
Development of In Vitro Potency Methods to Replace In Vivo Tests for Enterovirus 71 Inactivated Vaccine (Human Diploid Cell-Based/Vero Cell-Based)

 , , , ,
, , , ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Working Standard Development and Calibration
2.2. Development and Validation of the IVRP Method
2.2.1. ATP Setting
2.2.2. Risk Assessment
2.2.3. Optimization of ELISA Method
2.2.4. Model Selection
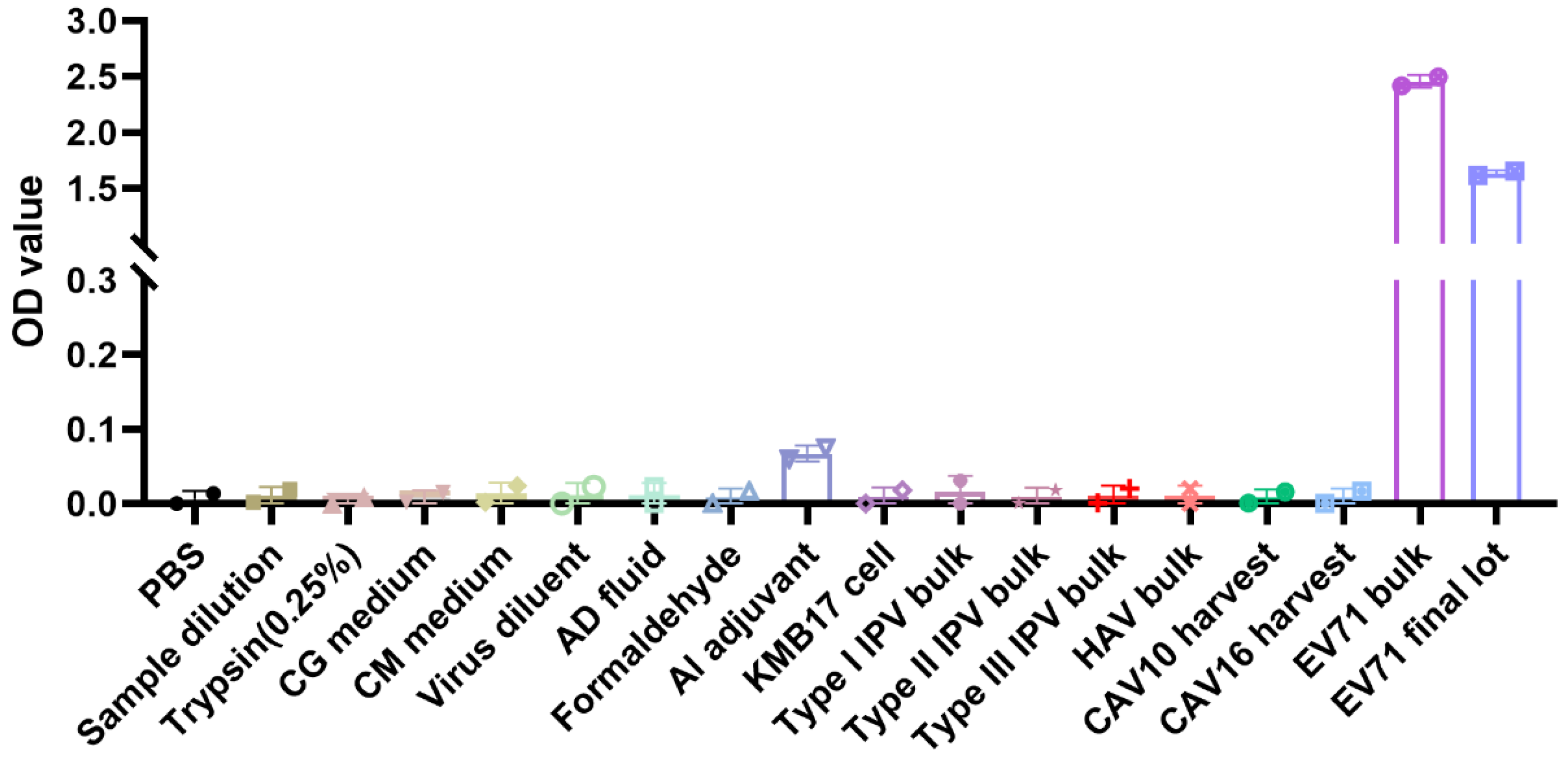
2.2.5. Specificity Validation
2.2.6. Precision and Accuracy Validation of IVRP
2.2.7. Method Capability Evaluation Indicator of IVRP
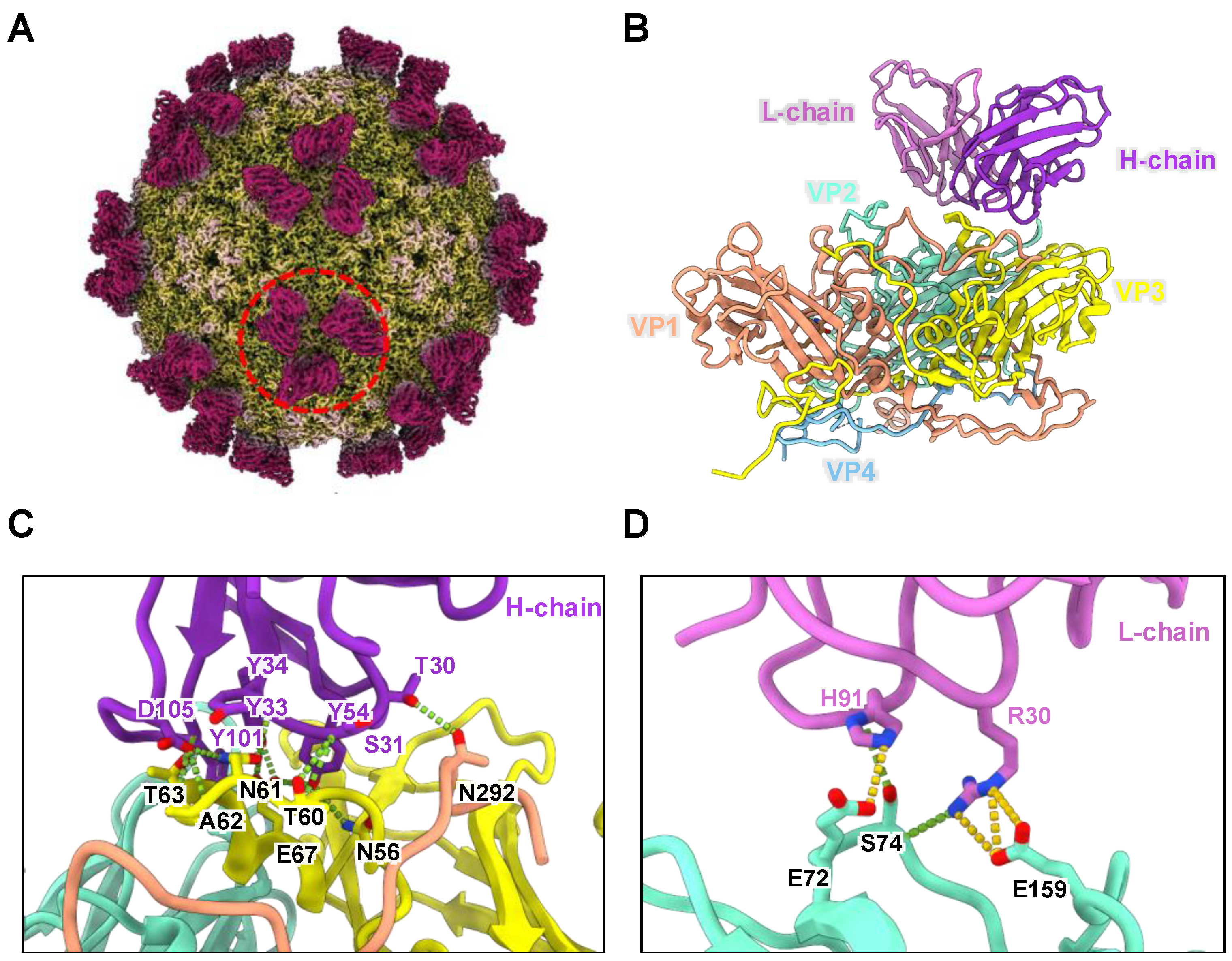
2.3. Epitope Identification of CT11F9 MAb
2.3.1. Cryo-EM Sample Preparation and Data Collection
2.3.2. Image Processing and Three-Dimensional Reconstruction
2.3.3. Model Building and Refinement
2.4. In Vivo Potency Assay
2.5. Neutralizing Assays
2.6. Western Blotting Analysis
2.7. Specification Establishment
2.8. Statistical Analysis
3. Results
3.1. Establishment of IVRP Working Standards
3.2. Establishment and Validation of an IVRP for EV71 Inactivated Vaccine Batch Testing
3.2.1. Establishment of an IVRP
Establishment of the ATP
Research on MAb
Risk Assessment
Method Optimization
Model Selection
3.2.2. Validation of the IVRP
Specificity
Accuracy and Precision
Method Capability Evaluation
