Precision Tools in Immuno-Oncology: Synthetic Gene Circuits for Cancer Immunotherapy



Abstract
:1. The Essential of Cancer Immunotherapies
2. T Cell-Based Therapies
3. Synthetic Biology for Immunotherapy
Synthetic Biology Approaches for Mammalian Cells Therapy and Cancer Immunotherapy
4. Synthetic Biology Approaches to Boost CAR-T Cell Treatment’s Efficacy
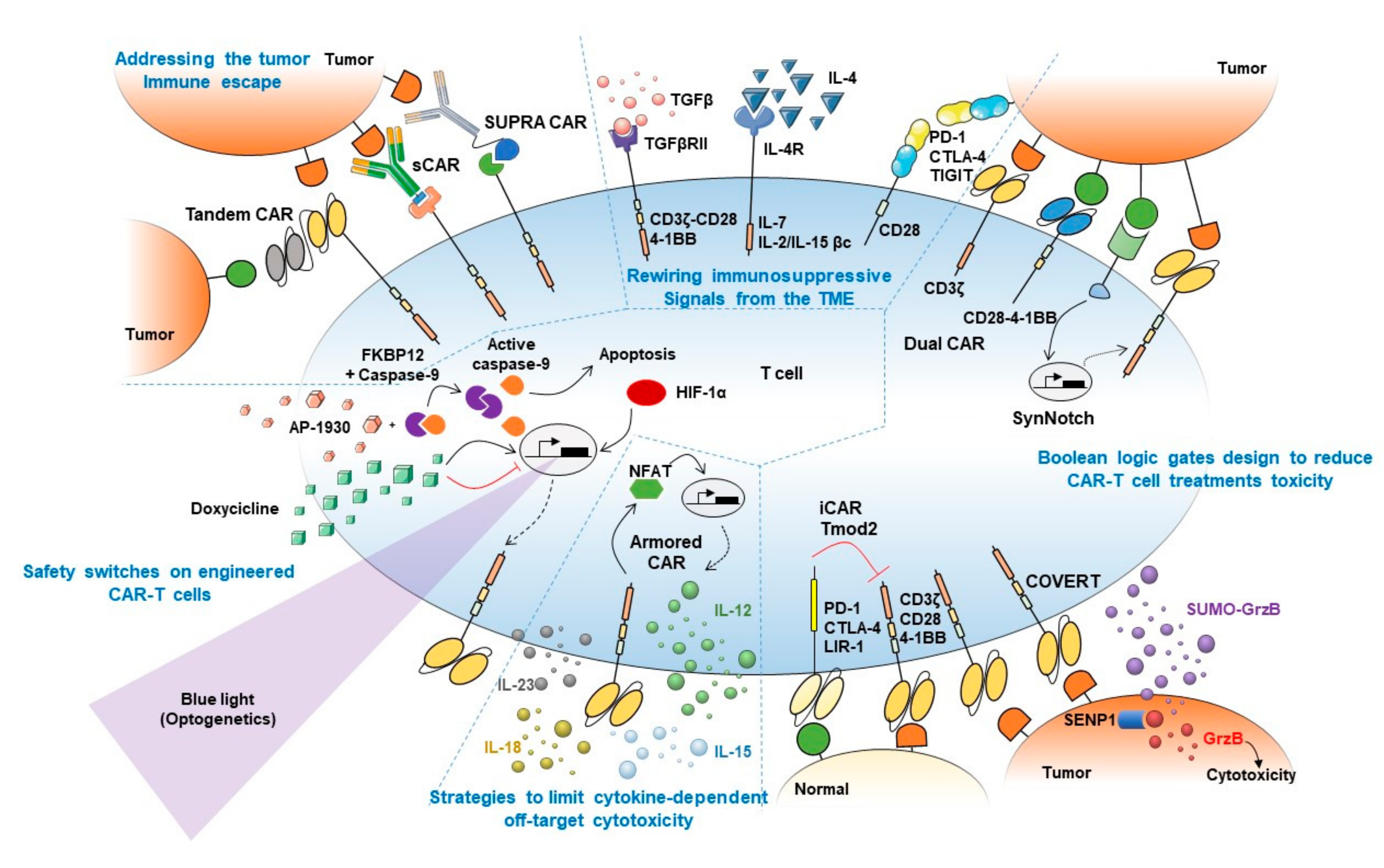
4.1. Addressing the Tumor Immune Escape
4.2. Rewiring Immunosuppressive Signals from the TME
4.3. Boolean Logic Gates Design to Reduce CAR-T Cell Treatments Toxicity
4.4. Strategies to Limit Cytokine-Dependent Off-Target Cytotoxicity
4.5. Safety Switches on Engineered CAR-T Cells
5. What’s Next? Future Targets to Increase Long-Term Efficacy of CAR-T Therapy and Unleash T Cells Cytotoxic Potential
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ACT | Adoptive cell-based therapy |
| ADCC | Antibody-dependent cellular cytotoxicity |
| BiTEs | Bispecific T-cell engagers |
| CAR | Chimeric antigen receptor |
| CBL-B | Casitas B-lineage lymphoma proto-oncogene-B |
| CISH | Cytokine inducible SH2 containing protein |
| COVERT | Cytoplasmic oncoprotein verifier and response trigger |
| CRS | Cytokine release syndrome |
| CTLA-4 | Cytotoxic T-lymphocyte antigen 4 |
| dCas9 | Dead CRISPR associated protein 9 |
| DGK | Diacylglycerol kinase |
| EGFR | Epidermal growth factor receptor |
| EpoR | Erythropoietin receptor |
| GEARs | Generalized engineered activation regulators |
| GEMS | Generalized extracellular molecules sensors |
| GM-CSF | Granulocyte-macrophage colony stimulating factor |
| GPCRs | G protein-coupled receptors |
| GrzB | Granzime B |
| HIF-1α | Hypoxia-inducible factor-1α |
| iCAR | Inactivating CAR |
| ICOS | Inducible T cell co-stimulator |
| IDO-1 | Indoleamine 2,3-dioxygenase 1 |
| IFN | Interferon |
| irAEs | Immune-related adverse events |
| ITAM | Immunoreceptor tyrosine-based activation motif |
| ITIM | Immunoreceptor tyrosine-based inhibitory motif |
| JAK/STAT | Janus kinase/signal transducer and activator of transcription |
| LAG-3 | Lymphocyte-activation gene-3 |
| LOH | Loss of heterozygosity |
| MAPK | Mitogen-activated protein kinase |
| MCP | MS2 bacteriophage coat protein |
| MDSC | Myeloid-derived suppressor cells |
| MHC | Major histocompatibility complex |
| MRSA | Methicillin resistant Staphylococcus aureus |
| NR4A | Nuclear receptor 4 A |
| PD-1/PD-L1 | Programmed cell death protein-1/Programmed cell death protein-ligand 1 |
| PI3K/Akt | Phosphatidylinositol 3-kinase/Protein kinase B |
| PLC-γ | Phospholipase C gamma |
| PSMA | Prostate-specific membrane antigen |
| PTPN22 | Protein tyrosine phosphatase, non-receptor type 22 |
