

Targeting PI3K Signaling to Overcome Tumor Immunosuppression: Synergistic Strategies to Enhance Cancer Vaccine Efficacy
 ,
,
Abstract
:1. Introduction
2. The Relationship Between the PI3K Signaling Pathway and Tumor Immunity
3. PI3K Inhibitors as a Monotherapy
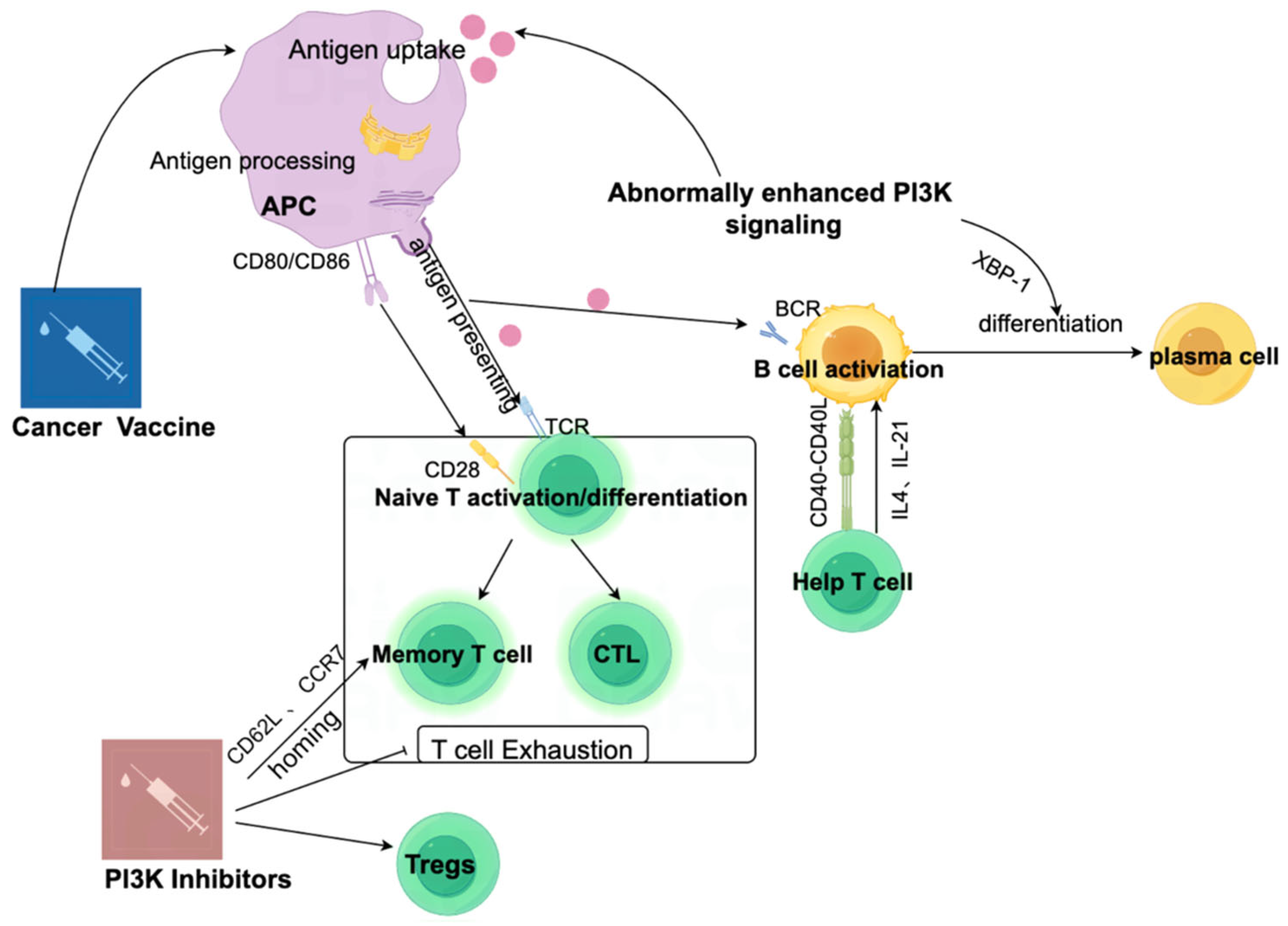
4. PI3K Inhibitors and Cancer Vaccines: A Promising Combination Strategy to Enhance Antitumor Immunity
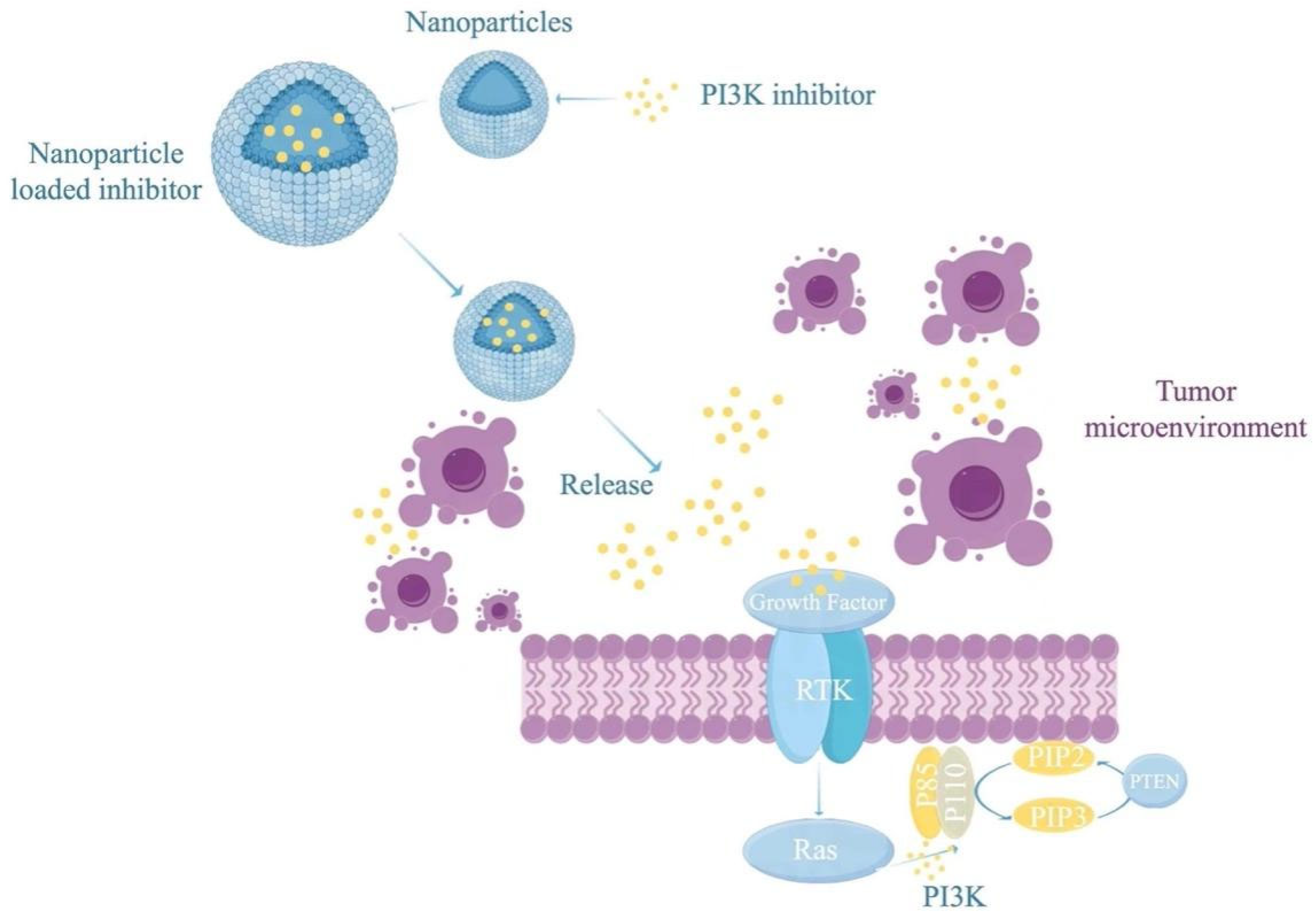
5. Improving PI3K Inhibitors for Combination with Cancer Vaccines
6. Clinical Translation and Challenges
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ott, P.A.; Hu, Z.; Keskin, D.B.; Shukla, S.A.; Sun, J.; Bozym, D.J.; Zhang, W.; Luoma, A.; Giobbie-Hurder, A.; Peter, L.; et al. An immunogenic personal neoantigen vaccine for patients with melanoma. Nature 2017, 547, 217–221. [Google Scholar] [CrossRef] [PubMed]
- Sahin, U.; Derhovanessian, E.; Miller, M.; Kloke, B.P.; Simon, P.; Lower, M.; Bukur, V.; Tadmor, A.D.; Luxemburger, U.; Schrors, B.; et al. Personalized RNA mutanome vaccines mobilize poly-specific therapeutic immunity against cancer. Nature 2017, 547, 222–226. [Google Scholar] [CrossRef] [PubMed]
- Joyce, J.A.; Fearon, D.T. T cell exclusion, immune privilege, and the tumor microenvironment. Science 2015, 348, 74–80. [Google Scholar] [CrossRef] [PubMed]
- Beatty, G.L.; Gladney, W.L. Immune escape mechanisms as a guide for cancer immunotherapy. Clin. Cancer Res. 2015, 21, 687–692. [Google Scholar] [CrossRef]
- Binnewies, M.; Roberts, E.W.; Kersten, K.; Chan, V.; Fearon, D.F.; Merad, M.; Coussens, L.M.; Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Hedrick, C.C.; et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat. Med. 2018, 24, 541–550. [Google Scholar] [CrossRef]
- Sai, J.; Owens, P.; Novitskiy, S.V.; Hawkins, O.E.; Vilgelm, A.E.; Yang, J.; Sobolik, T.; Lavender, N.; Johnson, A.C.; McClain, C.; et al. PI3K Inhibition Reduces Mammary Tumor Growth and Facilitates Antitumor Immunity and Anti-PD1 Responses. Clin. Cancer Res. 2017, 23, 3371–3384. [Google Scholar] [CrossRef]
- Glaviano, A.; Foo, A.S.C.; Lam, H.Y.; Yap, K.C.H.; Jacot, W.; Jones, R.H.; Eng, H.; Nair, M.G.; Makvandi, P.; Geoerger, B.; et al. PI3K/AKT/mTOR signaling transduction pathway and targeted therapies in cancer. Mol. Cancer 2023, 22, 138. [Google Scholar] [CrossRef]
- Chang, C.H.; Pearce, E.L. Emerging concepts of T cell metabolism as a target of immunotherapy. Nat. Immunol. 2016, 17, 364–368. [Google Scholar] [CrossRef]
- Chang, C.H.; Qiu, J.; O’Sullivan, D.; Buck, M.D.; Noguchi, T.; Curtis, J.D.; Chen, Q.; Gindin, M.; Gubin, M.M.; van der Windt, G.J.; et al. Metabolic Competition in the Tumor Microenvironment Is a Driver of Cancer Progression. Cell 2015, 162, 1229–1241. [Google Scholar] [CrossRef]
- Gabrilovich, D.I.; Nagaraj, S. Myeloid-derived suppressor cells as regulators of the immune system. Nat. Rev. Immunol. 2009, 9, 162–174. [Google Scholar] [CrossRef]
- Pompura, S.L.; Dominguez-Villar, M. The PI3K/AKT signaling pathway in regulatory T-cell development, stability, and function. J. Leukoc. Biol. 2018, 103, 1065–1076. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Shi, H.; Zhang, B.; Ou, X.; Ma, Q.; Chen, Y.; Shu, P.; Li, D.; Wang, Y. Myeloid-derived suppressor cells as immunosuppressive regulators and therapeutic targets in cancer. Signal Transduct. Target. Ther. 2021, 6, 362. [Google Scholar] [CrossRef] [PubMed]
- Beurel, E.; Jope, R.S. Differential regulation of STAT family members by glycogen synthase kinase-3. J. Biol. Chem. 2008, 283, 21934–21944. [Google Scholar] [CrossRef] [PubMed]
- Fruman, D.A.; Chiu, H.; Hopkins, B.D.; Bagrodia, S.; Cantley, L.C.; Abraham, R.T. The PI3K Pathway in Human Disease. Cell 2017, 170, 605–635. [Google Scholar] [CrossRef]