3.3. Evaluated the IVRP Methods with Different Concentrations and Heat-Treated Vaccines
3.3.1. Testing of EV71 Vaccines with Different Antigen Concentrations
3.3.2. Testing of Heat-Treated Samples
3.4. Establishment of Potency Specifications
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- WHO Regional Office for the Western Pacific. A Guide to Clinical Management and Public Health Response for Hand, Foot and Mouth Disease (HFMD); WHO Regional Office for the Western Pacific: Manila, Philippines, 2011. [Google Scholar]
- Liang, Z.; Wang, J. EV71 vaccine, an invaluable gift for children. Clin. Transl. Immunol. 2014, 3, e28. [Google Scholar] [CrossRef]
- Liang, Z.; Mao, Q.; Wang, Y.; Zhu, F.; Li, J.; Yao, X.; Gao, F.; Wu, X.; Xu, M.; Wang, J.Z. Progress on the research and development of inactivated EV71 whole-virus vaccines. Hum. Vaccines Immunother. 2013, 9, 1701–1705. [Google Scholar] [CrossRef]
- Zhu, F.; Xu, W.; Xia, J.; Liang, Z.; Liu, Y.; Zhang, X.; Tan, X.; Wang, L.; Mao, Q.; Wu, J.; et al. Efficacy, safety, and immunogenicity of an enterovirus 71 vaccine in China. N. Engl. J. Med. 2014, 370, 818–828. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Xie, F.; Lin, G.; Zhang, D. Immune Efficacy of the EV71 Vaccine in Fujian Province, China: A Real-World Analysis of HFMD. Vaccines 2023, 11, 944. