1. Introduction
Duck hepatitis A virus (DHAV) causes highly contagious and acute duck viral hepatitis (DVH) to ducklings within 3 weeks old, concomitant with liver necrosis and hemorrhage, and high mortality rate. As one of the pathogens that seriously jeopardize duck industry in Southeast Asia, an outbreak of DHAV infection will usually be followed by great economy losses [
1,
2]. DHAV has been classified into three serotypes according to results of neutralization tests, namely the classical serotype 1 (DHAV-1) [
3,
4,
5], the serotype isolated in Taiwan only (DHAV-2) [
6], and the serotype firstly identified in South Korea (DHAV-3) [
7]. No cross-neutralization was found between DHAV-1 and DHAV-2, and limited cross-neutralization has been reported between DHAV-1 and DHAV-3 [
6,
7,
8]. In recent years, the mixed infections of different DHAVs had happened with increasing frequency in eastern Asia [
1,
2,
6], intensifying the difficulty of DVH prevention and control.
DHAV belongs to genus
Avihepatovirus within the family
Picornaviridae. Excluding the poly(A) tail, which is irregular in length, the single-stranded positive-sense RNA of the complete DHAV genome is approximately 7700 nucleotides. It is encapsulated in an icosahedral structure, which is assembled with the structural proteins, namely VP0, VP3, and VP1 [
3,
6]. Among them, VP1 is considered as the external and dominant antigen with several conserved linear epitopes [
8,
9,
10].
Different from linear epitopes, conformational epitopes are formed when discontinuous amino acid in the primary protein sequence are folded into close proximity in a high level structure [
11,
12]. Thus, it is relatively different to map discontinuous epitopes using synthetic peptides or through amino acid mutant selections [
13]. As an in vitro screening technique for identifying ligands for proteins and other macromolecules, the phage displayed technology is a critical tool for protein-protein interaction studies [
14]. The phage displayed library is comprised of abundant bacteriophages expressing exogenous peptide or protein sequences in their coat proteins, allowing the selection of binding partners for myriad target types by iterative rounds of in vitro panning and amplification, followed by DNA sequencing [
13,
14]. It offers an efficient alternative to more traditional methods of epitope mapping, especially the conformational ones.
In this study, we identified a conformational epitope, which could be effectively recognized by monoclonal antibody (mAb) 4E6 of DHAV through a 12-mer random peptide phage display library. The results of neutralization assay showed that the mimitope was capable of blocking the virus recognition by mAb. Further animal tests confirmed the peptide 1GLTWKLPPSM10, which was truncated from the isolated 12-mer peptide 1GLTWKLPPSMVH12, could inhibit the virus replication in ducklings, suggesting its potential as an epitope vaccine against DVH.
2. Materials and Methods
2.1. Phage Library, Virus, and Antibodies
The phage library (1.5 × 1012 phages, 100 µL) prepared with the Ph.D.-12 Phage Display Peptide Library Kit (cat. no. E8110S; New England Biolabs, USA) was preserved in 50% glycerol in Tris-buffered saline, pH 7.5. The Escherichia coli ER2738 strain, which was supplied with the kit, was used to propagate the phages.
The DHAV-1 LY0801 (FJ436047, median embryo lethal dose, ELD
50 = 10
−5.7/0.2 mL), DHAV-1 FC16115 (MG515248, ELD
50 = 10
−5.75/0.2 mL), DHAV-3 SD1201 strain (KC993890, ELD
50 = 10
−3.7/0.2 mL), and DHAV-3 JN1203 strains (KP715480, ELD
50 = 10
−4.7/0.2 mL) were individually isolated from the livers of sick cherry valley ducks from Shandong Province, China, and stored in our laboratory [
8,
15,
16].
Using purified DHAV-1 LY0801 strain to immunize BALB/c mice, we finally got a hybridoma cell line secreting IgG1/kappa type mAb, named 4E6, with high affinity to both DHAV-1 and DHAV-3. Horseradish peroxidase (HRP) labeled goat anti-duck antibody, HRP labeled goat anti-rabbit antibody and HRP labeled goat anti-mouse antibody were purchased from KPL (Gaithersburg, MD, USA). Fluorescein 5-Isothiocyanate (FITC) labeled goat anti-mouse antibody, HRP labeled anti-GST (Glutathione S Transferase) mAb and HRP labeled anti-His (histidine) mAb were both purchased from Beyotime Biotechnology (Beijing, China). HRP labeled anti-M13 mAb was purchased from GE Healthcare (München, Germany).
2.2. Neutralization Assay for mAb 4E6 against DHAV-1 and DHAV-3
The protein content in ascites was quantitatively determined with a spectrophotometer (DS-11 Series, DeNovix, USA) and then diluted to 23, 24, 25, 26, 27, and 28 in saline. For each embryo, 0.1 mL of diluted mAb 4E6 was incubated with quantitative DHAV-1 or DHAV-3 (100 ELD50, 0.1 mL) at 37 °C for 1 h before the injection into embryos via the allantoic route. The control embryos received virus in the absence of mAb incubation. The embryos were allowed to hatch.
2.3. Preparation of Viral Antigens
DHAV-1 LY0801 and DHAV-3 SD1201 strains were used as templates to amplify the entire VP1, VP0 and VP3 genes of DHAV-1 and DHAV-3. The fragments were digested with EcoRI and XhoI and then inserted into the corresponding region of pFastBacHTB to generate pFastBac-1VP0, pFastBac-1VP3, pFastBac-1VP1, pFastBac-3VP0, pFastBac-3VP3, and pFastBac-3VP1 constructs for subsequent transformation into DH10Bac E. coli cells to obtain the recombinant bacmids. Meanwhile, a pFastBacHT-CAT vector was transformed and served as the positive control. According to the baculovirus expression system instructions, two rounds of blue-white screening were designed to isolate the recombinant bacmids, and the positive colonies were re-cultured to obtain b1VP0, b1VP3, b1VP1, b3VP0, b3VP3, and b3VP1 bacmids.