- Wang, Y.; Xiang, Y.; Xin, V.W.; Wang, X.W.; Peng, X.C.; Liu, X.Q.; Wang, D.; Li, N.; Cheng, J.T.; Lyv, Y.N.; et al. Dendritic cell biology and its role in tumor immunotherapy. J. Hematol. Oncol. 2020, 13, 107. [Google Scholar] [CrossRef]
- Jafari, M.; Ghadami, E.; Dadkhah, T.; Akhavan-Niaki, H. PI3k/AKT signaling pathway: Erythropoiesis and beyond. J. Cell Physiol. 2019, 234, 2373–2385. [Google Scholar] [CrossRef]
- He, Y.; Sun, M.M.; Zhang, G.G.; Yang, J.; Chen, K.S.; Xu, W.W.; Li, B. Targeting PI3K/Akt signal transduction for cancer therapy. Signal Transduct. Target. Ther. 2021, 6, 425. [Google Scholar] [CrossRef]
- Carnevalli, L.S.; Sinclair, C.; Taylor, M.A.; Gutierrez, P.M.; Langdon, S.; Coenen-Stass, A.M.L.; Mooney, L.; Hughes, A.; Jarvis, L.; Staniszewska, A.; et al. PI3Kα/δ inhibition promotes anti-tumor immunity through direct enhancement of effector CD8(+) T-cell activity. J. Immunother. Cancer 2018, 6, 158. [Google Scholar] [CrossRef]
- Li, P.; Zhou, D.; Chen, D.; Cheng, Y.; Chen, Y.; Lin, Z.; Zhang, X.; Huang, Z.; Cai, J.; Huang, W.; et al. Tumor-secreted IFI35 promotes proliferation and cytotoxic activity of CD8(+) T cells through PI3K/AKT/mTOR signaling pathway in colorectal cancer. J. Biomed. Sci. 2023, 30, 47. [Google Scholar] [CrossRef]
- Zhang, X.; Zeng, Y.; Qu, Q.; Zhu, J.; Liu, Z.; Ning, W.; Zeng, H.; Zhang, N.; Du, W.; Chen, C.; et al. PD-L1 induced by IFN-γ from tumor-associated macrophages via the JAK/STAT3 and PI3K/AKT signaling pathways promoted progression of lung cancer. Int. J. Clin. Oncol. 2017, 22, 1026–1033. [Google Scholar] [CrossRef]
- Noh, K.H.; Kang, T.H.; Kim, J.H.; Pai, S.I.; Lin, K.Y.; Hung, C.F.; Wu, T.C.; Kim, T.W. Activation of Akt as a mechanism for tumor immune evasion. Mol. Ther. 2009, 17, 439–447. [Google Scholar] [CrossRef] [PubMed]
- Yan, C.; Yang, J.; Saleh, N.; Chen, S.C.; Ayers, G.D.; Abramson, V.G.; Mayer, I.A.; Richmond, A. Inhibition of the PI3K/mTOR Pathway in Breast Cancer to Enhance Response to Immune Checkpoint Inhibitors in Breast Cancer. Int. J. Mol. Sci. 2021, 22, 5207. [Google Scholar] [CrossRef] [PubMed]
- Fu, Z.Y.; Huang, Y.; Lian, L.S.; Huang, H.T.; Zhan, S.F.; Cai, Y.; Li, J.X.; Liu, X.H. Potential of semen coicis in enhancing the anti-tumor effects of PD-1 inhibitor on A549 cell lines by blocking the PI3K-AKT-mTOR pathway. Clin. Transl. Oncol. 2024, 26, 2250–2261. [Google Scholar] [CrossRef] [PubMed]
- Tufail, M.; Wan, W.D.; Jiang, C.; Li, N. Targeting PI3K/AKT/mTOR signaling to overcome drug resistance in cancer. Chem. Biol. Interact. 2024, 396, 111055. [Google Scholar] [CrossRef]
- Laplante, M.; Sabatini, D.M. mTOR signaling in growth control and disease. Cell 2012, 149, 274–293. [Google Scholar] [CrossRef]
- Chen, Y.; Xu, Z.; Sun, H.; Ouyang, X.; Han, Y.; Yu, H.; Wu, N.; Xie, Y.; Su, B. Regulation of CD8(+) T memory and exhaustion by the mTOR signals. Cell Mol. Immunol. 2023, 20, 1023–1039. [Google Scholar] [CrossRef]
- Ma, S.; Ming, Y.; Wu, J.; Cui, G. Cellular metabolism regulates the differentiation and function of T-cell subsets. Cell Mol. Immunol. 2024, 21, 419–435. [Google Scholar] [CrossRef]
- Jellusova, J.; Rickert, R.C. The PI3K pathway in B cell metabolism. Crit. Rev. Biochem. Mol. Biol. 2016, 51, 359–378. [Google Scholar] [CrossRef]
- Lanahan, S.M.; Wymann, M.P.; Lucas, C.L. The role of PI3Kgamma in the immune system: New insights and translational implications. Nat. Rev. Immunol. 2022, 22, 687–700. [Google Scholar] [CrossRef]
- Haddadi, N.; Lin, Y.; Travis, G.; Simpson, A.M.; Nassif, N.T.; McGowan, E.M. PTEN/PTENP1: ‘Regulating the regulator of RTK-dependent PI3K/Akt signalling’, new targets for cancer therapy. Mol. Cancer 2018, 17, 37. [Google Scholar] [CrossRef]
