIRTUIN1/EZH2 Axis Mitigates Cytotoxic CD8 T Cell Function and Identifies Patients with SLE Prone to Infections. Cell Rep. 2020, 30, 112–123.e4. [Google Scholar] [CrossRef]
- Vaisitti, T.; Audrito, V.; Serra, S.; Buonincontri, R.; Sociali, G.; Mannino, E.; Pagnani, A.; Zucchetto, A.; Tissino, E.; Vitale, C.; et al. The enzymatic activities of CD38 enhance CLL growth and trafficking: Implications for therapeutic targeting. Leukemia 2015, 29, 356–368. [Google Scholar] [CrossRef]
- Becherini, P.; Soncini, D.; Ravera, S.; Gelli, E.; Martinuzzi, C.; Giorgetti, G.; Cagnetta, A.; Guolo, F.; Ivaldi, F.; Miglino, M.; et al. CD38-Induced Metabolic Dysfunction Primes Multiple Myeloma Cells for NAD(+)-Lowering Agents. Antioxidants 2023, 12, 494. [Google Scholar] [CrossRef]
- Wang, W.; Hu, Y.; Yang, C.; Zhu, S.; Wang, X.; Zhang, Z.; Deng, H. Decreased NAD Activates STAT3 and Integrin Pathways to Drive Epithelial-Mesenchymal Transition. Mol. Cell Proteomics 2018, 17, 2005–2017. [Google Scholar] [CrossRef]
- Verdin, E. NAD+ in aging, metabolism, and neurodegeneration. Science 2015, 350, 1208–1213. [Google Scholar] [CrossRef]
- Chini, C.C.S.; Zeidler, J.D.; Kashyap, S.; Warner, G.; Chini, E.N. Evolving concepts in NAD(+) metabolism. Cell Metab. 2021, 33, 1076–1087. [Google Scholar] [CrossRef]
- Imai, S.; Guarente, L. NAD+ and sirtuins in aging and disease. Trends Cell Biol. 2014, 24, 464–471. [Google Scholar] [CrossRef] [PubMed]
- Wallace, D.C. Bioenergetic origins of complexity and disease. Cold Spring Harb. Symp. Quant. Biol. 2011, 76, 1–16. [Google Scholar] [CrossRef]
- Wilson, N.; Kataura, T.; Korsgen, M.E.; Sun, C.; Sarkar, S.; Korolchuk, V.I. The autophagy-NAD axis in longevity and disease. Trends Cell Biol. 2023, 33, 788–802. [Google Scholar] [CrossRef]
- Ji, Z.; Liu, G.H.; Qu, J. Mitochondrial sirtuins, metabolism, and aging. J. Genet. Genomics 2022, 49, 287–298. [Google Scholar] [CrossRef]
- Perico, L.; Remuzzi, G.; Benigni, A. Sirtuins in kidney health and disease. Nat. Rev. Nephrol. 2024, 20, 313–329. [Google Scholar] [CrossRef] [PubMed]
- Kaeberlein, M.; McVey, M.; Guarente, L. The SIR2/3/4 complex and SIR2 alone promote longevity in Saccharomyces cerevisiae by two different mechanisms. Genes. Dev. 1999, 13, 2570–2580. [Google Scholar] [CrossRef] [PubMed]
- He, M.; Chiang, H.H.; Luo, H.; Zheng, Z.; Qiao, Q.; Wang, L.; Tan, M.; Ohkubo, R.; Mu, W.C.; Zhao, S.; et al. An Acetylation Switch of the NLRP3 Inflammasome Regulates Aging-Associated Chronic Inflammation and Insulin Resistance. Cell Metab. 2020, 31, 580–591.e5. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wang, X.; Li, X.K.; Lv, S.J.; Wang, H.P.; Liu, Y.; Zhou, J.; Gong, H.; Chen, X.F.; Ren, S.C.; et al. Sirtuin 2 deficiency aggravates ageing-induced vascular remodelling in humans and mice. Eur. Heart J. 2023, 44, 2746–2759. [Google Scholar] [CrossRef] [PubMed]
- Fang, E.F.; Hou, Y.; Palikaras, K.; Adriaanse, B.A.; Kerr, J.S.; Yang, B.; Lautrup, S.; Hasan-Olive, M.M.; Caponio, D.; Dan, X.; et al. Mitophagy inhibits amyloid-β and tau pathology and reverses cognitive deficits in models of Alzheimer’s disease. Nat. Neurosci. 2019, 22, 401–412. [Google Scholar] [CrossRef] [PubMed]