| RASAL1 | RAS protein activator like 1 |
| sCARs | Switchable CAR-T-cells |
| scFv | Single chain variable fragment of an antibody |
| SENP1 | Sentrin-specific protease 1 |
| sgRNA | Synthetic guide RNA |
| SHP1 | Src homology 2 domain-containing protein tyrosine phosphatase 1 |
| SOCS1 | Suppressor of cytokine signaling 1 |
| SUMO | Small ubiquitin-like modifier |
| SUPRA CAR | Split universal programmable CAR |
| SynNotch | Synthetic Notch |
| TAAs | Tumor-associated antigens |
| TCE | T cell exhaustion |
| TCM | T central memory |
| TCR | T cell receptor |
| TEV | Tobacco etch virus |
| TGFβ | Transforming growth factor beta |
| TGFβRII | Transforming growth factor beta receptor 2 |
| TIGIT | T cell immunoreceptor with Ig and ITIM domains |
| TILs | Tumor infiltrating lymphocytes |
| TIM-3 | T-cell immunoglobulin and mucin domain-3 |
| TLRs | Toll-like receptors |
| TME | Tumor microenvironment |
| TNFR | Tumor necrosis factor receptor |
| TOX | Thymocyte selection-associated high mobility group box protein |
| TRUCKs | T cells redirected for universal cytokine-mediated killing |
| VEGF | Vascular endothelial growth factor |
References
- Larkin, J.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.J.; Cowey, C.L.; Lao, C.D.; Schadendorf, D.; Dummer, R.; Smylie, M.; Rutkowski, P.; et al. Combined Nivolumab and Ipilimumab or Monotherapy in Untreated Melanoma. N. Engl. J. Med. 2015, 373, 23–34. [Google Scholar] [CrossRef] [Green Version]
- Houot, R.; Schultz, L.M.; Marabelle, A.; Kohrt, H. T-cell-based Immunotherapy: Adoptive Cell Transfer and Checkpoint Inhibition. Cancer Immunol. Res. 2015, 3, 1115–1122. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, C. The benefits of immunotherapy combinations. Nature 2017, 552, S67–S69. [Google Scholar] [CrossRef] [Green Version]
- Riley, R.S.; June, C.H.; Langer, R.; Mitchell, M.J. Delivery technologies for cancer immunotherapy. Nat. Rev. Drug Discov. 2019, 18, 175–196. [Google Scholar] [CrossRef]
- Wang, P.-F.; Chen, Y.; Song, S.-Y.; Wang, T.-J.; Ji, W.-J.; Li, S.-W.; Liu, N.; Yan, C.-X. Immune-Related Adverse Events Associated with Anti-PD-1/PD-L1 Treatment for Malignancies: A Meta-Analysis. Front. Pharmacol. 2017, 8, 1–20. [Google Scholar] [CrossRef]
- Chen, T.W.; Razak, A.R.; Bedard, P.L.; Siu, L.L.; Hansen, A.R. A systematic review of immune-related adverse event reporting in clinical trials of immune checkpoint inhibitors. Ann. Oncol. 2015, 26, 1824–1829. [Google Scholar] [CrossRef] [Green Version]
- June, C.H.; Warshauer, J.T.; Bluestone, J.A. Is autoimmunity the Achilles’ heel of cancer immunotherapy? Nat. Med. 2017, 23, 540–547. [Google Scholar] [CrossRef] [Green Version]