- Vergadi, E.; Ieronymaki, E.; Lyroni, K.; Vaporidi, K.; Tsatsanis, C. Akt Signaling Pathway in Macrophage Activation and M1/M2 Polarization. J. Immunol. 2017, 198, 1006–1014. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Russell, S.N.; Steiner, K.; O’Neill, E.; Jones, K.I. Targeting PI3K-gamma in myeloid driven tumour immune suppression: A systematic review and meta-analysis of the preclinical literature. Cancer Immunol. Immunother. 2024, 73, 204. [Google Scholar] [CrossRef] [PubMed]
- So, L.; Yea, S.S.; Oak, J.S.; Lu, M.; Manmadhan, A.; Ke, Q.H.; Janes, M.R.; Kessler, L.V.; Kucharski, J.M.; Li, L.S.; et al. Selective inhibition of phosphoinositide 3-kinase p110alpha preserves lymphocyte function. J. Biol. Chem. 2013, 288, 5718–5731. [Google Scholar] [CrossRef] [PubMed]
- Guillermet-Guibert, J.; Bjorklof, K.; Salpekar, A.; Gonella, C.; Ramadani, F.; Bilancio, A.; Meek, S.; Smith, A.J.; Okkenhaug, K.; Vanhaesebroeck, B. The p110beta isoform of phosphoinositide 3-kinase signals downstream of G protein-coupled receptors and is functionally redundant with p110gamma. Proc. Natl. Acad. Sci. USA 2008, 105, 8292–8297. [Google Scholar] [CrossRef]
- Vanhaesebroeck, B.; Whitehead, M.A.; Pineiro, R. Molecules in medicine mini-review: Isoforms of PI3K in biology and disease. J. Mol. Med. 2016, 94, 5–11. [Google Scholar] [CrossRef]
- Mantovani, A.; Sica, A.; Sozzani, S.; Allavena, P.; Vecchi, A.; Locati, M. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol. 2004, 25, 677–686. [Google Scholar] [CrossRef]
- Liang, X.H.; Chen, X.Y.; Yan, Y.; Cheng, A.Y.; Lin, J.Y.; Jiang, Y.X.; Chen, H.Z.; Jin, J.M.; Luan, X. Targeting metabolism to enhance immunotherapy within tumor microenvironment. Acta Pharmacol. Sin. 2024, 45, 2011–2022. [Google Scholar] [CrossRef]
- Brown, J.R.; Byrd, J.C.; Coutre, S.E.; Benson, D.M.; Flinn, I.W.; Wagner-Johnston, N.D.; Spurgeon, S.E.; Kahl, B.S.; Bello, C.; Webb, H.K.; et al. Idelalisib, an inhibitor of phosphatidylinositol 3-kinase p110delta, for relapsed/refractory chronic lymphocytic leukemia. Blood 2014, 123, 3390–3397. [Google Scholar] [CrossRef]
- Eschweiler, S.; Ramirez-Suastegui, C.; Li, Y.; King, E.; Chudley, L.; Thomas, J.; Wood, O.; von Witzleben, A.; Jeffrey, D.; McCann, K.; et al. Intermittent PI3Kdelta inhibition sustains anti-tumour immunity and curbs irAEs. Nature 2022, 605, 741–746. [Google Scholar] [CrossRef]
- Hong, D.S.; Postow, M.; Chmielowski, B.; Sullivan, R.; Patnaik, A.; Cohen, E.E.W.; Shapiro, G.; Steuer, C.; Gutierrez, M.; Yeckes-Rodin, H.; et al. Eganelisib, a First-in-Class PI3Kgamma Inhibitor, in Patients with Advanced Solid Tumors: Results of the Phase 1/1b MARIO-1 Trial. Clin. Cancer Res. 2023, 29, 2210–2219. [Google Scholar] [CrossRef]
- Matasar, M.J.; Capra, M.; Ozcan, M.; Lv, F.; Li, W.; Yanez, E.; Sapunarova, K.; Lin, T.; Jin, J.; Jurczak, W.; et al. Copanlisib plus rituximab versus placebo plus rituximab in patients with relapsed indolent non-Hodgkin lymphoma (CHRONOS-3): A double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Oncol. 2021, 22, 678–689. [Google Scholar] [CrossRef] [PubMed]
- Baharom, F.; Hermans, D.; Delamarre, L.; Seder, R.A. Vax-Innate: Improving therapeutic cancer vaccines by modulating T cells and the tumour microenvironment. Nat. Rev. Immunol. 2024, 25, 195–211. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Nie, J.; Ma, X.; Wei, Y.; Peng, Y.; Wei, X. Targeting PI3K in cancer: Mechanisms and advances in clinical trials. Mol. Cancer 2019, 18, 26. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Chen, J.; Xu, Z.; Yang, B.; He, Q.; Luo, P.; Yan, H.; Yang, X. Development and safety of PI3K inhibitors in cancer. Arch. Toxicol. 2023, 97, 635–650. [Google Scholar] [CrossRef]