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Song, Y.; Wang, L.; Zhang, X.; Hu, Y.; Hu, Y.; Xia, J.; Li, J.; Zhu, F. Two-year efficacy and immunogenicity of Sinovac Enterovirus 71 vaccine against hand, foot and mouth disease in children. Expert Rev. Vaccines 2016, 15, 129–137. [Google Scholar] [CrossRef] [PubMed]
- EDQM. General Text: Substitution of In Vivo Methods by In Vitro Methods for the Quality Control of Vaccines 01/2018:50214; European Pharmacopoeia: Strasbourg, France, 2019. [Google Scholar]
- WHO. Guidelines for Independent Lot Release of Vaccines by Regulatory Authorities, Annex 2, TRS No 978. Available online: https://www.who.int/publications/m/item/guidelines-for-independent-lot-release-of-vaccines-annex-2-trs-no-978 (accessed on 19 February 2025).
- WHO. Guidelines on the Phasing Out of Animal Tests for the Quality Control of Biological Products. Available online: https://cdn.who.int/media/docs/default-source/biologicals/call-for-comments/who-guideline-on-3rs---draft-1-version-29-nov-2024-pc_clean.pdf?sfvrsn=7809e18d_1 (accessed on 22 February 2025).
- WHO. Recommendations to Assure the Quality, Safety and Efficacy of Pneumococcal Conjugate Vaccines. Available online: https://cdn.who.int/media/docs/default-source/biologicals/vaccine-standardization/pneumococcus/trs_977_annex_3.pdf?sfvrsn=344f81e_3&download=true (accessed on 28 February 2025).