- Berraondo, P.; Sanmamed, M.F.; Ochoa, M.C.; Etxeberria, I.; Aznar, M.A.; Pérez-Gracia, J.L.; Rodríguez-Ruiz, M.E.; Ponz-Sarvise, M.; Castañón, E.; Melero, I. Cytokines in clinical cancer immunotherapy. Br. J. Cancer 2019, 120, 6–15. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.; Margolin, K. Cytokines in Cancer Immunotherapy. Cancers 2011, 3, 3856–3893. [Google Scholar] [CrossRef]
- Peggs, K.S.; Quezada, S.A.; Allison, J.P. Cancer immunotherapy: Co-stimulatory agonists and co-inhibitory antagonists. Clin. Exp. Immunol. 2009, 157, 9–19. [Google Scholar] [CrossRef]
- Guo, C.; Manjili, M.H.; Subjeck, J.R.; Sarkar, D.; Fisher, P.B.; Wang, X.-Y. Therapeutic Cancer Vaccines. In Advances in Cancer Research; Academic Press: Cambridge, MA, USA, 2013; Volume 119, pp. 421–475. ISBN 9780124071902. [Google Scholar]
- Bhatt, D.; Daemen, T. Therapeutic Vaccines and Cancer Immunotherapy. Vaccines 2020, 8, 596. [Google Scholar] [CrossRef]
- Geukes Foppen, M.H.; Donia, M.; Svane, I.M.; Haanen, J.B.A.G. Tumor-infiltrating lymphocytes for the treatment of metastatic cancer. Mol. Oncol. 2015, 9, 1918–1935. [Google Scholar] [CrossRef]
- Williams, A.D.; Payne, K.K.; Posey, A.D.; Hill, C.; Conejo-Garcia, J.; June, C.H.; Tchou, J. Immunotherapy for Breast Cancer: Current and Future Strategies. Curr. Surg. Rep. 2017, 5, 31. [Google Scholar] [CrossRef] [Green Version]
- Menon, S.; Shin, S.; Dy, G. Advances in Cancer Immunotherapy in Solid Tumors. Cancers 2016, 8, 106. [Google Scholar] [CrossRef] [Green Version]
- Ti, D.; Bai, M.; Li, X.; Wei, J.; Chen, D.; Wu, Z.; Wang, Y.; Han, W. Adaptive T cell immunotherapy in cancer. Sci. China Life Sci. 2020, 468. [Google Scholar] [CrossRef]
- Dafni, U.; Michielin, O.; Lluesma, S.M.; Tsourti, Z.; Polydoropoulou, V.; Karlis, D.; Besser, M.J.; Haanen, J.; Svane, I.-M.; Ohashi, P.S.; et al. Efficacy of adoptive therapy with tumor-infiltrating lymphocytes and recombinant interleukin-2 in advanced cutaneous melanoma: A systematic review and meta-analysis. Ann. Oncol. 2019, 30, 1902–1913. [Google Scholar] [CrossRef] [Green Version]
- Chmielewski, M.; Hombach, A.A.; Abken, H. Antigen-Specific T-Cell Activation Independently of the MHC: Chimeric Antigen Receptor-Redirected T Cells. Front. Immunol. 2013, 4, 371. [Google Scholar] [CrossRef] [Green Version]
- Barbari, C.; Fontaine, T.; Parajuli, P.; Lamichhane, N.; Jakubski, S.; Lamichhane, P.; Deshmukh, R.R. Immunotherapies and Combination Strategies for Immuno-Oncology. Int. J. Mol. Sci. 2020, 21, 5009. [Google Scholar] [CrossRef]
- Zhao, Z.; Chen, Y.; Francisco, N.M.; Zhang, Y.; Wu, M. The application of CAR-T cell therapy in hematological malignancies: Advantages and challenges. Acta Pharm. Sin. B 2018, 8, 539–551. [Google Scholar] [CrossRef]