- Curigliano, G.; Shah, R.R. Safety and Tolerability of Phosphatidylinositol-3-Kinase (PI3K) Inhibitors in Oncology. Drug Saf. 2019, 42, 247–262. [Google Scholar] [CrossRef]
- Castel, P.; Toska, E.; Engelman, J.A.; Scaltriti, M. The present and future of PI3K inhibitors for cancer therapy. Nat. Cancer 2021, 2, 587–597. [Google Scholar] [CrossRef]
- Hanker, A.B.; Kaklamani, V.; Arteaga, C.L. Challenges for the Clinical Development of PI3K Inhibitors: Strategies to Improve Their Impact in Solid Tumors. Cancer Discov. 2019, 9, 482–491. [Google Scholar] [CrossRef]
- Zhang, Y.; Yan, H.; Xu, Z.; Yang, B.; Luo, P.; He, Q. Molecular basis for class side effects associated with PI3K/AKT/mTOR pathway inhibitors. Expert Opin. Drug Metab. Toxicol. 2019, 15, 767–774. [Google Scholar] [CrossRef]
- Mishra, R.; Patel, H.; Alanazi, S.; Kilroy, M.K.; Garrett, J.T. PI3K Inhibitors in Cancer: Clinical Implications and Adverse Effects. Int. J. Mol. Sci. 2021, 22, 3464. [Google Scholar] [CrossRef]
- Ku, A.T.; Young, A.I.J.; Ibrahim, A.A.; Bu, W.; Jiang, W.; Lin, M.; Williams, L.C.; McCue, B.L.; Miles, G.; Nagi, C.; et al. Short-term PI3K Inhibition Prevents Breast Cancer in Preclinical Models. Cancer Prev. Res. 2023, 16, 65–73. [Google Scholar] [CrossRef]
- Bertho, M.; Patsouris, A.; Augereau, P.; Robert, M.; Frenel, J.S.; Blonz, C.; Campone, M. A pharmacokinetic evaluation of alpelisib for the treatment of HR+, HER2-negative, PIK3CA-mutated advanced or metastatic breast cancer. Expert Opin. Drug Metab. Toxicol. 2021, 17, 139–152. [Google Scholar] [CrossRef] [PubMed]
- Perez, C.R.; De Palma, M. Engineering dendritic cell vaccines to improve cancer immunotherapy. Nat. Commun. 2019, 10, 5408. [Google Scholar] [CrossRef] [PubMed]
- Zelenetz, A.D.; Barrientos, J.C.; Brown, J.R.; Coiffier, B.; Delgado, J.; Egyed, M.; Ghia, P.; Illes, A.; Jurczak, W.; Marlton, P.; et al. Idelalisib or placebo in combination with bendamustine and rituximab in patients with relapsed or refractory chronic lymphocytic leukaemia: Interim results from a phase 3, randomised, double-blind, placebo-controlled trial. Lancet Oncol. 2017, 18, 297–311. [Google Scholar] [CrossRef] [PubMed]
- Kuttke, M.; Hromadova, D.; Yildirim, C.; Brunner, J.S.; Vogel, A.; Paar, H.; Peters, S.; Weber, M.; Hofmann, M.; Kerndl, M.; et al. PI3K Signaling in Dendritic Cells Aggravates DSS-Induced Colitis. Front. Immunol. 2022, 13, 695576. [Google Scholar] [CrossRef]
- Lee, D.; Kozurek, E.C.; Abdullah, M.; Wong, E.J.; Li, R.; Liu, Z.S.; Nguyen, H.D.; Dickerson, E.B.; Kim, J.H. PIK3CA mutation fortifies molecular determinants for immune signaling in vascular cancers. Cancer Gene Ther. 2024, 32, 254–267. [Google Scholar] [CrossRef]
- Zhao, T.; Cai, Y.; Jiang, Y.; He, X.; Wei, Y.; Yu, Y.; Tian, X. Vaccine adjuvants: Mechanisms and platforms. Signal Transduct. Target. Ther. 2023, 8, 283. [Google Scholar] [CrossRef]
- Pulendran, B.S.; Arunachalam, P.; O’Hagan, D.T. Emerging concepts in the science of vaccine adjuvants. Nat. Rev. Drug Discov. 2021, 20, 454–475. [Google Scholar] [CrossRef]
- Wang, X.; Ding, J.; Meng, L.H. PI3K isoform-selective inhibitors: Next-generation targeted cancer therapies. Acta Pharmacol. Sin. 2015, 36, 1170–1176. [Google Scholar] [CrossRef]
- Hu, J.; Fu, S.; Zhan, Z.; Zhang, J. Advancements in dual-target inhibitors of PI3K for tumor therapy: Clinical progress, development strategies, prospects. Eur. J. Med. Chem. 2024, 265, 116109. [Google Scholar] [CrossRef]
- Chellappa, S.; Kushekhar, K.; Munthe, L.A.; Tjonnfjord, G.E.; Aandahl, E.M.; Okkenhaug, K.; Tasken, K. The PI3K p110delta Isoform Inhibitor Idelalisib Preferentially Inhibits Human Regulatory T Cell Function. J. Immunol. 2019, 202, 1397–1405. [Google Scholar] [CrossRef]