- Zhang, X.; Wu, X.; He, Q.; Wang, J.; Mao, Q.; Liang, Z.; Xu, M. Research progress on substitution of in vivo method(s) by in vitro method(s) for human vaccine potency assays. Expert Rev. Vaccines 2023, 22, 270–277. [Google Scholar] [CrossRef]
- van den Biggelaar, R.; Hoefnagel, M.H.N.; Vandebriel, R.J.; Sloots, A.; Hendriksen, C.F.M.; van Eden, W.; Rutten, V.; Jansen, C.A. Overcoming scientific barriers in the transition from in vivo to non-animal batch testing of human and veterinary vaccines. Expert Rev. Vaccines 2021, 20, 1221–1233. [Google Scholar] [CrossRef]
- Morgeaux, S.; Poirier, B.; Ragan, C.I.; Wilkinson, D.; Arabin, U.; Guinet-Morlot, F.; Levis, R.; Meyer, H.; Riou, P.; Shaid, S.; et al. Replacement of in vivo human rabies vaccine potency testing by in vitro glycoprotein quantification using ELISA—Results of an international collaborative study. Vaccine 2017, 35, 966–971. [Google Scholar] [CrossRef]
- Signorazzi, A.; Etna, M.P.; Coccia, E.M.; Huckriede, A. In vitro assessment of tick-borne encephalitis vaccine: Suitable human cell platforms and potential biomarkers. ALTEX 2021, 38, 431–441. [Google Scholar] [CrossRef]
- Vermeulen, M.; Feck, I.; Francotte, A.; Hassall, L.; Tesolin, L.; Van Molle, W.; Pizzato, R.; Laurent, T.; Hoebreck, C.; Stickings, P.; et al. Development of a multiplex-based immunoassay for the characterization of diphtheria, tetanus and acellular pertussis antigens in human combined DTaP vaccines. J. Immunol. Methods 2023, 517, 113483. [Google Scholar] [CrossRef]
- Hassall, L.; Yara, D.A.; Riches-Duit, R.; Rigsby, P.; Dobly, A.; Vermeulen, M.; Francotte, A.; Faber, B.; Stickings, P. Development of a monoclonal antibody sandwich ELISA for the quality control of human and animal tetanus vaccines. ALTEX 2024, 41, 588–604. [Google Scholar] [CrossRef] [PubMed]
- Hassall, L.; Yara, D.A.; Riches-Duit, R.; Rigsby, P.; Dobly, A.; Vermeulen, M.; Francotte, A.; Stickings, P. Development of a monoclonal antibody sandwich ELISA for the determination of antigen content and quality in diphtheria vaccines. ALTEX 2024, 41, 57–68. [Google Scholar] [CrossRef] [PubMed]
- International Council for Harmonization of Technical Requirements for Pharmaceuticals for Human Use Q14 Analytical Procedure Development. Geneva, CH. 2023. Available online: https://database.ich.org/sites/default/files/ICH_Q14_Guideline_2023_1116.pdf (accessed on 28 February 2025).
- International Council for Harmonization of Technical Requirements for Pharmaceuticals for Human Use Q2, R2 Harmonised Guideline Validation of Analytical Procedure. Geneva, CH. 2023. Available online: https://database.ich.org/sites/default/files/ICH_Q2%28R2%29_Guideline_2023_1130.pdf (accessed on 28 February 2025).
- 9101 Public Release of Revised Draft Guidelines for Validation of Analytical Methods (Second Edition). Available online: https://pharmcube-bydrug.oss-cn-beijing.aliyuncs.com/info/message_cn_file/b28748ed23da656050bdb1b52ec67cb9.pdf (accessed on 28 February 2025).
- WHO. WHO Manual for the Establishment of National and Other Secondary Standards for Vaccines. Available online: https://iris.who.int/bitstream/handle/10665/70669/WHO_IVB_11.03_eng.pdf?sequence=1 (accessed on 28 February 2025).