- Membrez, M.; Migliavacca, E.; Christen, S.; Yaku, K.; Trieu, J.; Lee, A.K.; Morandini, F.; Giner, M.P.; Stiner, J.; Makarov, M.V.; et al. Trigonelline is an NAD(+) precursor that improves muscle function during ageing and is reduced in human sarcopenia. Nat. Metab. 2024, 6, 433–447. [Google Scholar] [CrossRef] [PubMed]
- Kartsonaki, C.; Baillie, J.K.; Barrio, N.G.; Baruch, J.; Beane, A.; Blumberg, L.; Bozza, F.; Broadley, T.; Burrell, A.; Carson, G.; et al. Characteristics and outcomes of an international cohort of 600,000 hospitalized patients with COVID-19. Int. J. Epidemiol. 2023, 52, 355–376. [Google Scholar] [CrossRef]
- Romero Starke, K.; Reissig, D.; Petereit-Haack, G.; Schmauder, S.; Nienhaus, A.; Seidler, A. The isolated effect of age on the risk of COVID-19 severe outcomes: A systematic review with meta-analysis. BMJ Glob. Health 2021, 6, e006434. [Google Scholar] [CrossRef]
- Wiersinga, W.J.; Rhodes, A.; Cheng, A.C.; Peacock, S.J.; Prescott, H.C. Pathophysiology, Transmission, Diagnosis, and Treatment of Coronavirus Disease 2019 (COVID-19): A Review. JAMA 2020, 324, 782–793. [Google Scholar] [CrossRef]
- O’Driscoll, M.; Ribeiro Dos Santos, G.; Wang, L.; Cummings, D.A.T.; Azman, A.S.; Paireau, J.; Fontanet, A.; Cauchemez, S.; Salje, H. Age-specific mortality and immunity patterns of SARS-CoV-2. Nature 2021, 590, 140–145. [Google Scholar] [CrossRef] [PubMed]
- CDC. Available online: https://www.cdc.gov/mmwr/volumes/73/wr/mm7331a1.htm?s_cid=mm7331a1_w (accessed on 22 September 2024).
- Minchella, P.A.; Chanda, D.; Hines, J.Z.; Fwoloshi, S.; Itoh, M.; Kampamba, D.; Chirwa, R.; Sivile, S.; Zyambo, K.D.; Agolory, S.; et al. Clinical Characteristics and Outcomes of Patients Hospitalized With COVID-19 During the First 4 Waves in Zambia. JAMA Netw. Open 2022, 5, e2246152. [Google Scholar] [CrossRef]
- Kaml, M.; Weiskirchner, I.; Keller, M.; Luft, T.; Hoster, E.; Hasford, J.; Young, L.; Bartlett, B.; Neuner, C.; Fischer, K.H.; et al. Booster vaccination in the elderly: Their success depends on the vaccine type applied earlier in life as well as on pre-vaccination antibody titers. Vaccine 2006, 24, 6808–6811. [Google Scholar] [CrossRef] [PubMed]
- Yoshikawa, T.T. Epidemiology and unique aspects of aging and infectious diseases. Clin. Infect. Dis. 2000, 30, 931–933. [Google Scholar] [CrossRef] [PubMed]
- Zinatizadeh, M.R.; Zarandi, P.K.; Zinatizadeh, M.; Yousefi, M.H.; Amani, J.; Rezaei, N. Efficacy of mRNA, adenoviral vector, and perfusion protein COVID-19 vaccines. Biomed. Pharmacother. 2022, 146, 112527. [Google Scholar] [CrossRef] [PubMed]
- Franceschi, C. Inflammaging as a major characteristic of old people: Can it be prevented or cured? Nutr. Rev. 2007, 65 Pt 2, S173–S176. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Liang, Q.; Ren, Y.; Guo, C.; Ge, X.; Wang, L.; Cheng, Q.; Luo, P.; Zhang, Y.; Han, X. Immunosenescence: Molecular mechanisms and diseases. Signal Transduct. Target. Ther. 2023, 8, 200. [Google Scholar] [CrossRef] [PubMed]