- Mizrachi, A.; Shamay, Y.; Shah, J.; Brook, S.; Soong, J.; Rajasekhar, V.K.; Humm, J.L.; Healey, J.H.; Powell, S.N.; Baselga, J.; et al. Tumour-specific PI3K inhibition via nanoparticle-targeted delivery in head and neck squamous cell carcinoma. Nat. Commun. 2017, 8, 14292. [Google Scholar] [CrossRef] [PubMed]
- Bordbar-Khiabani, A.; Gasik, M. Smart Hydrogels for Advanced Drug Delivery Systems. Int. J. Mol. Sci. 2022, 23, 3665. [Google Scholar] [CrossRef] [PubMed]
- Talekar, M.; Ganta, S.; Amiji, M.; Jamieson, S.; Kendall, J.; Denny, W.A.; Garg, S. Development of PIK-75 nanosuspension formulation with enhanced delivery efficiency and cytotoxicity for targeted anti-cancer therapy. Int. J. Pharm. 2013, 450, 278–289. [Google Scholar] [CrossRef]
- Whitehead, C.E.; Ziemke, E.K.; Frankowski-McGregor, C.L.; Mumby, R.A.; Chung, J.; Li, J.; Osher, N.; Coker, O.; Baladandayuthapani, V.; Kopetz, S.; et al. A first-in-class selective inhibitor of EGFR and PI3K offers a single-molecule approach to targeting adaptive resistance. Nat. Cancer 2024, 5, 1250–1266. [Google Scholar] [CrossRef] [PubMed]
- Zhavoronkov, A.; Ivanenkov, Y.A.; Aliper, A.; Veselov, M.S.; Aladinskiy, V.A.; Aladinskaya, A.V.; Terentiev, V.A.; Polykovskiy, D.A.; Kuznetsov, M.D.; Asadulaev, A.; et al. Deep learning enables rapid identification of potent DDR1 kinase inhibitors. Nat. Biotechnol. 2019, 37, 1038–1040. [Google Scholar] [CrossRef]
- Lee, R.Y.; Wu, Y.; Goh, D.; Tan, V.; Ng, C.W.; Lim, J.C.T.; Lau, M.C.; Yeong, J.P.S. Application of Artificial Intelligence to In Vitro Tumor Modeling and Characterization of the Tumor Microenvironment. Adv. Healthc. Mater. 2023, 12, e2202457. [Google Scholar] [CrossRef]
- Weigelt, B.; Warne, P.H.; Downward, J. PIK3CA mutation, but not PTEN loss of function, determines the sensitivity of breast cancer cells to mTOR inhibitory drugs. Oncogene 2011, 30, 3222–3233. [Google Scholar] [CrossRef]
- Janku, F.; Hong, D.S.; Fu, S.; Piha-Paul, S.A.; Naing, A.; Falchook, G.S.; Tsimberidou, A.M.; Stepanek, V.M.; Moulder, S.L.; Lee, J.J.; et al. Assessing PIK3CA and PTEN in early-phase trials with PI3K/AKT/mTOR inhibitors. Cell Rep. 2014, 6, 377–387. [Google Scholar] [CrossRef]
- Janku, F.; Tsimberidou, A.M.; Garrido-Laguna, I.; Wang, X.; Luthra, R.; Hong, D.S.; Naing, A.; Falchook, G.S.; Moroney, J.W.; Piha-Paul, S.A.; et al. PIK3CA mutations in patients with advanced cancers treated with PI3K/AKT/mTOR axis inhibitors. Mol. Cancer Ther. 2011, 10, 558–565. [Google Scholar] [CrossRef]
- Andre, F.; Ciruelos, E.; Rubovszky, G.; Campone, M.; Loibl, S.; Rugo, H.S.; Iwata, H.; Conte, P.; Mayer, I.A.; Kaufman, B.; et al. Alpelisib for PIK3CA-Mutated, Hormone Receptor-Positive Advanced Breast Cancer. N. Engl. J. Med. 2019, 380, 1929–1940. [Google Scholar] [CrossRef]
- Li, J.; Wu, S.; Zhang, K.; Sun, X.; Lin, W.; Wang, C.; Lin, S. Clustered Regularly Interspaced Short Palindromic Repeat/CRISPR-Associated Protein and Its Utility All at Sea: Status, Challenges, and Prospects. Microorganisms 2024, 12, 118. [Google Scholar] [CrossRef] [PubMed]
- Shalem, O.; Sanjana, N.E.; Zhang, F. High-throughput functional genomics using CRISPR-Cas9. Nat. Rev. Genet. 2015, 16, 299–311. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Saand, M.A.; Huang, L.; Abdelaal, W.B.; Zhang, J.; Wu, Y.; Li, J.; Sirohi, M.H.; Wang, F. Applications of Multi-Omics Technologies for Crop Improvement. Front. Plant Sci. 2021, 12, 563953. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez Reyes, C.D.; Alejo-Jacuinde, G.; Perez Sanchez, B.; Chavez Reyes, J.; Onigbinde, S.; Mogut, D.; Hernandez-Jasso, I.; Calderon-Vallejo, D.; Quintanar, J.L.; Mechref, Y. Multi Omics Applications in Biological Systems. Curr. Issues Mol. Biol. 2024, 46, 5777–5793. [Google Scholar] [CrossRef]