2.4. Indirect Immunofluorescence (IFA) and Western Blotting Assay
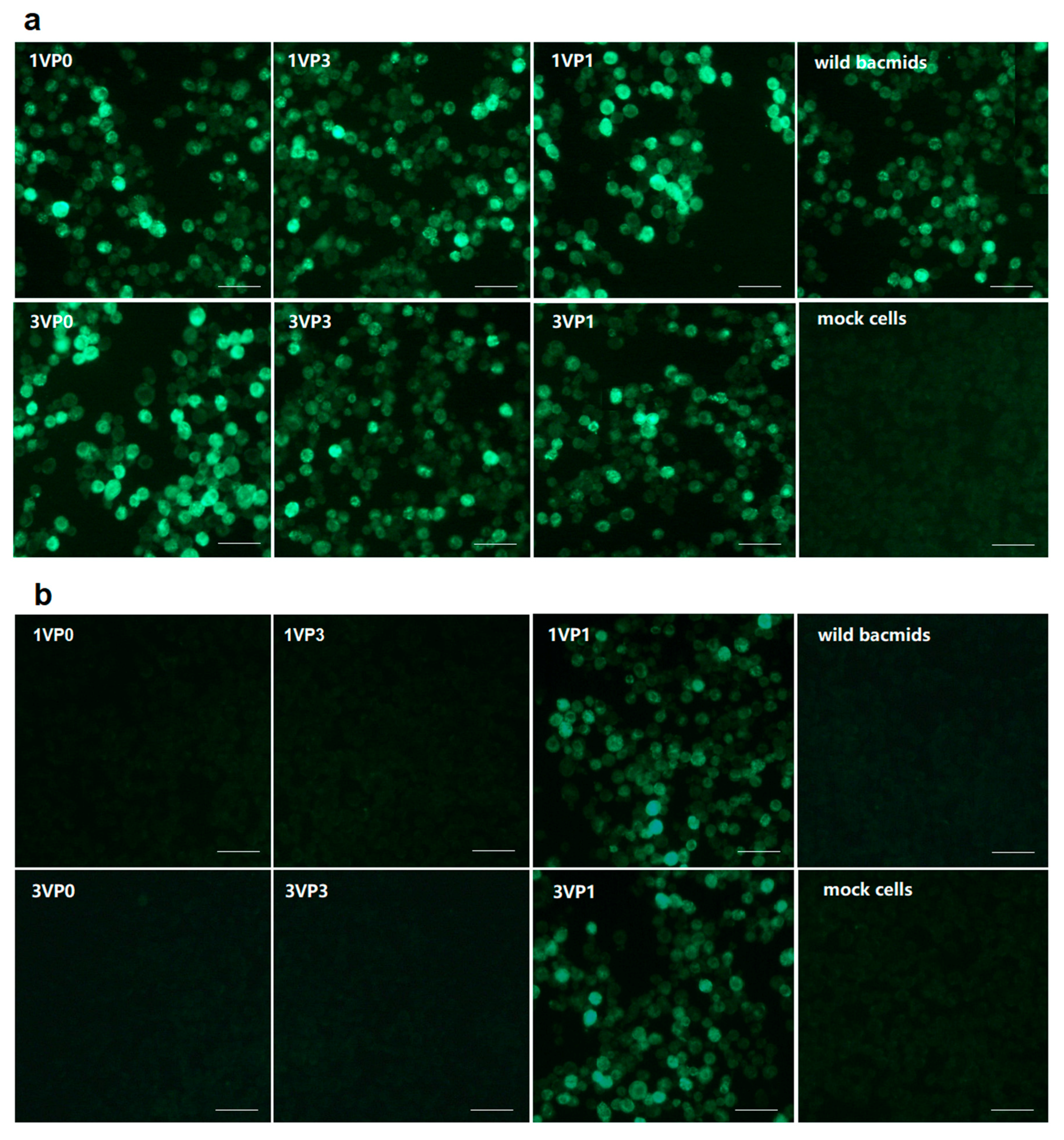
Spodoptera frugiperda 9 (Sf9) cells were homogenously seeded in 12-well plates and then transfected with 2.0 μg of recombinant bacmid using 5 μL of X-tremeGENE HP DNA Transfection Reagent. Sf9 cells infected with wild baculovirus and mock cells served as positive and negative controls, respectively. At 72 hours post infection (hpi), expressed proteins in cells were analyzed by IFA and western blotting assays.
For IFA, the cells were washed with phosphate-buffered saline (PBS) three times and then fixed with ice-chilled fixative (acetone-methanol, 1:1 v/v) for 10 min at room temperature. The cells infected with different bacmids were individually incubated with mAb 4E6 (1:200) at 37 °C for 1 h. Subsequently, the cells were washed three times and incubated with a secondary antibody, FITC-labeled goat anti-mouse immunoglobulin G (IgG) (1:500), for 1 h in dark conditions. The cells were washed and viewed under an Olympus IMT 2 fluorescence micro-scope (Olympus, Japan). Images were acquired with an identical exposure time and digitally documented.
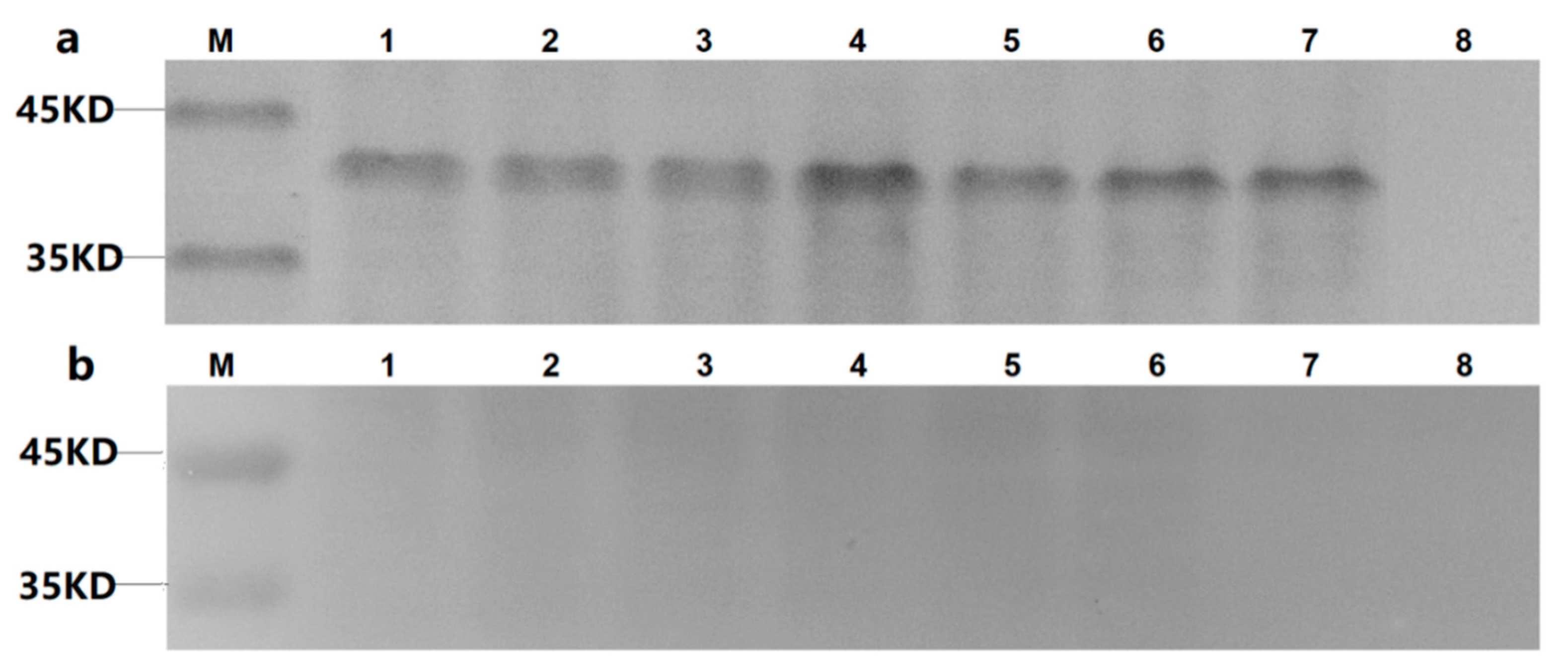
For western blotting analysis, the Sf9 monolayer underwent three rounds of infection to amplify the bacmids. The cells were washed three times with PBS, and the virus-infected cells were lysed with cell lysis buffer and sonicated on ice. Sodium dodecyl polyacrylamide gel electrophoresis (SDS-PAGE) and western blotting analysis were successively carried out. Proteins were electro-transferred onto the polyvinylidene difluoride (PVDF) membranes (Millipore, Bedford, MA, USA). The membranes were blocked with 1% bovine serum albumin (BSA) in PBS and washed with PBS containing 0.01% phosphate-buffered saline Tween-20 (PBST), followed by incubation with the anti-His mAb or mAb 4E6 at 37 °C for 2 h. The membranes were washed three times with PBST and then incubated with HRP-conjugated goat anti-mouse IgG (1:1000) for 1 h at 37 °C. The membranes were washed three times, and the reaction was visualized with the Diaminobenzidine Tertrahydrochloride (DAB) Horseradish Peroxidase Color Development Kit (Beyotime, China).