- Tang, H.; Qiao, J.; Fu, Y.-X. Immunotherapy and tumor microenvironment. Cancer Lett. 2016, 370, 85–90. [Google Scholar] [CrossRef]
- Rodriguez-Garcia, A.; Palazon, A.; Noguera-Ortega, E.; Powell, D.J.; Guedan, S. CAR-T Cells Hit the Tumor Microenvironment: Strategies to Overcome Tumor Escape. Front. Immunol. 2020, 11. [Google Scholar] [CrossRef]
- Lanitis, E.; Irving, M.; Coukos, G. Targeting the tumor vasculature to enhance T cell activity. Curr. Opin. Immunol. 2015, 33, 55–63. [Google Scholar] [CrossRef] [Green Version]
- Galon, J.; Bruni, D. Approaches to treat immune hot, altered and cold tumours with combination immunotherapies. Nat. Rev. Drug Discov. 2019, 18, 197–218. [Google Scholar] [CrossRef]
- Whiteside, T.L.; Demaria, S.; Rodriguez-Ruiz, M.E.; Zarour, H.M.; Melero, I. Emerging Opportunities and Challenges in Cancer Immunotherapy. Clin. Cancer Res. 2016, 22, 1845–1855. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Han, W. Biomarkers of cytokine release syndrome and neurotoxicity related to CAR-T cell therapy. Biomark. Res. 2018, 6, 4. [Google Scholar] [CrossRef] [Green Version]
- Hunter, B.D.; Jacobson, C.A. CAR T-Cell Associated Neurotoxicity: Mechanisms, Clinicopathologic Correlates, and Future Directions. JNCI J. Natl. Cancer Inst. 2019, 111, 646–654. [Google Scholar] [CrossRef]
- Caliendo, F.; Dukhinova, M.; Siciliano, V. Engineered Cell-Based Therapeutics: Synthetic Biology Meets Immunology. Front. Bioeng. Biotechnol. 2019, 7. [Google Scholar] [CrossRef] [Green Version]
- Kopniczky, M.B.; Canavan, C.; McClymont, D.W.; Crone, M.A.; Suckling, L.; Goetzmann, B.; Siciliano, V.; MacDonald, J.T.; Jensen, K.; Freemont, P.S. Cell-Free Protein Synthesis as a Prototyping Platform for Mammalian Synthetic Biology. ACS Synth. Biol. 2020, 9, 144–156. [Google Scholar] [CrossRef]
- Jeong, D.; Klocke, M.; Agarwal, S.; Kim, J.; Choi, S.; Franco, E.; Kim, J. Cell-Free Synthetic Biology Platform for Engineering Synthetic Biological Circuits and Systems. Methods Protoc. 2019, 2, 39. [Google Scholar] [CrossRef] [Green Version]
- Scheller, L.; Fussenegger, M. From synthetic biology to human therapy: Engineered mammalian cells. Curr. Opin. Biotechnol. 2019, 58, 108–116. [Google Scholar] [CrossRef]
- Di Bernardo, D.; Marucci, L.; Menolascina, F.; Siciliano, V. Predicting Synthetic Gene Networks. In Methods in Molecular Biology; Humana Press: Totowa, NJ, USA, 2012; Volume 813, pp. 57–81. ISBN 9781617794117. [Google Scholar]
- MacDonald, J.T.; Siciliano, V. Computational Sequence Design with R2oDNA Designer. In Methods in Molecular Biology; Humana: New York, NY, USA, 2017; Volume 1651, pp. 249–262. [Google Scholar]