- Cao, X.; Li, W.; Wang, T.; Ran, D.; Davalos, V.; Planas-Serra, L.; Pujol, A.; Esteller, M.; Wang, X.; Yu, H. Accelerated biological aging in COVID-19 patients. Nat. Commun. 2022, 13, 2135. [Google Scholar] [CrossRef] [PubMed]
- Gioia, U.; Tavella, S.; Martínez-Orellana, P.; Cicio, G.; Colliva, A.; Ceccon, M.; Cabrini, M.; Henriques, A.C.; Fumagalli, V.; Paldino, A.; et al. SARS-CoV-2 infection induces DNA damage, through CHK1 degradation and impaired 53BP1 recruitment, and cellular senescence. Nat. Cell Biol. 2023, 25, 550–564. [Google Scholar] [CrossRef] [PubMed]
- Tsuji, S.; Minami, S.; Hashimoto, R.; Konishi, Y.; Suzuki, T.; Kondo, T.; Sasai, M.; Torii, S.; Ono, C.; Shichinohe, S.; et al. SARS-CoV-2 infection triggers paracrine senescence and leads to a sustained senescence-associated inflammatory response. Nat. Aging 2022, 2, 115–124. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, C.A.; Tchkonia, T.; Niedernhofer, L.J.; Robbins, P.D.; Kirkland, J.L.; Lee, S. COVID-19 and cellular senescence. Nat. Rev. Immunol. 2023, 23, 251–263. [Google Scholar] [CrossRef]
- Prata, L.; Ovsyannikova, I.G.; Tchkonia, T.; Kirkland, J.L. Senescent cell clearance by the immune system: Emerging therapeutic opportunities. Semin. Immunol. 2018, 40, 101275. [Google Scholar] [CrossRef] [PubMed]
- Lekva, T.; Ueland, T.; Halvorsen, B.; Murphy, S.L.; Dyrhol-Riise, A.M.; Tveita, A.; Finbråten, A.K.; Mathiessen, A.; Müller, K.E.; Aaløkken, T.M.; et al. Markers of cellular senescence is associated with persistent pulmonary pathology after COVID-19 infection. Infect. Dis. 2022, 54, 918–923. [Google Scholar] [CrossRef]
- Lieberman, N.A.P.; Peddu, V.; Xie, H.; Shrestha, L.; Huang, M.L.; Mears, M.C.; Cajimat, M.N.; Bente, D.A.; Shi, P.Y.; Bovier, F.; et al. In vivo antiviral host transcriptional response to SARS-CoV-2 by viral load, sex, and age. PLoS Biol. 2020, 18, e3000849. [Google Scholar] [CrossRef]
- Heer, C.D.; Sanderson, D.J.; Voth, L.S.; Alhammad, Y.M.O.; Schmidt, M.S.; Trammell, S.A.J.; Perlman, S.; Cohen, M.S.; Fehr, A.R.; Brenner, C. Coronavirus infection and PARP expression dysregulate the NAD metabolome: An actionable component of innate immunity. J. Biol. Chem. 2020, 295, 17986–17996. [Google Scholar] [CrossRef]
- Freeberg, K.A.; Ludwig, K.R.; Chonchol, M.; Seals, D.R.; Rossman, M.J. NAD(+)-boosting compounds enhance nitric oxide production and prevent oxidative stress in endothelial cells exposed to plasma from patients with COVID-19. Nitric Oxide 2023, 140–141, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Deng, Y.; Pang, H.; Ma, T.; Ye, Q.; Chen, Q.; Chen, H.; Hu, Z.; Qin, C.F.; Xu, Z. Treatment of SARS-CoV-2-induced pneumonia with NAD(+) and NMN in two mouse models. Cell Discov. 2022, 8, 38. [Google Scholar] [CrossRef] [PubMed]
- Huizenga, R. Dramatic Clinical Improvement in Nine Consecutive Acutely Ill Elderly COVID-19 Patients Treated with a Nicotinamide Mononucleotide Cocktail: A Case Series. SSRN 2020. Available online: https://ssrn.com/abstract=3677428 (accessed on 22 September 2024).