- Greenwell, I.B.; Ip, A.; Cohen, J.B. PI3K Inhibitors: Understanding Toxicity Mechanisms and Management. Oncology 2017, 31, 821–828. [Google Scholar]
- Di Leo, A.; Johnston, S.; Lee, K.S.; Ciruelos, E.; Lonning, P.E.; Janni, W.; O’Regan, R.; Mouret-Reynier, M.A.; Kalev, D.; Egle, D.; et al. Buparlisib plus fulvestrant in postmenopausal women with hormone-receptor-positive, HER2-negative, advanced breast cancer progressing on or after mTOR inhibition (BELLE-3): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2018, 19, 87–100. [Google Scholar] [CrossRef]
- Baselga, J.; Im, S.A.; Iwata, H.; Cortes, J.; De Laurentiis, M.; Jiang, Z.; Arteaga, C.L.; Jonat, W.; Clemons, M.; Ito, Y.; et al. Buparlisib plus fulvestrant versus placebo plus fulvestrant in postmenopausal, hormone receptor-positive, HER2-negative, advanced breast cancer (BELLE-2): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2017, 18, 904–916. [Google Scholar] [CrossRef]
- Fan, T.; Zhang, M.; Yang, J.; Zhu, Z.; Cao, W.; Dong, C. Therapeutic cancer vaccines: Advancements, challenges, and prospects. Signal Transduct. Target. Ther. 2023, 8, 450. [Google Scholar] [CrossRef]
- Juric, D.; Janku, F.; Rodón, J.; Burris, H.A.; Mayer, I.A.; Schuler, M.; Seggewiss-Bernhardt, R.; Gil-Martin, M.; Middleton, M.R.; Baselga, J.; et al. Alpelisib Plus Fulvestrant in PIK3CA-Altered and PIK3CA-Wild-Type Estrogen Receptor-Positive Advanced Breast Cancer: A Phase 1b Clinical Trial. JAMA Oncol. 2019, 5, e184475. [Google Scholar] [CrossRef]
- Mayer, I.A.; Prat, A.; Egle, D.; Blau, S.; Fidalgo, J.A.P.; Gnant, M.; Fasching, P.A.; Colleoni, M.; Wolff, A.C.; Winer, E.P.; et al. A Phase II Randomized Study of Neoadjuvant Letrozole Plus Alpelisib for Hormone Receptor-Positive, Human Epidermal Growth Factor Receptor 2-Negative Breast Cancer (NEO-ORB). Clin. Cancer Res. 2019, 25, 2975–2987. [Google Scholar] [CrossRef]
- Rugo, H.S.; Seneviratne, L.; Beck, J.T.; Glaspy, J.A.; Peguero, J.A.; Pluard, T.J.; Dhillon, N.; Hwang, L.C.; Nangia, C.; Mayer, I.A.; et al. Prevention of everolimus-related stomatitis in women with hormone receptor-positive, HER2-negative metastatic breast cancer using dexamethasone mouthwash (SWISH): A single-arm, phase 2 trial. Lancet Oncol. 2017, 18, 654–662. [Google Scholar] [CrossRef] [PubMed]
- Flinn, I.W.; Miller, C.B.; Ardeshna, K.M.; Tetreault, S.; Assouline, S.E.; Mayer, J.; Merli, M.; Lunin, S.D.; Pettitt, A.R.; Nagy, Z.; et al. DYNAMO: A Phase II Study of Duvelisib (IPI-145) in Patients With Refractory Indolent Non-Hodgkin Lymphoma. J. Clin. Oncol. 2019, 37, 912–922. [Google Scholar] [CrossRef] [PubMed]
- Tobeigei, F.H.; Gahtani, R.M.; Shaikh, A.; Al Ali, A.; Kameli, N.; Kamli, H.; Rajagopalan, P. Computational high-throughput screening and in vitro approaches identify CB-006-3; A novel PI3K-BRAF(V600E) dual targeted inhibitor against melanoma. Oncol. Res. 2021, 29, 305–318. [Google Scholar] [CrossRef] [PubMed]
- Tzenaki, N.; Xenou, L.; Goulielmaki, E.; Tsapara, A.; Voudouri, I.; Antoniou, A.; Valianatos, G.; Tzardi, M.; De Bree, E.; Berdiaki, A.; et al. A combined opposite targeting of p110δ PI3K and RhoA abrogates skin cancer. Commun. Biol. 2024, 7, 26. [Google Scholar] [CrossRef]
- Ueyama, A.; Nogami, W.; Nashiki, K.; Haruna, M.; Miwa, H.; Hagiwara, M.; Nagira, M.; Wada, H.; Nagira, Y. Immunotherapy Targeting CCR8+ Regulatory T Cells Induces Antitumor Effects via Dramatic Changes to the Intratumor CD8+ T Cell Profile. J. Immunol. 2023, 211, 673–682. [Google Scholar] [CrossRef]
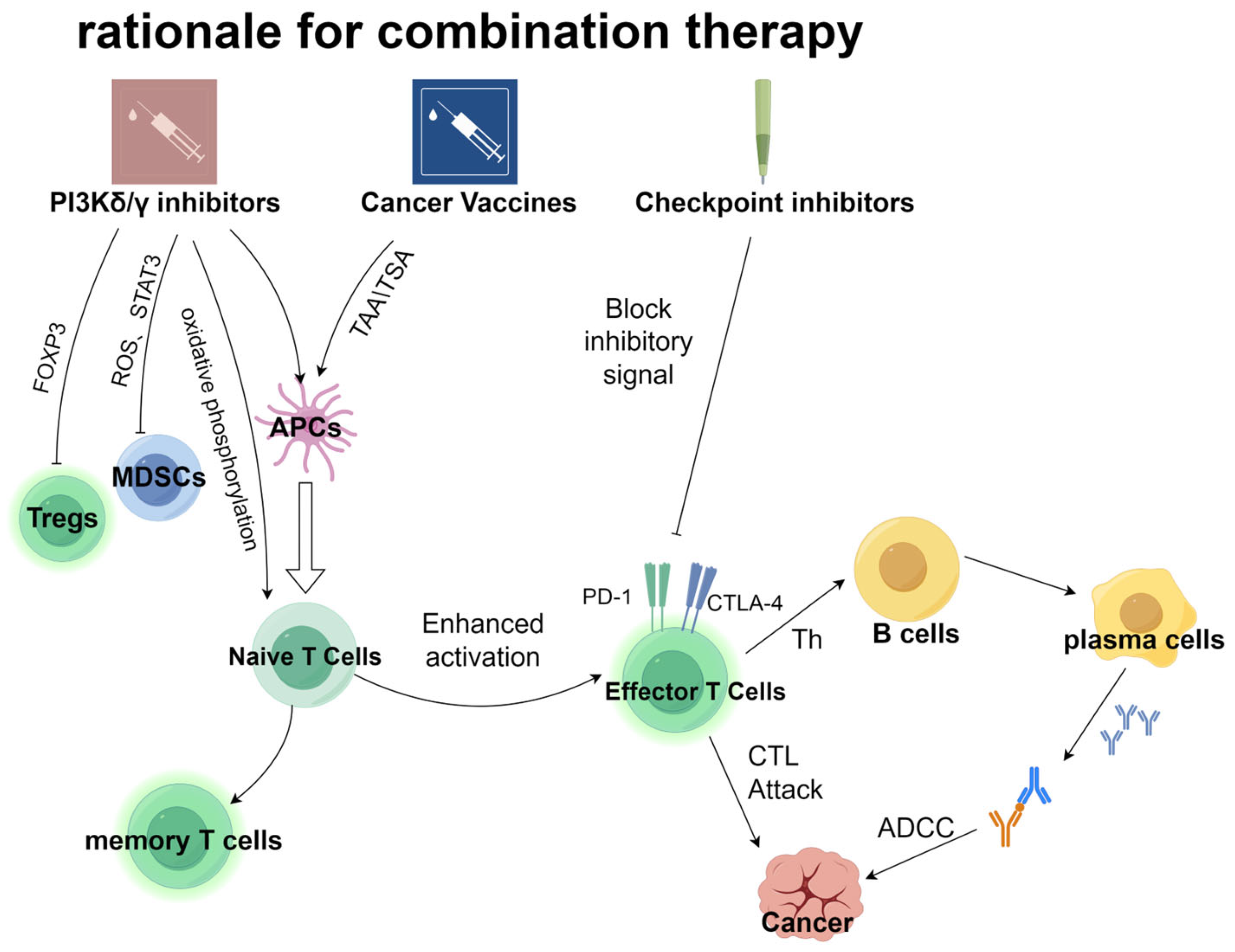
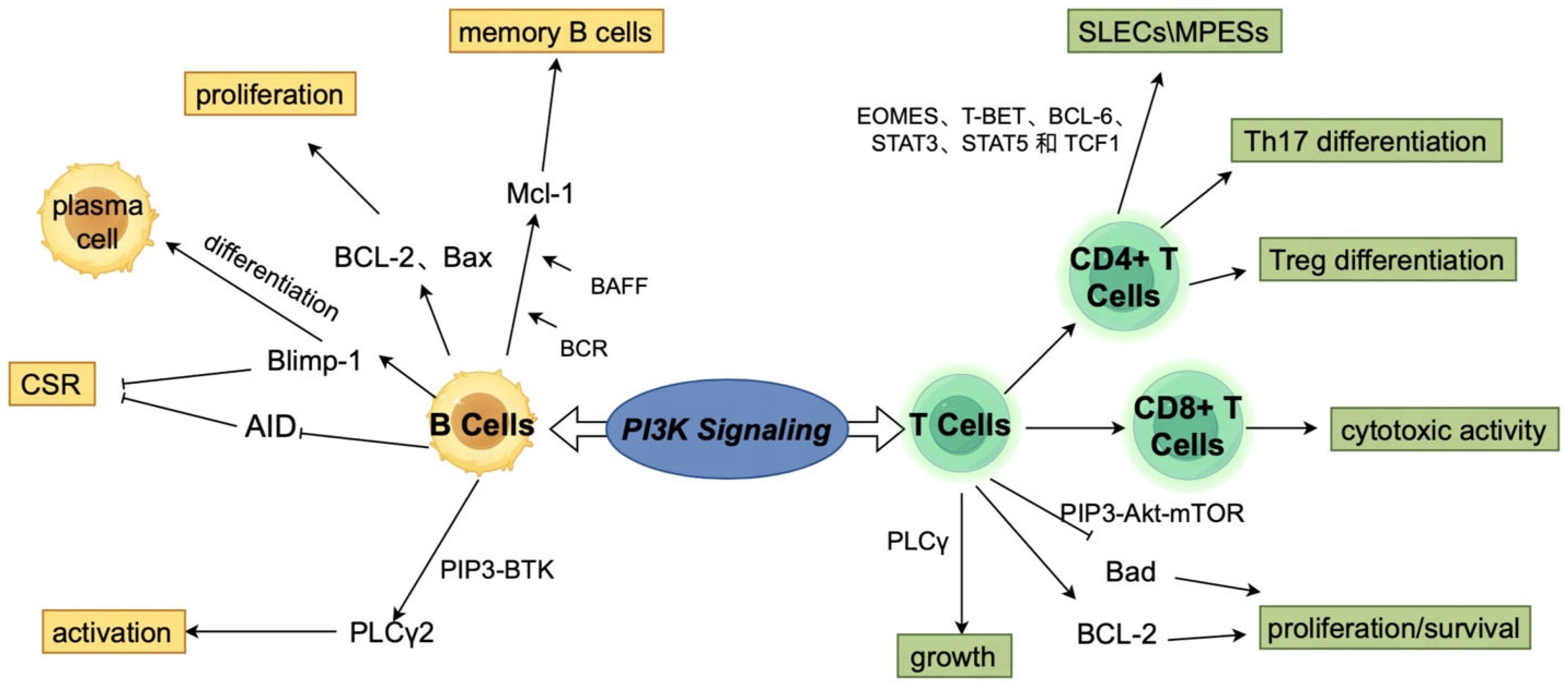
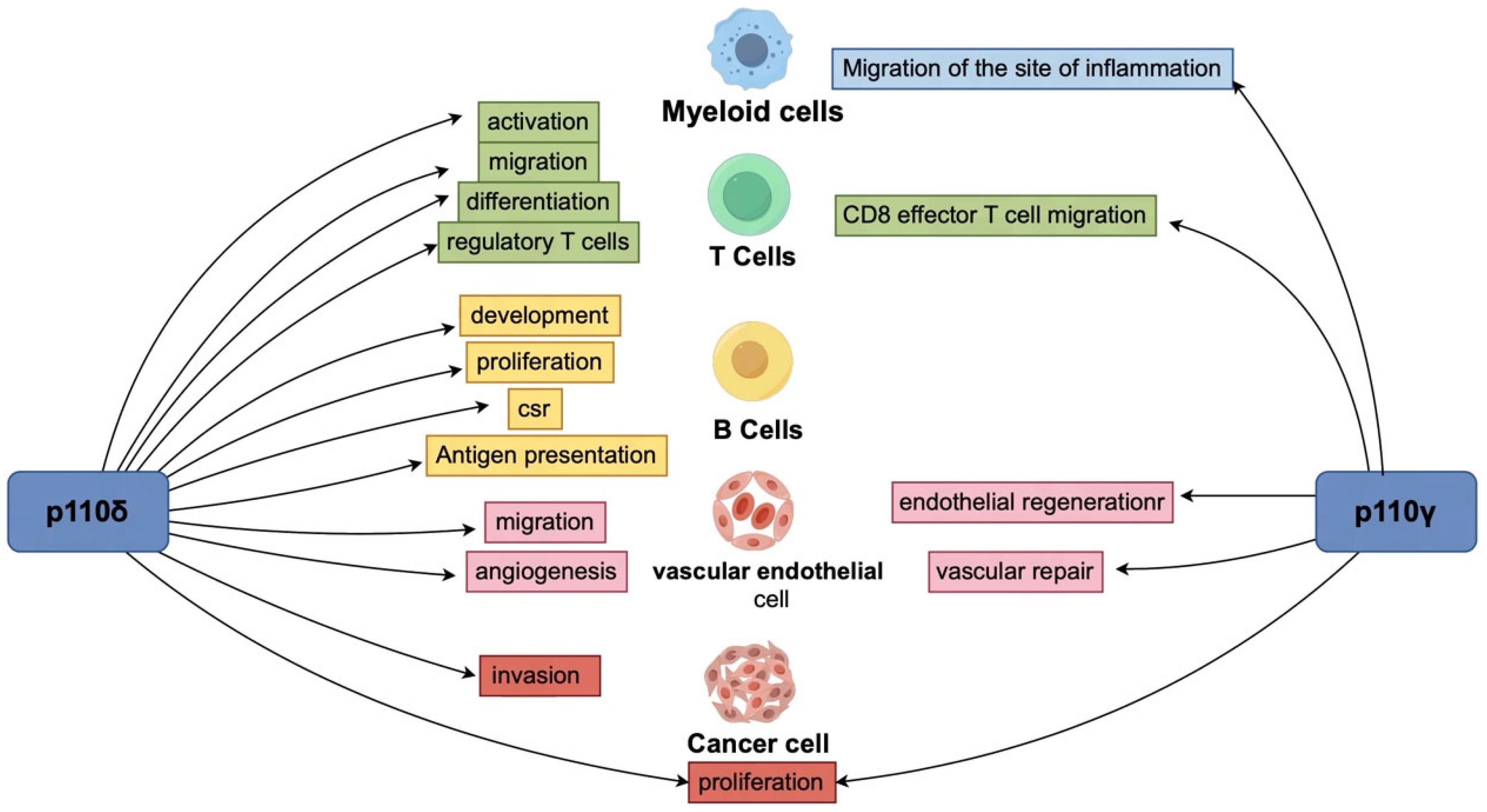
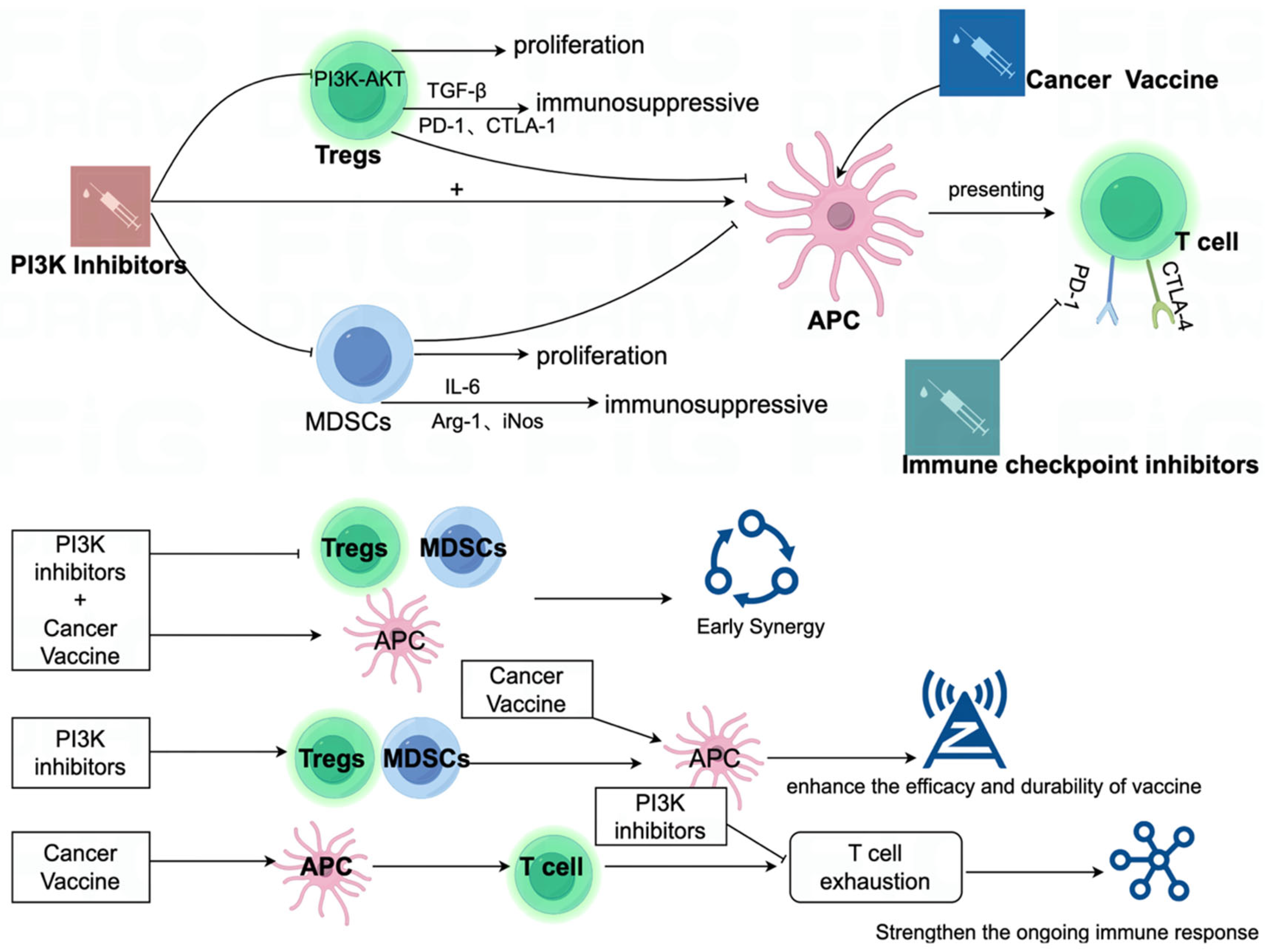

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| NCI ID (Trial) | Phase | Type of Tumor | Type of PIK3CG | Target Enrollment | Primary Endpoint |