- Cella, F.; Siciliano, V. Protein-based parts and devices that respond to intracellular and extracellular signals in mammalian cells. Curr. Opin. Chem. Biol. 2019, 52, 47–53. [Google Scholar] [CrossRef]
- Kojima, R.; Aubel, D.; Fussenegger, M. Building sophisticated sensors of extracellular cues that enable mammalian cells to work as “doctors” in the body. Cell. Mol. Life Sci. 2020, 77, 3567–3581. [Google Scholar] [CrossRef] [Green Version]
- Schukur, L.; Geering, B.; Charpin-El Hamri, G.; Fussenegger, M. Implantable synthetic cytokine converter cells with AND-gate logic treat experimental psoriasis. Sci. Transl. Med. 2015, 7, 318ra201. [Google Scholar] [CrossRef]
- Liu, Y.; Charpin-El Hamri, G.; Ye, H.; Fussenegger, M. A synthetic free fatty acid-regulated transgene switch in mammalian cells and mice. Nucleic Acids Res. 2018, 46, 9864–9874. [Google Scholar] [CrossRef]
- Scheller, L.; Strittmatter, T.; Fuchs, D.; Bojar, D.; Fussenegger, M. Generalized extracellular molecule sensor platform for programming cellular behavior. Nat. Chem. Biol. 2018, 14, 723–729. [Google Scholar] [CrossRef]
- Krawczyk, K.; Scheller, L.; Kim, H.; Fussenegger, M. Rewiring of endogenous signaling pathways to genomic targets for therapeutic cell reprogramming. Nat. Commun. 2020, 11, 608. [Google Scholar] [CrossRef] [Green Version]
- Siciliano, V.; DiAndreth, B.; Monel, B.; Beal, J.; Huh, J.; Clayton, K.L.; Wroblewska, L.; McKeon, A.; Walker, B.D.; Weiss, R. Engineering modular intracellular protein sensor-actuator devices. Nat. Commun. 2018, 9, 1881. [Google Scholar] [CrossRef]
- Duhkinova, M.; Crina, C.; Weiss, R.; Siciliano, V. Engineering Intracellular Protein Sensors in Mammalian Cells. J. Vis. Exp. 2020, 2020. [Google Scholar] [CrossRef]
- June, C.H.; O’Connor, R.S.; Kawalekar, O.U.; Ghassemi, S.; Milone, M.C. CAR T cell immunotherapy for human cancer. Science 2018, 359, 1361–1365. [Google Scholar] [CrossRef] [Green Version]
- Chmielewski, M.; Hombach, A.A.; Abken, H. Of CARs and TRUCKs: Chimeric antigen receptor (CAR) T cells engineered with an inducible cytokine to modulate the tumor stroma. Immunol. Rev. 2014, 257, 83–90. [Google Scholar] [CrossRef]
- Chmielewski, M.; Abken, H. TRUCKs: The fourth generation of CARs. Expert Opin. Biol. Ther. 2015, 15, 1145–1154. [Google Scholar] [CrossRef]
- Zah, E.; Lin, M.-Y.; Silva-Benedict, A.; Jensen, M.C.; Chen, Y.Y. T Cells Expressing CD19/CD20 Bispecific Chimeric Antigen Receptors Prevent Antigen Escape by Malignant B Cells. Cancer Immunol. Res. 2016, 4, 498–508. [Google Scholar] [CrossRef] [Green Version]
- Zah, E.; Nam, E.; Bhuvan, V.; Tran, U.; Ji, B.Y.; Gosliner, S.B.; Wang, X.; Brown, C.E.; Chen, Y.Y. Systematically optimized BCMA/CS1 bispecific CAR-T cells robustly control heterogeneous multiple myeloma. Nat. Commun. 2020, 11, 2283. [Google Scholar] [CrossRef]