2.5. Bio-Panning and Selection of Phages
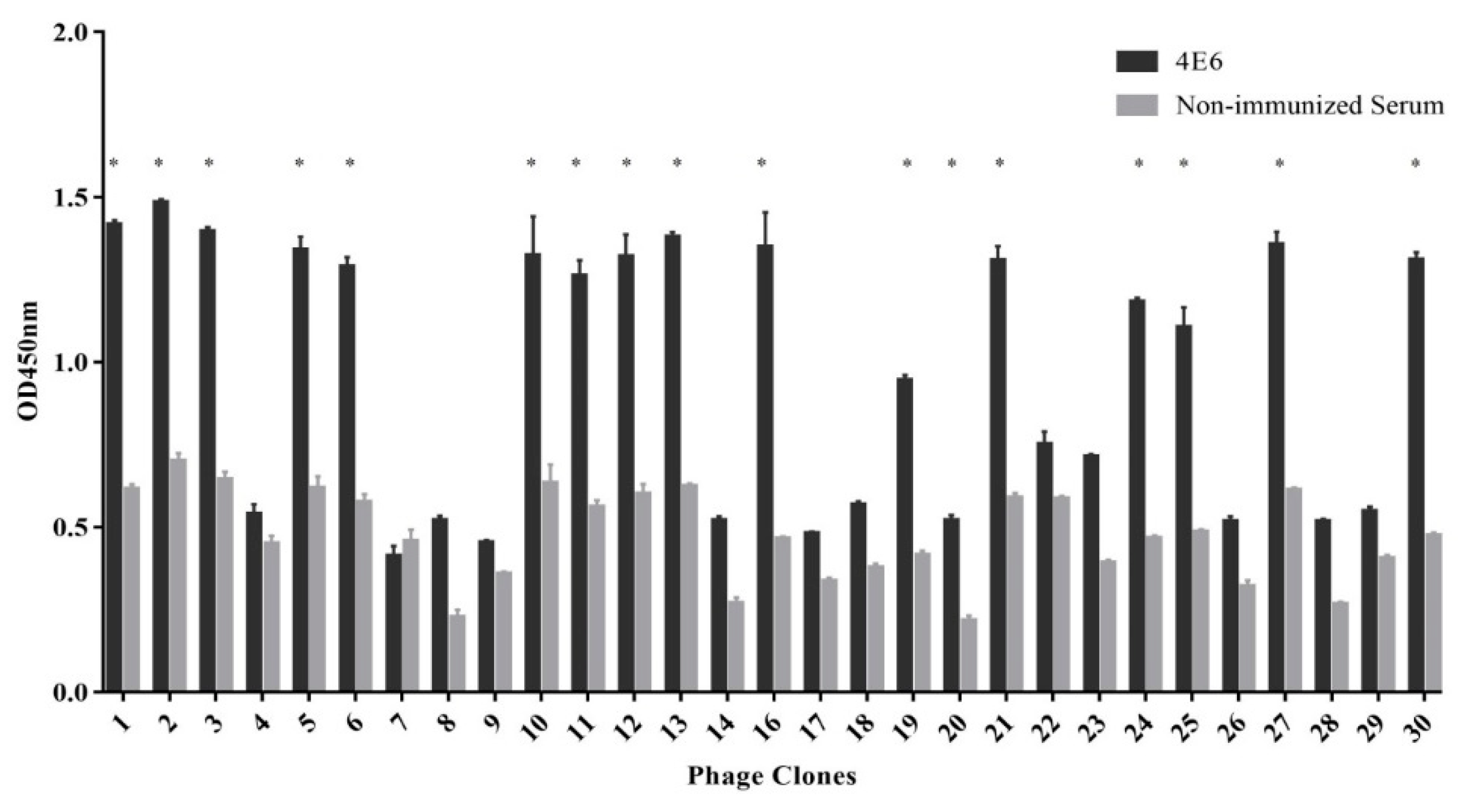
The mAb 4E6 in ascites was prepared by ammonium sulfate precipitation and then purified using protein G agarose (Roche, Mannheim, Germany). The screening procedure of the phage display library was carried out according to the manufacturer’s instructions. For the first step, the 96-well plate was coated with purified 4E6 (100 µg/mL in 0.1 M NaHCO3, pH 8.6) at 4 °C overnight. The plate was washed with TBST (50 mM Tris-HCl, pH7.5, 150 mM NaCl, 0.1% v/v Tween 20), and the coated wells were blocked with 1 mg/mL BSA in 0.1 M NaHCO3 buffer. After incubation with the phage library (4 × 108 phages/well) for 1 h at room temperature, the unbound phages were removed by washing ten times with TBST. The bound phages were eluted with 0.2 M glycine-HCl, pH 2.2 containing 1 mg/mL BSA) and immediately neutralized with 1 M Tris-HCl, pH 9.1. The eluted phage was amplified in E. coli (ER2738) and titered on LB/IPTG/Xgal plates. The subsequent two rounds of selection were performed similarly to the first step, with the mAb 4E6 concentration reduced to 50 µg/mL and the Tween 20 concentration increased to 0.5%. Individual clones were randomly selected for phage enzyme linked immunosorbent assay (ELISA) and DNA sequencing.