|---|---|---|---|---|---|
| NCT05455619 | II | Breast Cancer | Evexomostat | 52 | AEs |
| NCT06530550 | II | Lymphoma, NK-LGL Leukemia, T-LGL Leukemia | Linperlisib; Duvelisib | 51 | ORR |
| NCT05082025 | II | Endometrial Cancer, Ovarian Cancer | copanlisib in combination with fulvestrant | 7 | To establish the use of copanlisib in combination with fulvestrant administered to subjects with selected estrogen receptor-positive (ER+). |
| NCT02164006 | I | Hodgkin’s Lymphoma | TGR-1202 + brentuximab vedotin | 16 | ORR |
| NCT05306041 | II | HER2-positive Breast Cancer | Inavolisib | 170 | ORR |
| NCT05676710 | I | Relapsed/Refractory Large Granular T Lymphocytic Leukemia | Linperlisib | 8 | AEs |
| NCT02389842 | I | Breast Cancer | Palbociclib + Taselisib/Pictilisib | 79 | AEs |
| NCT03131908 | II | Melanoma | GSK2636771 | 27 | ORR |
| NCT03581942 | II | Primary Central Nervous System Lymphoma (PCNSL) | Copanlisib+ Ibrutinib | 18 | PFS |
| NCT01920061 | I | Neoplasm | PF-05212384; Docetaxel; Cisplatin; Dacomitinib | 110 | AEs |
| NCT05387616 | II | Follicular Lymphoma | Copanlisib + Obinutuzumab | 98 | PFS |
| NCT03730142 | I | Advanced Cancer | WXFL10030390 | 82 | ORR |
| NCT01660451 | II | Lymphoma | Copanlisib | 227 | ORR |
| NCT03065062 | I | Lung Cancer Squamous Cell; Solid Tumors; Head and Neck Cancer; Pancreatic Cancer | Palbociclib; Gedatolisib | 96 | PFS |
| NCT05387616 | II | Follicular Lymphoma | Copanlisib + Obinutuzumab | 98 | PFS |
| NCT03711578 | II | Tenalisib | Non-Hodgkin Lymphoma | 20 | ORR |
| NCT04495621 | II | Metastatic Colorectal Cancer | MEN1611 + Cetuximab | 29 | ORR |
| NCT01791478 | I | Breast Cancer | BYL719 | 46 | PFS |
| NCT03767335 | I | Breast Cancer | MEN1611 + Trastuzumab +/− Fulvestrant | 62 | MTD |
| NCT04439188 | II | Lymphoma | GSK2636771 | 35 | ORR |
| NCT06132932 | II | PIK3CA Mutation-Related Tumors | WX390 | 38 | ORR |
| NCT06224257 | II | Large Granular T Lymphocytic Leukemia | Linperlisib | 41 | AEs |
| NCT01737450 | II | Progressive Disease | BKM120 | 58 | OS |
| NCT05683418 | I | Squamous Cell Carcinoma of Head and Neck; Urothelial Carcinoma; Endometrial Cancer; HR+/HER2-negative Breast Cancer | TOS-358 | 241 | AEs |
| NCT02268851 | I | Lymphoma | Ibrutinib; TGR-1202 | 45 | ORR |
| NCT01882803 | II | Duvelisib | Indolent Non-Hodgkin Lymphoma | 129 | ORR |
| NCT04439149 | II | Lymphoma | GSK2636771 | 35 | ORR |
| NCT05508906 | I | Breast Cancer | Drug: OP-1250 Ribociclib; Alpelisib; Everolimus | 155 | TEAEs |
| NCT06189209 | II | Triple Negative Breast Cancer | Tenalisib | 40 | ORR |
| NCT04038359 | II | Indolent Non-Hodgkin Lymphoma | Duvelisib | 103 | ORR |
| NCT04843098 | II | Head and Neck Squamous Cell Carcinoma | TL117; Paclitaxel | 108 | DLT |
| NCT03126019 | II | Lymphoma | Parsaclisib | 126 | PFS |
| NCT05021900 | II | Breast Cancer | Tenalisib | 40 | ORR |
| NCT02307240 | I | Triple-Negative Breast Cancer; High-grade Serous Ovarian Cancer; Solid Tumors; NUT Midline Carcinoma | CUDC-907 | 43 | AEs |
| NCT03538041 | II | Autoimmune Hemolytic Anemia | Parsaclisib | 25 | AEs |
| NCT04204057 | II | Leukemia, Lymphocytic, Chronic, B Cell | Tenalisib | 21 | AEs |
| NCT03770000 | II | T Cell Lymphoma | Tenalisib + Romidepsin | 33 | ORR |
| NCT05073250 | II | Inert Non-Hodgkin’s Lymphoma | IBI376; Rituximab | 40 | ORR |
| NCT03235544 | II | Lymphoma | Parsaclisib | 162 | ORR |
| NCT03586661 | I | Adenocarcinoma | niraparib; copanlisib | 31 | Maximum tolerated dose |
| NCT06239467 | I | Food Allergy | linvoseltamab; dupilumab | 6 | TEAEs |
| NCT03144674 | II | Lymphoma | Parsaclisib | 110 | ORR |
| NCT03522298 | II | Glioblastoma | Paxalisib | 30 | TEAEs |
| NCT01836861 | I | Healthy | IPI-145 | 6 | PK parameters of IPI-145 in plasma |