- Qin, H.; Ramakrishna, S.; Nguyen, S.; Fountaine, T.J.; Ponduri, A.; Stetler-Stevenson, M.; Yuan, C.M.; Haso, W.; Shern, J.F.; Shah, N.N.; et al. Preclinical Development of Bivalent Chimeric Antigen Receptors Targeting Both CD19 and CD22. Mol. Ther. Oncolytics 2018, 11, 127–137. [Google Scholar] [CrossRef] [Green Version]
- Rodgers, D.T.; Mazagova, M.; Hampton, E.N.; Cao, Y.; Ramadoss, N.S.; Hardy, I.R.; Schulman, A.; Du, J.; Wang, F.; Singer, O.; et al. Switch-mediated activation and retargeting of CAR-T cells for B-cell malignancies. Proc. Natl. Acad. Sci. USA 2016, 113, E459–E468. [Google Scholar] [CrossRef] [Green Version]
- Ma, J.S.Y.; Kim, J.Y.; Kazane, S.A.; Choi, S.; Yun, H.Y.; Kim, M.S.; Rodgers, D.T.; Pugh, H.M.; Singer, O.; Sun, S.B.; et al. Versatile strategy for controlling the specificity and activity of engineered T cells. Proc. Natl. Acad. Sci. USA 2016, 113, E450–E458. [Google Scholar] [CrossRef] [Green Version]
- Tamada, K.; Geng, D.; Sakoda, Y.; Bansal, N.; Srivastava, R.; Li, Z.; Davila, E. Redirecting Gene-Modified T Cells toward Various Cancer Types Using Tagged Antibodies. Clin. Cancer Res. 2012, 18, 6436–6445. [Google Scholar] [CrossRef] [Green Version]
- Urbanska, K.; Lanitis, E.; Poussin, M.; Lynn, R.C.; Gavin, B.P.; Kelderman, S.; Yu, J.; Scholler, N.; Powell, D.J. A Universal Strategy for Adoptive Immunotherapy of Cancer through Use of a Novel T-cell Antigen Receptor. Cancer Res. 2012, 72, 1844–1852. [Google Scholar] [CrossRef] [Green Version]
- Cho, J.H.; Collins, J.J.; Wong, W.W. Universal Chimeric Antigen Receptors for Multiplexed and Logical Control of T Cell Responses. Cell 2018, 173, 1426–1438.e11. [Google Scholar] [CrossRef] [Green Version]
- Chang, Z.L.; Lorenzini, M.H.; Chen, X.; Tran, U.; Bangayan, N.J.; Chen, Y.Y. Rewiring T-cell responses to soluble factors with chimeric antigen receptors. Nat. Chem. Biol. 2018, 14, 317–324. [Google Scholar] [CrossRef]
- Sukumaran, S.; Watanabe, N.; Bajgain, P.; Raja, K.; Mohammed, S.; Fisher, W.E.; Brenner, M.K.; Leen, A.M.; Vera, J.F. Enhancing the Potency and Specificity of Engineered T Cells for Cancer Treatment. Cancer Discov. 2018, 8, 972–987. [Google Scholar] [CrossRef] [Green Version]
- Leen, A.M.; Sukumaran, S.; Watanabe, N.; Mohammed, S.; Keirnan, J.; Yanagisawa, R.; Anurathapan, U.; Rendon, D.; Heslop, H.E.; Rooney, C.M.; et al. Reversal of Tumor Immune Inhibition Using a Chimeric Cytokine Receptor. Mol. Ther. 2014, 22, 1211–1220. [Google Scholar] [CrossRef] [Green Version]