2.6. Phage ELISA and DNA Sequencing
A 96-well plate was coated with purified mAb 4E6 (approximately 10 µg/well mAb in 0.1 M NaHCO3, pH 8.6) overnight at 4 °C. The plates were washed three times with TBST, followed by blocking with 3% BSA in PBS for 2 h at room temperature. Subsequently, phages (1010 pfu/well) were added into the wells, and the plate was incubated for 1 h at room temperature. The plates were washed ten times with TBST, followed by the addition of HRP-conjugated anti-M13 mAb (1:5000) and incubation at 37 °C for 1 h. Subsequently, tetramethylbenzidine (TMB) substrate was added and the plates were incubated at 37 °C for 15 min. The reaction was stopped with 2 M H2SO4 and the absorbance was measured at 450 nm. Monophages with relatively high absorbance values were selected for sequencing.
Single-stranded DNA was isolated from the monophages according to the instructions supplied with the phage display peptide library kit and sequenced using the 96gIII sequencing primer.
2.7. Phage Design and Sequence Analysis
Peptides Pep0 and PepN were synthesized (purity >95%) by GenScript China Inc (Nanjing, Jiangsu, China). The amino acid sequence of Pep0 was 1GLTWKLPPSMVH12, and the amino acid sequence of PepN, which served as the negative control, was 1IGENITNPIKPN12. Ten DHAV-1 strains and ten DHAV-3 strains from NCBI were selected for the VP1 sequence alignment with the corresponding epitope region using the Lasergene tool (DNASTAR Inc., Madison, WT, USA).
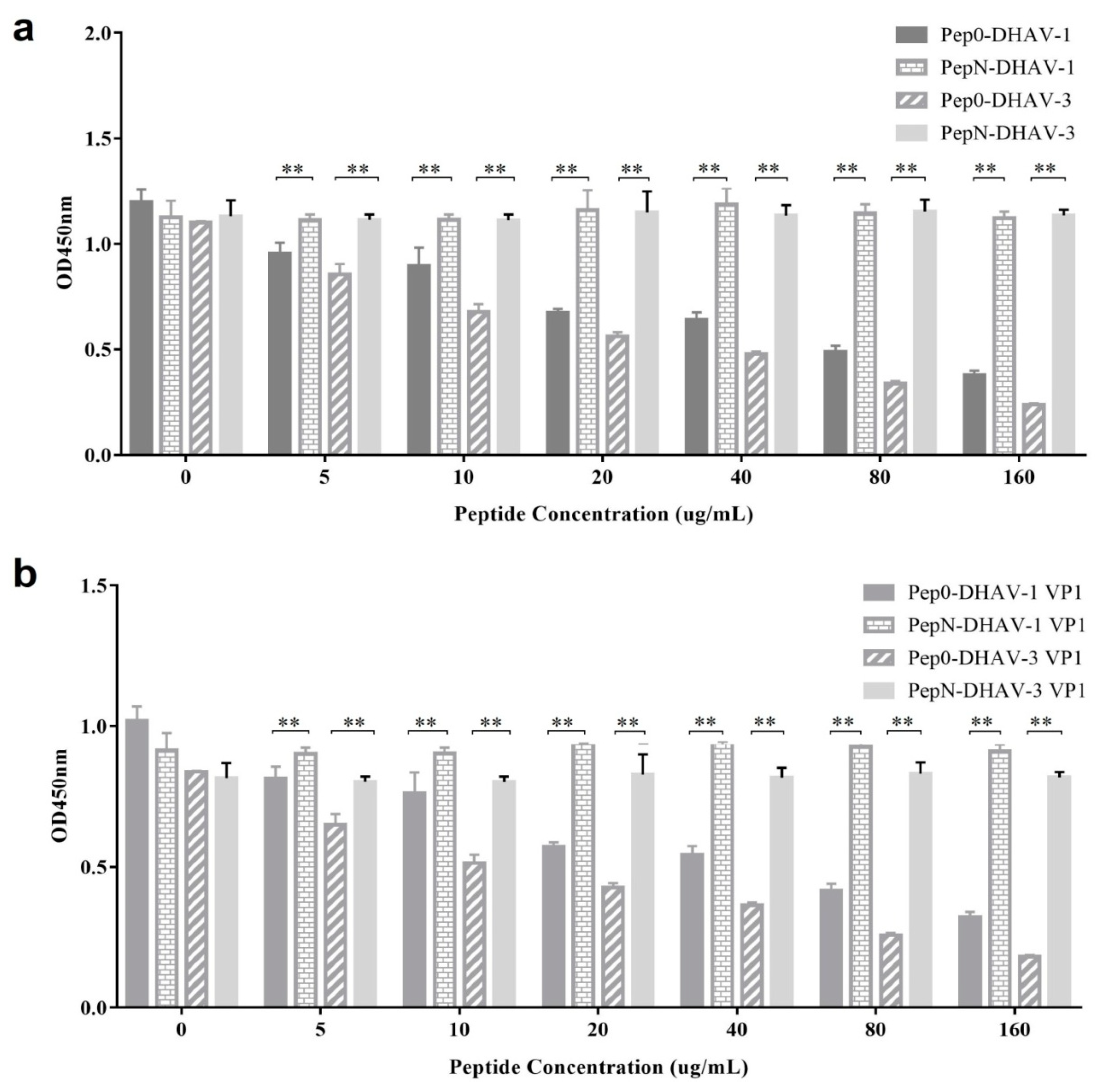
2.8. Competitive ELISA of Synthetic Peptides
The 96-well plates were coated with 100 µL of DHAV-1, DHAV-3, 1VP1-Sf9 expressed lysate, or 3VP1-Sf9 expressed lysate at an optimal dilution at 4 °C overnight (a preliminary experiment which was designed to determine the most appropriate amount of the antigens used in the competition ELISA test and it was not shown here). The plates were washed three times, followed by blocking with 3% BSA in PBS for 1.5 h at 37 °C. Thereafter, the synthetic peptide Pep0 or PepN (0, 5, 10, 20, 40, 80 and 160 μg/mL) was combined with mAb 4E6 (0.2 μg/mL in PBS) and incubated at 37 °C for 1 h. The peptide/antibody combinations were added to the DHAV-coated 96-well plates and incubated at 37 °C for 1 h. The plates were washed three times with PBST, followed by the addition of HRP-labeled goat anti-mouse IgG (1:4000) and incubation at 37 °C for 1 h. Subsequently, TMB substrate was added and the plates were incubated at 37 °C for 15 min. The reaction was stopped with 2 M H2SO4 and the absorbance was measured at 450 nm. All the statistical analysis in this study was performed using GraphPad software.