- Altay, O.; Arif, M.; Li, X.; Yang, H.; Aydın, M.; Alkurt, G.; Kim, W.; Akyol, D.; Zhang, C.; Dinler-Doganay, G.; et al. Combined Metabolic Activators Accelerates Recovery in Mild-to-Moderate COVID-19. Adv. Sci. 2021, 8, e2101222. [Google Scholar] [CrossRef] [PubMed]
- Raines, N.H.; Cheung, M.D.; Wilson, L.S.; Edberg, J.C.; Erdmann, N.B.; Schmaier, A.A.; Berryhill, T.F.; Manickas-Hill, Z.; Li, J.Z.; Yu, X.G.; et al. Nicotinamide Adenine Dinucleotide Biosynthetic Impairment and Urinary Metabolomic Alterations Observed in Hospitalized Adults With COVID-19-Related Acute Kidney Injury. Kidney Int. Rep. 2021, 6, 3002–3013. [Google Scholar] [CrossRef]
- Raines, N.H.; Ganatra, S.; Nissaisorakarn, P.; Pandit, A.; Morales, A.; Asnani, A.; Sadrolashrafi, M.; Maheshwari, R.; Patel, R.; Bang, V.; et al. Niacinamide May Be Associated with Improved Outcomes in COVID-19-Related Acute Kidney Injury: An Observational Study. Kidney360 2021, 2, 33–41. [Google Scholar] [CrossRef] [PubMed]
- Stone, N.E.; Jaramillo, S.A.; Jones, A.N.; Vazquez, A.J.; Martz, M.; Versluis, L.M.; Raniere, M.O.; Nunnally, H.E.; Zarn, K.E.; Nottingham, R.; et al. Stenoparib, an Inhibitor of Cellular Poly(ADP-Ribose) Polymerase, Blocks Replication of the SARS-CoV-2 and HCoV-NL63 Human Coronaviruses In Vitro. mBio 2021, 12, e03495-20. [Google Scholar] [CrossRef]
- Papp, H.; Tóth, E.; Bóvári-Biri, J.; Bánfai, K.; Juhász, P.; Mahdi, M.; Russo, L.C.; Bajusz, D.; Sipos, A.; Petri, L.; et al. The PARP inhibitor rucaparib blocks SARS-CoV-2 virus binding to cells and the immune reaction in models of COVID-19. Br. J. Pharmacol. 2024, 181, 4782–4803. [Google Scholar] [CrossRef] [PubMed]
- Zarn, K.E.; Jaramillo, S.A.; Zapata, A.R.; Stone, N.E.; Jones, A.N.; Nunnally, H.E.; Settles, E.W.; Ng, K.; Keim, P.S.; Knudsen, S.; et al. Stenoparib, an inhibitor of cellular poly (ADP-ribose) polymerases (PARPs), blocks in vitro replication of SARS-CoV-2 variants. PLoS ONE 2022, 17, e0272916. [Google Scholar] [CrossRef] [PubMed]
- Pasquereau, S.; Nehme, Z.; Haidar Ahmad, S.; Daouad, F.; Van Assche, J.; Wallet, C.; Schwartz, C.; Rohr, O.; Morot-Bizot, S.; Herbein, G. Resveratrol Inhibits HCoV-229E and SARS-CoV-2 Coronavirus Replication In Vitro. Viruses 2021, 13, 354. [Google Scholar] [CrossRef]
- Russo, C.; Valle, M.S.; Malaguarnera, L.; Romano, I.R.; Malaguarnera, L. Comparison of Vitamin D and Resveratrol Performances in COVID-19. Nutrients 2023, 15, 2639. [Google Scholar] [CrossRef]
- Ramdani, L.H.; Bachari, K. Potential therapeutic effects of Resveratrol against SARS-CoV-2. Acta Virol. 2020, 64, 276–280. [Google Scholar] [CrossRef]
- McCreary, M.R.; Schnell, P.M.; Rhoda, D.A. Randomized double-blind placebo-controlled proof-of-concept trial of resveratrol for outpatient treatment of mild coronavirus disease (COVID-19). Sci. Rep. 2022, 12, 10978. [Google Scholar] [CrossRef] [PubMed]
- Saeedi-Boroujeni, A.; Mahmoudian-Sani, M.R. Anti-inflammatory potential of Quercetin in COVID-19 treatment. J. Inflamm. 2021, 18, 3. [Google Scholar] [CrossRef] [PubMed]
- Terao, T.; Naduka, T.; Ikeda, D.; Fukumoto, A.; Kamura, Y.; Kuzume, A.; Tabata, R.; Tsushima, T.; Miura, D.; Narita, K.; et al. Depletion of CD38-positive regulatory T cells by anti-CD38 monoclonal antibodies induces a durable response to SARS-CoV-2 vaccination in patients with plasma cell dyscrasia. Br. J. Haematol. 2022, 197, 417–421. [Google Scholar] [CrossRef] [PubMed]