- Wilkie, S.; Burbridge, S.E.; Chiapero-Stanke, L.; Pereira, A.C.P.; Cleary, S.; van der Stegen, S.J.C.; Spicer, J.F.; Davies, D.M.; Maher, J. Selective Expansion of Chimeric Antigen Receptor-targeted T-cells with Potent Effector Function using Interleukin-4. J. Biol. Chem. 2010, 285, 25538–25544. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Ranganathan, R.; Jiang, S.; Fang, C.; Sun, J.; Kim, S.; Newick, K.; Lo, A.; June, C.H.; Zhao, Y.; et al. A Chimeric Switch-Receptor Targeting PD1 Augments the Efficacy of Second-Generation CAR T Cells in Advanced Solid Tumors. Cancer Res. 2016, 76, 1578–1590. [Google Scholar] [CrossRef] [Green Version]
- Shin, J.H.; Park, H.B.; Oh, Y.M.; Lim, D.P.; Lee, J.E.; Seo, H.H.; Lee, S.J.; Eom, H.S.; Kim, I.-H.; Lee, S.H.; et al. Positive conversion of negative signaling of CTLA4 potentiates antitumor efficacy of adoptive T-cell therapy in murine tumor models. Blood 2012, 119, 5678–5687. [Google Scholar] [CrossRef] [Green Version]
- Kloss, C.C.; Condomines, M.; Cartellieri, M.; Bachmann, M.; Sadelain, M. Combinatorial antigen recognition with balanced signaling promotes selective tumor eradication by engineered T cells. Nat. Biotechnol. 2013, 31, 71–75. [Google Scholar] [CrossRef]
- Roybal, K.T.; Williams, J.Z.; Morsut, L.; Rupp, L.J.; Kolinko, I.; Choe, J.H.; Walker, W.J.; McNally, K.A.; Lim, W.A. Engineering T Cells with Customized Therapeutic Response Programs Using Synthetic Notch Receptors. Cell 2016, 167, 419–432.e16. [Google Scholar] [CrossRef] [Green Version]
- Ho, P.; Ede, C.; Chen, Y.Y. Modularly Constructed Synthetic Granzyme B Molecule Enables Interrogation of Intracellular Proteases for Targeted Cytotoxicity. ACS Synth. Biol. 2017, 6, 1484–1495. [Google Scholar] [CrossRef]
- Fedorov, V.D.; Themeli, M.; Sadelain, M. PD-1- and CTLA-4-Based Inhibitory Chimeric Antigen Receptors (iCARs) Divert Off-Target Immunotherapy Responses. Sci. Transl. Med. 2013, 5, 215ra172. [Google Scholar] [CrossRef] [Green Version]
- Hamburger, A.E.; DiAndreth, B.; Cui, J.; Daris, M.E.; Munguia, M.L.; Deshmukh, K.; Mock, J.-Y.; Asuelime, G.E.; Lim, E.D.; Kreke, M.R.; et al. Engineered T cells directed at tumors with defined allelic loss. Mol. Immunol. 2020. [Google Scholar] [CrossRef]
- Sockolosky, J.T.; Trotta, E.; Parisi, G.; Picton, L.; Su, L.L.; Le, A.C.; Chhabra, A.; Silveria, S.L.; George, B.M.; King, I.C.; et al. Selective targeting of engineered T cells using orthogonal IL-2 cytokine-receptor complexes. Science 2018, 359, 1037–1042. [Google Scholar] [CrossRef] [Green Version]
- Shum, T.; Omer, B.; Tashiro, H.; Kruse, R.L.; Wagner, D.L.; Parikh, K.; Yi, Z.; Sauer, T.; Liu, D.; Parihar, R.; et al. Constitutive Signaling from an Engineered IL7 Receptor Promotes Durable Tumor Elimination by Tumor-Redirected T Cells. Cancer Discov. 2017, 7, 1238–1247. [Google Scholar] [CrossRef] [Green Version]