| NCT05143229 | I | Breast Cancer | alpelisib + sacituzumab | 18 | ORR |
| NCT01155453 | I | Advanced and Selected Solid Tumors | BKM120 + GSK1120212 DE | 113 | AEs |
| NCT05501886 | III | Breast Cancer | Gedatolisib; Palbociclib; Fulvestrant; Alpelisib | 701 | PFS |
| NCT06764186 | III | Breast Cancer | Capivasertib + fulvestrant | 100 | ORR |
| NCT02437318 | III | Breast Cancer | Fulvestrant + alpelisib | 572 | OS |
| NCT05631795 | IV | Breast Cancer | Alpelisib + fulvestrant | 100 | AEs |
| NCT02049541 | I | Lymphoma | BKM120 | 18 | ORR |
| NCT05768139 | II | Breast Cancer; Gynecologic Cancer; HNSCC; Solid Tumors, Adult | Drug: STX-478 Fulvestrant; Ribociclib; Palbociclib | 400 | DLT |
| NCT03218826 | I | Advanced Breast Carcinoma; Advanced Malignant Solid Neoplasm; Advanced Prostate Carcinoma; Anatomic Stage III Breast Cancer AJCC v8 | Docetaxel; AZD8186 | 23 | DLT |
| NCT03696355 | I | Brain and Central Nervous System Tumors | GDC-0084 | 27 | OS |
| NCT01756118 | I | Leukemia | BEZ235 | 24 | AEs |
| NCT04282018 | II | Lymphoma | BGB-10188; Zanubrutinib; Tislelizumab | 9\7 | |
| NCT02285179 | II | Breast Cancer | GDC-0032+Tamoxifen | 189 | AEs |
| NCT03688152 | I | Lymphoma | INCB053914 + INCB050465 | 9 | TEAEs |
| NCT02998476 | II | Lymphoma | Parsaclisib | 60 | PFS |
| NCT03424122 | I | B cell Lymphoma | Parsaclisib + Rituximab | 50 | TEAEs |
| Cancer Type | Cell Strain | Animal Model | Detection Index |
|---|---|---|---|
| Breast Cancer | MDA-MB-468 | - | Downregulation of BRCA1/2 transcription |
| Melanoma | IRB 11-003254 | - | ERK phosphorylation level |
| Breast Cancer | MCF7, ZR75-1, SUM52 and CAMA-1 | - | Increase in MYC |
| Small Cell Lung Cancer | H69, H1048, H209 | Female Balb/c nude Mice | LKB1 and pAMPK α Expression rates |
| Small Cell Lung Cancer | JIMT1, HCC1954 | - | Tumor growth rate |
| Breast Cancer | BT474 | - | Tumor growth rate |
| Lymphoma | MEC1,GM06990, JeKo-1 and Mino | Wild-type mice | AID α expression rates |
| Breast Cancer | BT474 and MCF7 | Female mice | Average tumor volume |
| Glioblastoma | LN229, U87MG, GBM43, GBM5 and GBM12 | Tumor growth rate | |
| Breast Cancer | OCUB-F, SUM185PE and SUM190PT etl. | Unintentional NU/NU nude mice | Tumor growth rate |
| Breast Cancer | HR6 | Female mice | Tumor growth rate |
| Breast Cancer | Met-1 | FVB/N background Mice | Tumor growth rate |
| Prostate Cancers | Pten+/−, PtenLx/Lx and PtenLx/Lx;Trp53Lx/Lx MEFs | Mice | Apoptosis response of cells |
| Breast Cancer | BT-20, DLD-1, HEK293T, HCC1937, HeLa, MDA-MB-435, MDA-MB-231, NIH/3T3 and MCF7 | Female nude mice | Tumor growth rate |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cui, R.; Luo, Z.; Zhang, X.; Yu, X.; Yuan, G.; Li, X.; Xie, F.; Jiang, O. Targeting PI3K Signaling to Overcome Tumor Immunosuppression: Synergistic Strategies to Enhance Cancer Vaccine Efficacy. Vaccines 2025, 13, 292. https://doi.org/10.3390/vaccines13030292
Cui R, Luo Z, Zhang X, Yu X, Yuan G, Li X, Xie F, Jiang O. Targeting PI3K Signaling to Overcome Tumor Immunosuppression: Synergistic Strategies to Enhance Cancer Vaccine Efficacy. Vaccines. 2025; 13(3):292. https://doi.org/10.3390/vaccines13030292
Chicago/Turabian StyleCui, Ran, Zhongxiang Luo, Xialin Zhang, Xinlin Yu, Gang Yuan, Xingming Li, Fei Xie, and Ou Jiang. 2025. "Targeting PI3K Signaling to Overcome Tumor Immunosuppression: Synergistic Strategies to Enhance Cancer Vaccine Efficacy" Vaccines 13, no. 3: 292. https://doi.org/10.3390/vaccines13030292
APA StyleCui, R., Luo, Z., Zhang, X., Yu, X., Yuan, G., Li, X., Xie, F., & Jiang, O. (2025). Targeting PI3K Signaling to Overcome Tumor Immunosuppression: Synergistic Strategies to Enhance Cancer Vaccine Efficacy. Vaccines, 13(3), 292. https://doi.org/10.3390/vaccines13030292