2.9. Immunization of Rabbits with Synthetic Pep-Keyhole Limpet Hemocyanin (KLH)
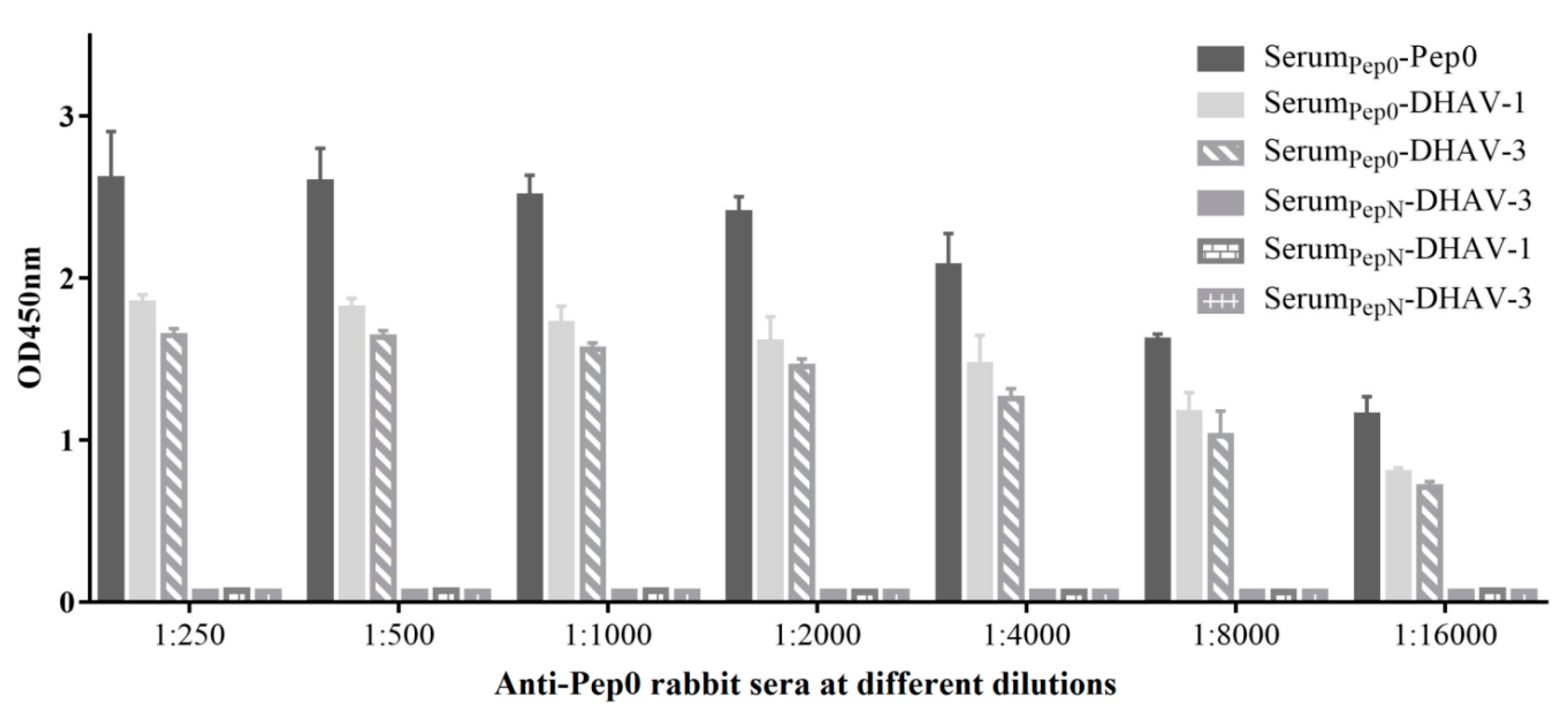
Pep0 and PepN were respectively synthesized and coupled to keyhole limpet hemocyanin (KLH), and named as Pep0-KLH and PepN-KLH. To prepare the polyclonal antibody, rabbits were injected with 200 μg of peptide emulsified in Freund’s complete adjuvant (FCA), boosted 2 weeks later, and re-boosted two additional times at 2-week intervals with 200 μg of peptide emulsified in Freund’s incomplete adjuvant (FIA). Sera were collected at 7 days after the third immunization and detected by ELISA.
The 96-well plates were respectively coated with the peptide Pep0 (stock concentration, 0.4 μg/mL; 100 μL/well), DHAV-1 and DHAV-3 (concentration was identical to that indicated above) at 4 °C overnight, and then blocked with 3% BSA. Immunized rabbit sera at different dilutions were added to the plates, which were then incubated at 37 °C for 2 h. Antibodies bound to the peptide Pep0, DHAV-1 and DHAV-3 were detected by ELISA with HRP-labeled goat anti-rabbit IgG (1:250) followed by the peroxidase substrate.
2.10. Blocking Assay of Pep0 Sera to DHAV
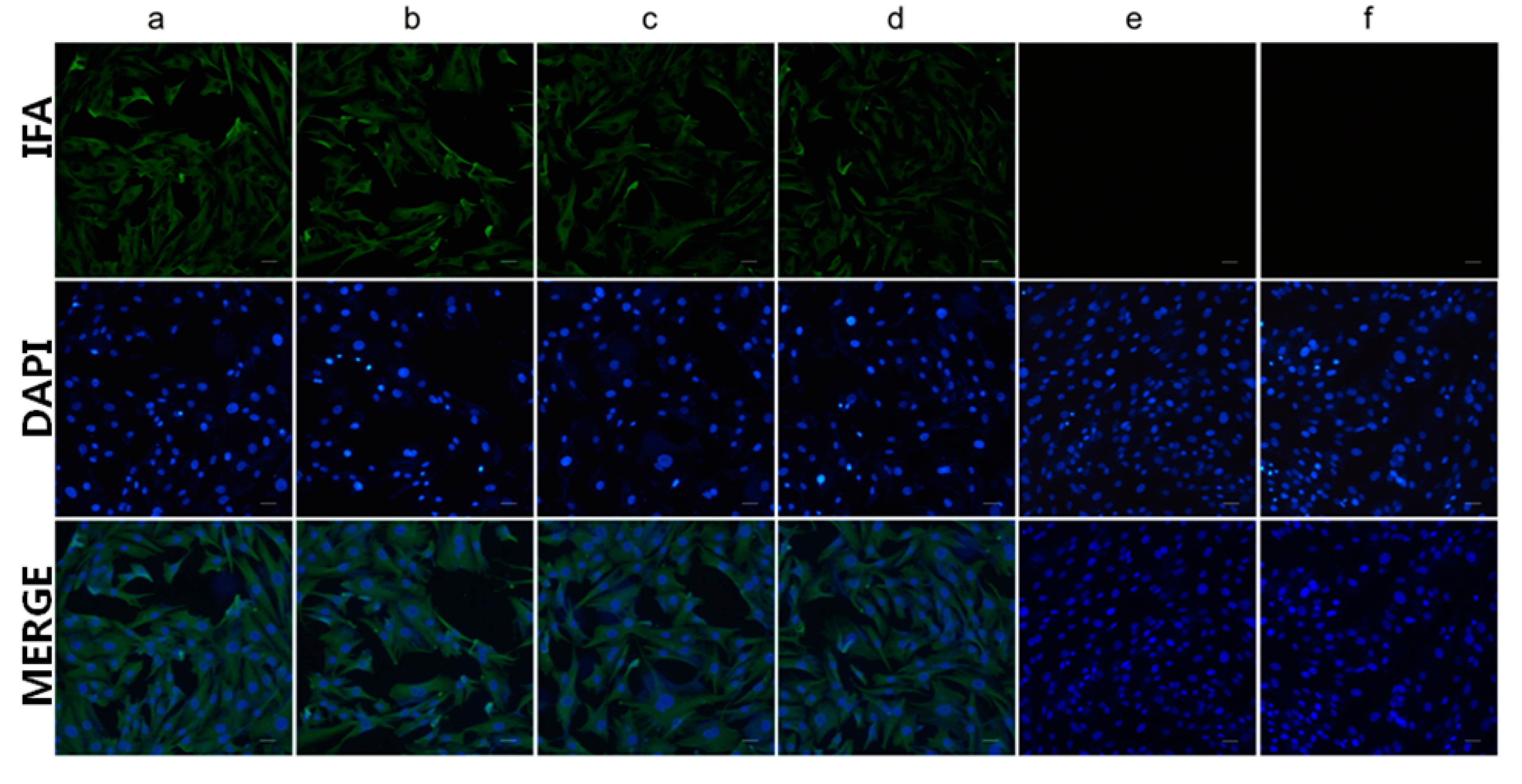
Monolayers of primary duck embryonic hepatocytes (DEH) cells were cultured and infected with DHAV-1 FC16115 and DHAV-3 JN1206 strains respectively. IFA was performed using mAb 4E6 and anti-Pep0 rabbit sera as the primary antibodies and FITC-conjugated goat anti-mouse IgG as the secondary antibody. Images were acquired with an identical exposure time and digitally documented.