- ChPC. General requirements: Requirements for preparation and calibration of national standard substances of biologicals. In Pharmacopoeia of the People’s Republic of China, 2020 ed.; Chinese Pharmacopoeia Commission: Beijing, China, 2020. [Google Scholar]
- The United States Pharmacopeial Convention. U S Pharmacopeia, 43-NF38 ed.; <1033> Biological assay validation; USA Pharmacopeia Convention: North Bethesda, MD, USA, 2020; pp. 7337–7350. [Google Scholar]
- Mastronarde, D.N. Automated electron microscope tomography using robust prediction of specimen movements. J. Struct. Biol. 2005, 152, 36–51. [Google Scholar] [CrossRef] [PubMed]
- Zheng, S.Q.; Palovcak, E.; Armache, J.-P.; Verba, K.A.; Cheng, Y.; Agard, D.A. MotionCor2: Anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat. Methods 2017, 14, 331–332. [Google Scholar] [CrossRef]
- Zhang, K. Gctf: Real-time CTF determination and correction. J. Struct. Biol. 2016, 193, 1–12. [Google Scholar] [CrossRef]
- Punjani, A.; Rubinstein, J.L.; Fleet, D.J.; Brubaker, M.A. cryoSPARC: Algorithms for rapid unsupervised cryo-EM structure determination. Nat. Methods 2017, 14, 290–296. [Google Scholar] [CrossRef]
- Scheres, S.H.; Chen, S. Prevention of overfitting in cryo-EM structure determination. Nat. Methods 2012, 9, 853–854. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- Emsley, P.; Lohkamp, B.; Scott, W.G.; Cowtan, K. Features and development of Coot. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66, 486–501. [Google Scholar] [CrossRef]
- Adams, P.D.; Afonine, P.V.; Bunkóczi, G.; Chen, V.B.; Davis, I.W.; Echols, N.; Headd, J.J.; Hung, L.W.; Kapral, G.J.; Grosse-Kunstleve, R.W.; et al. PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66, 213–221. [Google Scholar] [CrossRef]
- Chen, V.B.; Arendall, W.B., 3rd; Headd, J.J.; Keedy, D.A.; Immormino, R.M.; Kapral, G.J.; Murray, L.W.; Richardson, J.S.; Richardson, D.C. MolProbity: All-atom structure validation for macromolecular crystallography. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66, 12–21. [Google Scholar] [CrossRef] [PubMed]
- Goddard, T.D.; Huang, C.C.; Meng, E.C.; Pettersen, E.F.; Couch, G.S.; Morris, J.H.; Ferrin, T.E. UCSF ChimeraX: Meeting modern challenges in visualization and analysis. Protein Sci. Publ. Protein Soc. 2018, 27, 14–25. [Google Scholar] [CrossRef]
- Li, Z.; Xu, L.; He, D.; Yang, L.; Liu, C.; Chen, Y.; Shih, J.W.; Zhang, J.; Zhao, Q.; Cheng, T.; et al. In vivo time-related evaluation of a therapeutic neutralization monoclonal antibody against lethal enterovirus 71 infection in a mouse model. PLoS ONE 2014, 9, e109391. [Google Scholar] [CrossRef]
- Chen, Y.; Li, C.; He, D.; Cheng, T.; Ge, S.; Shih, J.W.; Zhao, Q.; Chen, P.J.; Zhang, J.; Xia, N. Antigenic analysis of divergent genotypes human Enterovirus 71 viruses by a panel of neutralizing monoclonal antibodies: Current genotyping of EV71 does not reflect their antigenicity. Vaccine 2013, 31, 425–430. [Google Scholar] [CrossRef]
- ICH Q6B Specifications: Test Procedures and Acceptance Criteria for Biotechnological/Biological Products—Scientific Guideline. Available online: https://www.ema.europa.eu/en/ich-q6b-specifications-test-procedures-acceptance-criteria-biotechnological-biological-products-scientific-guideline#:~:text=This%20document%20proposes%20a%20uniform%20set%20of%20international,and%20biological%20products%20to%20support%20new%20marketing%20applications (accessed on 28 February 2025).