- Nahi, H.; Chrobok, M.; Gran, C.; Lund, J.; Gruber, A.; Gahrton, G.; Ljungman, P.; Wagner, A.K.; Alici, E. Infectious complications and NK cell depletion following daratumumab treatment of Multiple Myeloma. PLoS ONE 2019, 14, e0211927. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
| Enzymes | Disease | Model | Source | Observations | References |
|---|---|---|---|---|---|
| PARPs | HBV | HBV-infected HepaRG/HepG2 | - | NAD+ consuming and PARP1 recognizes broken dsDNA and promotes DNA repair by (NHEJ) pathways | [53] |
| HIV | HIV-infected U937 cells with TNFa. treatment | Flow Laboratories, Irvine, UK | PARP1 overexpression and NAD+ consuming | [54] | |
| HIV-infected MDM with PARP inhibitor treatment | University of Nebraska Medical Center, Department of Pharmacology and Experimental Neuroscience | suppression of HIV-1 replication by obstructing HIV-1 LTR activation | [55] | ||
| MTB | Mtb H37Rv wt, NadE-DUC, and PpnK-DUC strains | - | NAD+ depletion and arrested growth of Mtb | [56] | |
| Parp9-/- mice | The Jackson Laboratory | increased susceptibility to Mtb infection mediated by type I IFN signaling | [57] | ||
| SIRTs | HCV | HCV core gene transfected HepG2 cells | - | decreasing NAD/NADH ratio and the activity of SIRT1, glucose, and lipid metabolism disorder | [59] |
| HBV | HBV-transfected HepG2 cells | - | the upregulation of SIRT1 augmented HBV replication | [60] | |
| KSHV | KSHV-infected primary effusion lymphoma (PEL) cell line BCBL-1 | - | Low NAD+ level disrupts viral latency by inhibiting SIRT1 function | [61] | |
| Sepsis | Septic mice induced by cecal ligation and puncture (CLP) | GemPharmatech Laboratory Animals (Nanjing, China) | Low NAD+ level and NAD+/SIRT1 pathway was inhibited | [62] | |
| MTB | J2 macrophages, BMDM, Sirt3-/- mice | University of Massachusetts Medical School and The Jackson Laboratory | SIRT3 downregulation, increased ROS, and cell death | [64] | |
| CD38 | HBV | CD8 T cells from patients with chronic active hepatitis | - | CD38 overexpression leads to NAD+ depletion and dysregulation of DNA repair mechanisms | [66] |
| HIV | CD8 T cells from HIV patients | - | Increased CD38 activity promotes NAD+ consumption and exacerbates mitochondrial oxidative stress | [67] | |
| RSV | RSV infected MDDC | - | The increased production of type I IFNs activates CD38 and CD38/cADPR pathway | [41] |
| Enzymes | Diseases | Models | Source | Observations | References |
|---|---|---|---|---|---|
| PARPs | AD | Aβ-Arc-expressing flies with PARP mutation | - | Increased NAD+ level and improved mitochondrial function | [89] |
| APP/PS1 mice with NR treatment | The Jackson Laboratory | Aberrant activation of DNA sensing pathways and the level of neuroinflammation are reduced | [90] | ||
| Ovarian cancer | OC cell lines OVCAR8 and SKOV3 exposed to CHK1 inhibition and PARG inhibitor | ATCC | Increased DNA damage, activation of PARP1/2, and decrease in NAD+ level | [91] | |
| SIRTs | IBD | Mice subjected to experimental colitis | The Jackson Laboratory | NAD+ depletion, decrease in SIRT1 activity, and mitochondrial dysfunction | [94] |