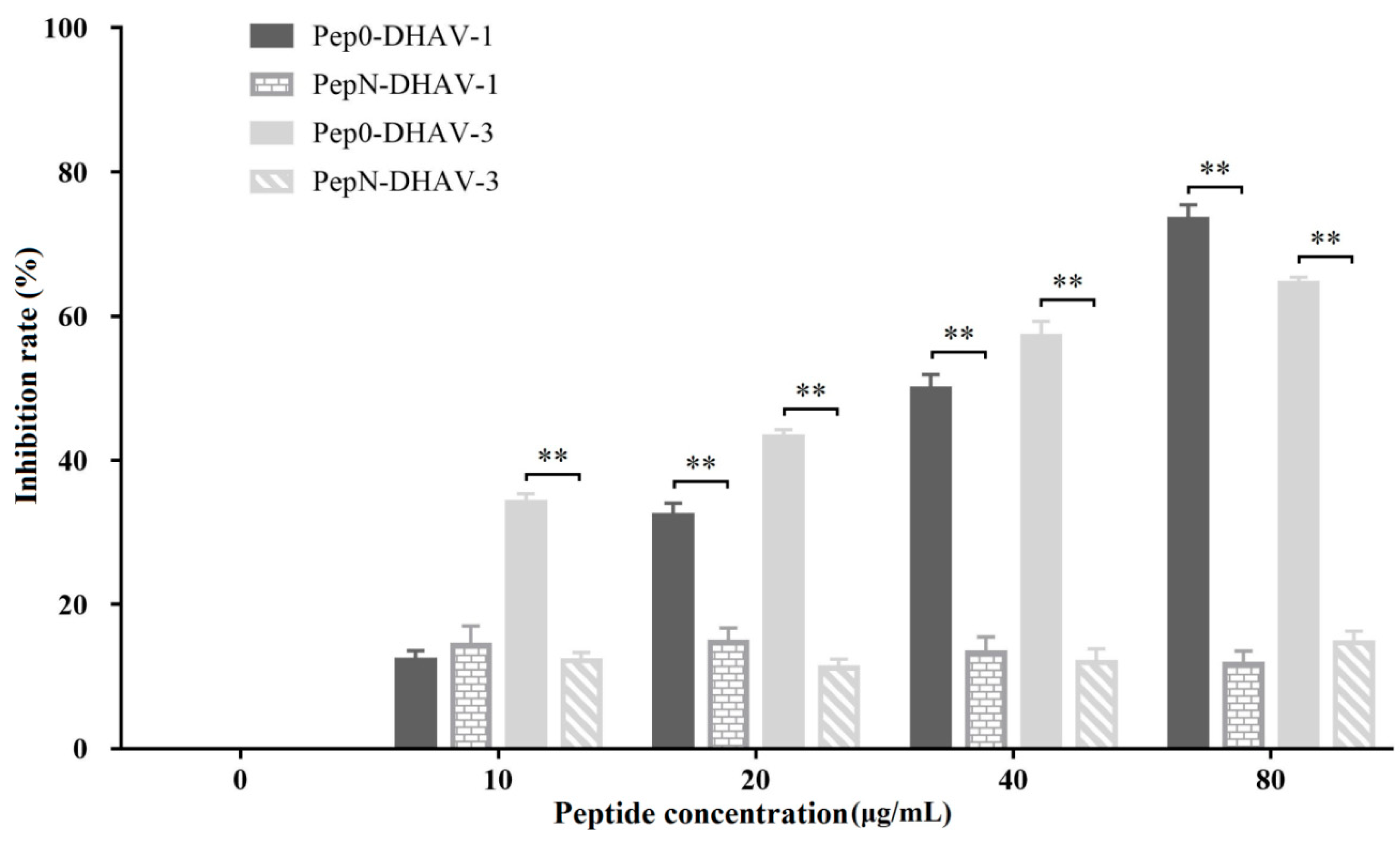
DEH cells were uniformly seeded in 12-well plates. Pep0 and PepN at 0, 10, 20, 40, and 80 μg/mL were respectively combined with an optimal concentration of DHAV and then added to the cells. After incubation for 2 h, the mixture was discarded, and the cells were washed. After incubation for 72 h at 37 °C, the cells were collected, and DHAV infection was assessed using real-time quantitative polymerase chain reaction (RT-qPCR) as described in our previous study [
17].