- EDQM Summary Report on the Statistics on the Use of Animals for Scientific Purposes in the Member States of the European Union and Norway in 2020. Available online: https://data.consilium.europa.eu/doc/document/ST-8083-2023-INIT/en/pdf (accessed on 28 February 2025).
- WHO Guidelines on Nonclinical Evaluation of Vaccines. Available online: https://cdn.who.int/media/docs/default-source/biologicals/vaccine-standardization/annex-1nonclinical.p31-63.pdf?sfvrsn=e87c28d8_3&download=true (accessed on 28 February 2025).
- Shank-Retzlaff, M.; Wang, F.; Morley, T.; Anderson, C.; Hamm, M.; Brown, M.; Rowland, K.; Pancari, G.; Zorman, J.; Lowe, R.; et al. Correlation between mouse potency and in vitro relative potency for human papillomavirus Type 16 virus-like particles and Gardasil vaccine samples. Hum. Vaccines 2005, 1, 191–197. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Sun, Y.; Wu, X.; Carroll, D.S.; Lv, W.; You, L.; Ji, Y.; Shi, J.; Yan, J.; Xu, G.; et al. Development of a relative potency test using ELISA for human rabies vaccines. Biol. J. Int. Assoc. Biol. Stand. 2018, 55, 59–62. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.C.; Kim, D.K.; Kim, H.J.; Hong, S.H.; Kim, Y.; Lim, J.M.; Hong, J.; Kim, C.H.; Park, Y.K.; Kim, J. A collaborative study of an alternative in vitro potency assay for the Japanese encephalitis vaccine. Virus Res. 2016, 223, 190–196. [Google Scholar] [CrossRef]
- Volokhov, D.V.; Fry, A.M.; Furtak, V.; Jones, R.M.; Musiychuk, K.; Norikane, J.; Green, B.J.; Srinivas, G.B.; Streatfield, S.J.; Yusibov, V. An ELISA-based antigenicity test of rabies recombinant glycoprotein cannot predict its protective potency in vivo. Mol. Cell. Probes 2022, 63, 101815. [Google Scholar] [CrossRef]
- Liang, Z.; Mao, Q.; Gao, Q.; Li, X.; Dong, C.; Yu, X.; Yao, X.; Li, F.; Yin, W.; Li, Q.; et al. Establishing China’s national standards of antigen content and neutralizing antibody responses for evaluation of enterovirus 71 (EV71) vaccines. Vaccine 2011, 29, 9668–9674. [Google Scholar] [CrossRef]
- WHO. Recommendations to Assure the Quality, Safety and Efficacy of Enterovirus 71 Vaccines (Inactivated). Available online: https://cdn.who.int/media/docs/default-source/biologicals/vaccine-standardization/enterovirus/who_trs_1030_annex-3_ev71_vaccines.pdf?sfvrsn=b250908f_11&download=true (accessed on 22 February 2025).
- Cooper, G.; Mao, Q.; Crawt, L.; Wang, Y.; Dougall, T.; Rigsby, P.; Liang, Z.; Xu, M.; The Collaborative Study Group; Minor, P.; et al. Establishment of the 1st WHO International Standard for anti-EV71 serum (Human). Biol. J. Int. Assoc. Biol. Stand. 2018, 53, 39–50. [Google Scholar] [CrossRef]
- The United States Pharmacopeial Convention. U. S. Pharmacopeia, USPNF Issue 1 ed.; <1220> Analytical Procedure Life Cycle; USA Pharmacopeia Convention: North Bethesda, MD, USA, 2022; Volume 2022. [Google Scholar]
- Han, L.; An, C.; Liu, D.; Wang, Z.; Bian, L.; He, Q.; Liu, J.; Wang, Q.; Liu, M.; Mao, Q.; et al. Development of an ELISA Assay for the Determination of SARS-CoV-2 Protein Subunit Vaccine Antigen Content. Viruses 2022, 15, 62. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Bai, Y.; Liu, M.; Tan, D.; Li, J.; Wang, Z.; Liang, Z.; Xu, M.; Wang, J.; Mao, Q. Standardized neutralization antibody analytical procedure for clinical samples based on the AQbD concept. Signal Transduct. Target. Ther. 2023, 8, 165. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| A | B | C | ||
|---|---|---|---|---|
| Comparison of working standard (WS) and Phase 3 vaccine (P3V) | The ratio of ED50 (WS/P3V) | 1.49 | 0.90 | 2.04 |