| T2D | High-fat diet-induced T2D mice and age-induced diabetic mice | The Jackson Laboratory | Decrease in NAD+ level, suppression of SIRT1 activity, and metabolic complications | [16] | |
| Obesity | High-fat diet-induced mice | Charles River | Decrease in NAD+ level, suppression of SIRT1/3 activity, and oxidative metabolism | [95] | |
| NAFLD | high-fat diet and methionine/choline-deficient diet-induced NAFLD mice with NR treatment | Sino-British SIPPR/BK Lab Animal Ltd. | Increase in NAD+ level and SIRT2 activity, reversion of hepatic steatosis, and steatohepatitis | [96] | |
| CD38 | SLE | CD8CD38high T cells from patients with SLE | ATCC | Decrease in MAD+ level and cytotoxicity | [97] |
| CLL | CLL cells from Peripheral blood samples of patients with CLL | - | NAD+ depletion, increase in Ca2+ concentrations, and CLL aggressiveness | [98] | |
| MM | MM cell lines HMCL with CD38 overexpressing | ATCC or DSMZ | NAD+ depletion, NAD+ depletion, and mitochondrial metabolism reprogramming | [99] | |
| NSCLC/Liver Cancer | CD38+ A549/CD38+ HepG2 | cell bank of the Chinese Academy of Sciences | Decrease in NAD+ level, promoting EMT | [100] |
| Trial ID | Interventions | Clinical Phase | Study Type | Results | References |
|---|---|---|---|---|---|
| NCT04573153 | NR + serine + L-carnitine tartrate + N-acetylcysteine + hydroxychloroquine (CMA) | II/III | Randomized, placebo-controlled | In patients using CMAs, the time to complete recovery is significantly shorter, and plasma levels of proteins and metabolites associated with inflammation and antioxidant metabolism are significantly improved. | [135] |
| NCT04407390 | NR 1 g/d | II | Randomized, double-blind, placebo-controlled | No results available yet. | |
| NCT04818216 | NR 1 g/d | II | Randomized, double-blind, placebo-controlled | Patients in the nicotinamide riboside group had higher levels of NAD+ in whole blood. | |
| NCT05175768 | NMN, NMN+L-leucine | Not applicable | Randomized, double-blind, placebo-controlled | No results available yet. | |
| NCT05038488 | MIB-626 | II | Randomized, double-blind, placebo-controlled | No results available yet. | |
| NCT04751604 | Nicotinamide | Not applicable | Randomized, double-blind, placebo-controlled | No results available yet. | |
| NCT04910230 | Nicotinamide | Not applicable | Randomized, double-blind, placebo-controlled | No clinical difference observed between therapy and placebo group. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Song, S.; Gan, J.; Long, Q.; Gao, Z.; Zheng, Y. Decoding NAD+ Metabolism in COVID-19: Implications for Immune Modulation and Therapy. Vaccines 2025, 13, 1. https://doi.org/10.3390/vaccines13010001
Song S, Gan J, Long Q, Gao Z, Zheng Y. Decoding NAD+ Metabolism in COVID-19: Implications for Immune Modulation and Therapy. Vaccines. 2025; 13(1):1. https://doi.org/10.3390/vaccines13010001
Chicago/Turabian StyleSong, Shixu, Jialing Gan, Qiuyue Long, Zhancheng Gao, and Yali Zheng. 2025. "Decoding NAD+ Metabolism in COVID-19: Implications for Immune Modulation and Therapy" Vaccines 13, no. 1: 1. https://doi.org/10.3390/vaccines13010001
APA StyleSong, S., Gan, J., Long, Q., Gao, Z., & Zheng, Y. (2025). Decoding NAD+ Metabolism in COVID-19: Implications for Immune Modulation and Therapy. Vaccines, 13(1), 1. https://doi.org/10.3390/vaccines13010001