2.11. Identification of the Essential Amino Acids in the Epitopes
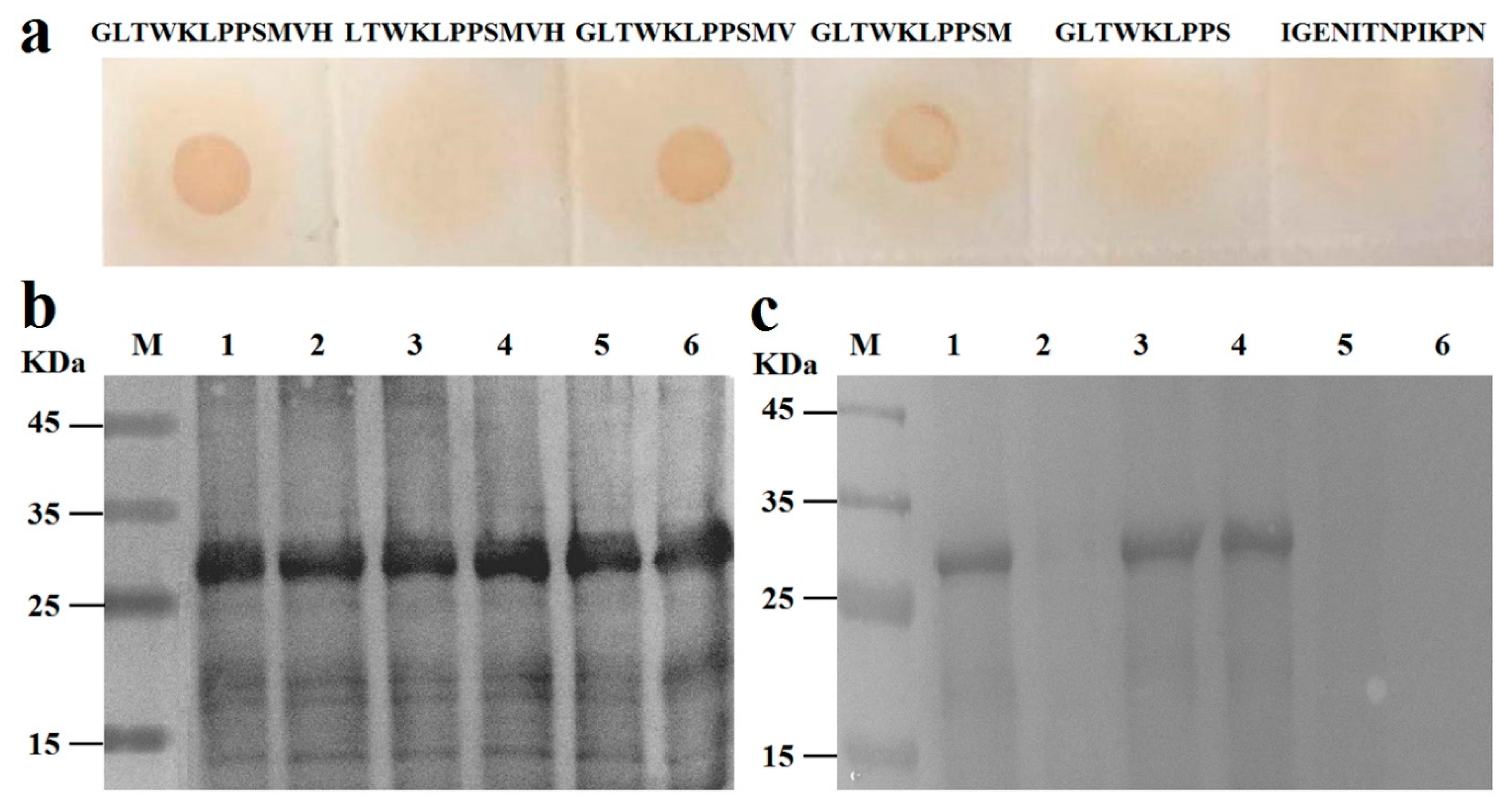
C- or N-terminal deletion mutants of the
1GLTWKLPPSMVH
12 peptide were synthesized (purity >95%) by GenScript China Inc (
Table 1), and approximately 1 µg of each mutant was transferred to a nitrocellulose membrane (Millipore, Bedford, MA, USA). Subsequently, anti-Pep0 rabbit sera (1:1000) were incubated with the membrane-bound peptides at 37 °C for 1 h. After washing three times with PBST, the membrane was probed with HRP-conjugated goat anti-rabbit IgG as secondary antibody.
To prepare the GST-Pep fusion protein, the sense and antisense oligonucleotide fragments encoding the corresponding Pep clone, with stop codon,
HindIII site, and sticky ends of
BamHI/
SalI, were synthesized by Sango Company (Shanghai, China) (
Table 1). The synthesized fragments were inserted at the
BamHI and
SalI sites of the pGEX-6P-1 vector to construct the recombinant vectors. The expressed fragments, which were purified with the GST Purification Kit (TaKaRa, Dalian, China), were subjected to 10% SDS-PAGE and finally transferred to nitrocellulose membranes. The membranes were probed with mAb 4E6 (1:1000) as the primary antibody described above.
2.12. Immunization of Ducks with Synthetic Pep1-KLH
A total of 90 ducklings were immunized by intramuscular injection with Pep1-KLH (25 μg/duckling) emulsified in FCA on day 1 and with Pep1-KLH (50 μg/duckling) emulsified in FIA on day 7. For the control group, 90 ducklings were immunized with PepN-KLH as described above. Serum samples were collected from live ducklings on day 10, before the challenge with DHAVs. Using HRP-labeled goat anti-duck IgG (1:500), antibodies bound to the peptide Pep0, DHAV-1, and DHAV-3 were detected by ELISA as described above.
For each group, on day 10, 30 ducklings were infected with the fifth generation duck embryo allantoic liquids of the DHAV-1 FC16115 (0.2 mL/duck) by intramuscular and intranasal injections, 30 ducklings were infected with the fifth generation duck embryo allantoic liquids of the DHAV-3 JN1206 (0.2 mL/duck) in the same way, and the rest, 30 ducklings, were infected with both DHAV-1 FC16115 (0.1 mL/duck) and DHAV-3 JN1206 (0.1 mL/duck). The mental state of each duckling was recorded daily for 7 consecutive days and then the ducklings were euthanized. The livers of all ducklings were collected and stored at −80 °C. The RNA viral copy number was determined by RT-qPCR amplification as described in our previous study [
17].
4. Discussion
For DHAV, a unique member of the
Avihepatovirus genus of the
Picornaviridae family, VP1 protein is the most important immunogenic antigen and induces protective neutralizing antibodies in ducks [
8,
9,
10]. Among three genotypes, DHAV-1 and DHAV-3 are considered as the most common continental segregation types [
1]. Although possessing VP1 proteins of different lengths (generally 238 and 240 amino acids residues), the results of sequence alignment show a certain degree of amino acid similarity between the two types [
4,
5]. In previous study, we identified two conserved neutralizing linear B-cell epitope with a mAb 4F8, namely
75GEIILT
80 in DHAV-1 VP1 and
75GEVILT
80 in DHAV-3 VP1 protein [
8], further confirming that limited cross neutralization existed between DHAV-1 and DHAV-3.