| Antigen content ratio after dissociation (WS/P3V) | 1.25 | 1.20 | 0.91 | |
| EV71 antigen content of WS (IU/mL) | 242 (95% CI 240–245) | 812 (95% CI 793–830) | 999 (95% CI 985–1012) | |
| Objectives | Results | |||
|---|---|---|---|---|
| Manufacturer A | Manufacturer B | Manufacturer C | ||
| Intended purpose | Establishing an in vitro relative potency method that can be used for batch testing and stability testing of the final lot of EV71 inactivated vaccine. | |||
| Critical quality attribute (CQA) and experimental principle | CQA: The antigen content of EV71 inactivated vaccines. Experimental principle: The EV71 polyclonal antibody was immobilized on the solid phase. An immune complex with the antigen in the sample would form. After washing, an enzyme-labeled MAb targeting conformational epitopes relevant to the protection offered by the vaccines was added. Thereby, an enzyme-labeled antibody–antigen–antibody complex–solid phase complex was formed. After subsequent washing, substrate was added for color development. The optical density in the microplate wells was directly proportional to the concentration of the analyte. The in vitro relative potency of the sample compared to the standard product is then calculated. | |||
| Specificity | No cross reaction with hepatitis A virus, influenza virus, and other enteroviruses | |||
| Accuracy (relative bias) | The relative bias of each known relative potency should be less than 15%, and its 90% confidence interval should be in the range of ±20% | In the known relative potency range of 0.5–2.0, the relative bias of each level was less than 2%, and the 90% confidence limit was not higher than ±5% | In the known relative potency range of 0.5–2.0, the relative bias of each level was less than 10%, and the 90% confidence limit was not higher than ±15% | In the known relative potency range of 0.5–2.0, the relative bias of each level was less than 10%, and the 90% confidence limit was not higher than ±15% |
| Intermediate precision | ≤15% | ≤9% | ≤8% | ≤8% |
| Total analytical error | ≤20% | ≤9% | ≤10% | ≤8% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, X.; Yi, L.; Yu, D.; Li, J.; Li, X.; Wu, X.; Gao, F.; He, Q.; Wang, W.; Wang, K.; et al. Development of In Vitro Potency Methods to Replace In Vivo Tests for Enterovirus 71 Inactivated Vaccine (Human Diploid Cell-Based/Vero Cell-Based). Vaccines 2025, 13, 404. https://doi.org/10.3390/vaccines13040404
Zhang X, Yi L, Yu D, Li J, Li X, Wu X, Gao F, He Q, Wang W, Wang K, et al. Development of In Vitro Potency Methods to Replace In Vivo Tests for Enterovirus 71 Inactivated Vaccine (Human Diploid Cell-Based/Vero Cell-Based). Vaccines. 2025; 13(4):404. https://doi.org/10.3390/vaccines13040404
Chicago/Turabian StyleZhang, Xuanxuan, Li Yi, Dan Yu, Jun Li, Xintian Li, Xing Wu, Fan Gao, Qian He, Wenhui Wang, Kaiwen Wang, and et al. 2025. "Development of In Vitro Potency Methods to Replace In Vivo Tests for Enterovirus 71 Inactivated Vaccine (Human Diploid Cell-Based/Vero Cell-Based)" Vaccines 13, no. 4: 404. https://doi.org/10.3390/vaccines13040404
APA StyleZhang, X., Yi, L., Yu, D., Li, J., Li, X., Wu, X., Gao, F., He, Q., Wang, W., Wang, K., Wang, Z., Liu, Z., Li, Y., Zhao, Y., Li, H., Ma, X., Zheng, Q., Xu, L., Cheng, T., ... Liang, Z. (2025). Development of In Vitro Potency Methods to Replace In Vivo Tests for Enterovirus 71 Inactivated Vaccine (Human Diploid Cell-Based/Vero Cell-Based). Vaccines, 13(4), 404. https://doi.org/10.3390/vaccines13040404