Epitopes are characterized as linear and conformational epitopes. Different from linear epitopes, which usually contain 6-10 linearized surface amino acids in the target protein, amino acids for the conformational epitopes are discontinuous and difficult to map [
18]. In previous reports, several linear epitopes were identified in DHAV with mAb [
8,
9,
10]. To date, there was no conformational epitope being identified in DHAV.
An abundant of phage display peptides constituted the library with all possible amino acid compositions. By screening the library, many mimotopes were identified, and the peptides have been reported to trigger immune responses as successful vaccines [
19,
20]. Using a nanobody (Nb) DHAV-1 VP1 protein, we previously screened the phage display library of DHAV-1 gene fragments and identified a conserved linear epitope
174PAPTST
179 in DHAV-1 VP1 protein [
10]. In this study, purified mAb 4E6 was used to screen a 12-mer phage display peptide library by bio-panning, and the peptide
1GLTWKLPPSMVH
12 was obtained. Further analysis demonstrated that the peptide could specifically block mAb 4E6 from binding DHAV (
Figure 4). Moreover, the results of immunization assays demonstrated that the shortened peptide,
1GLTWKLPPSM
10, could trigger an effective immune response to DHAV and inhibit virus infection in vivo (
Figure 7). Although sequence alignment showed no homology between the mimotopes and the DHAV VP1 sequence, these results strongly suggested that the peptide was identical to the antigen epitope of DHAV that could be recognized by mAb 4E6.
Mixed infections of DHAV-1 and DHAV-3 are widespread in China’s mainland [
21]. In this study, after intramuscular vaccination in ducklings, the synthetic Pep1-KLH displayed the ability of eliciting strong specific antibody against DHAV-1 and DHAV-3 in sera (
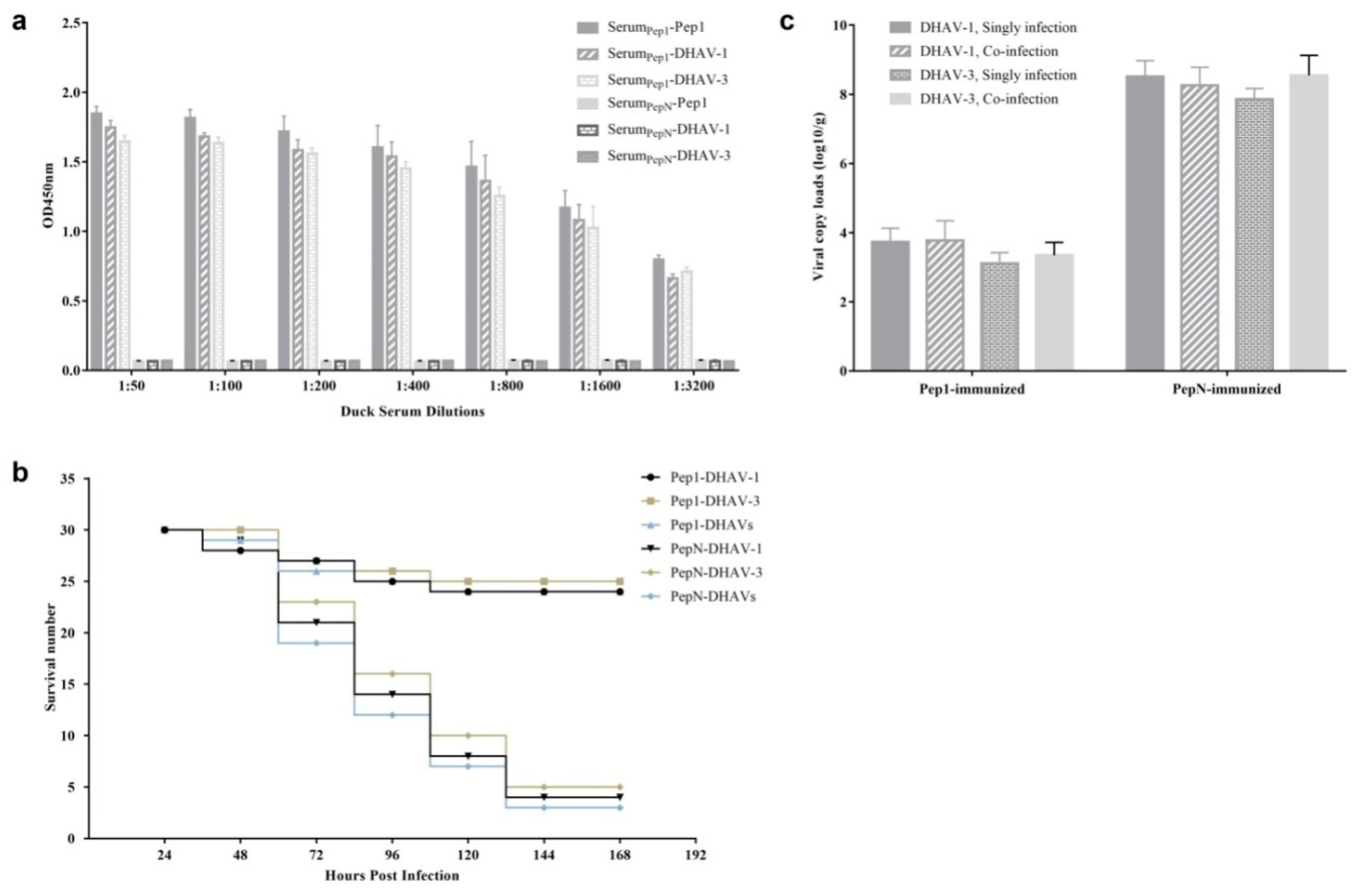
Figure 9a). The survival rate was 81.1% (73/90) for Pep1-KLH immunized group, whereas 13.3% (12/90) for PepN-KLH immunized group (
Figure 9b). The viral loads of DHAV-1 and DHAV-3 in the livers of live ducklings in Pep1-KLH immunized group were significantly lower than those in the livers of dead ducklings in PepN-KLH immunized group (
Figure 9c). These results indicated that the immunization of ducklings with the mimotope Pep1 could inhibit replication of both DHAV-1 and DHAV-3 and protect the ducklings from DVH, suggesting that the neutralizing conformational mimotope Pep1 might be a promising vaccine candidate for the prevention of DHAV infection.
In animal models of co-infection, the pathogenicity of one virus could be promoted or suppressed by another virus [
22,
23]. In current research, the viral loads of DHAV-1 and DHAV-3 in the livers of dead ducklings respectively had no significant difference between single-infected dead ducklings and co-infected ones (
Figure 9c), which was consistent with our previous study [
17]. In addition, the viral loads of DHAV-1 and DHAV-3 in the livers of live ducklings also respectively had no significant difference between the single infected group or co-infected group (
p > 0.05). These results indicated that the co-infection of DHAV-1 and DHAV-3 had no effect on the replication and the viral loads of the two viruses
in vivo.

 ,
, 










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}