- Kerkar, S.P.; Goldszmid, R.S.; Muranski, P.; Chinnasamy, D.; Yu, Z.; Reger, R.N.; Leonardi, A.J.; Morgan, R.A.; Wang, E.; Marincola, F.M.; et al. IL-12 triggers a programmatic change in dysfunctional myeloid-derived cells within mouse tumors. J. Clin. Investig. 2011, 121, 4746–4757. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Morgan, R.A.; Beane, J.D.; Zheng, Z.; Dudley, M.E.; Kassim, S.H.; Nahvi, A.V.; Ngo, L.T.; Sherry, R.M.; Phan, G.Q.; et al. Tumor-Infiltrating Lymphocytes Genetically Engineered with an Inducible Gene Encoding Interleukin-12 for the Immunotherapy of Metastatic Melanoma. Clin. Cancer Res. 2015, 21, 2278–2288. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Davies, J.S.; Serna, C.; Yu, Z.; Restifo, N.P.; Rosenberg, S.A.; Morgan, R.A.; Hinrichs, C.S. Enhanced efficacy and limited systemic cytokine exposure with membrane-anchored interleukin-12 T-cell therapy in murine tumor models. J. Immunother. Cancer 2020, 8, e000210. [Google Scholar] [CrossRef] [Green Version]
- Kunert, A.; Chmielewski, M.; Wijers, R.; Berrevoets, C.; Abken, H.; Debets, R. Intra-tumoral production of IL18, but not IL12, by TCR-engineered T cells is non-toxic and counteracts immune evasion of solid tumors. Oncoimmunology 2018, 7, e1378842. [Google Scholar] [CrossRef] [Green Version]
- Straathof, K.C.; Pulè, M.A.; Yotnda, P.; Dotti, G.; Vanin, E.F.; Brenner, M.K.; Heslop, H.E.; Spencer, D.M.; Rooney, C.M. An inducible caspase 9 safety switch for T-cell therapy. Blood 2005, 105, 4247–4254. [Google Scholar] [CrossRef]
- Di Stasi, A.; Tey, S.-K.; Dotti, G.; Fujita, Y.; Kennedy-Nasser, A.; Martinez, C.; Straathof, K.; Liu, E.; Durett, A.G.; Grilley, B.; et al. Inducible Apoptosis as a Safety Switch for Adoptive Cell Therapy. N. Engl. J. Med. 2011, 365, 1673–1683. [Google Scholar] [CrossRef] [Green Version]
- Mamonkin, M.; Mukherjee, M.; Srinivasan, M.; Sharma, S.; Gomes-Silva, D.; Mo, F.; Krenciute, G.; Orange, J.S.; Brenner, M.K. Reversible Transgene Expression Reduces Fratricide and Permits 4-1BB Costimulation of CAR T Cells Directed to T-cell Malignancies. Cancer Immunol. Res. 2018, 6, 47–58. [Google Scholar] [CrossRef] [Green Version]
- Sakemura, R.; Terakura, S.; Watanabe, K.; Julamanee, J.; Takagi, E.; Miyao, K.; Koyama, D.; Goto, T.; Hanajiri, R.; Nishida, T.; et al. A Tet-On Inducible System for Controlling CD19-Chimeric Antigen Receptor Expression upon Drug Administration. Cancer Immunol. Res. 2016, 4, 658–668. [Google Scholar] [CrossRef] [Green Version]
- Juillerat, A.; Marechal, A.; Filhol, J.M.; Valogne, Y.; Valton, J.; Duclert, A.; Duchateau, P.; Poirot, L. An oxygen sensitive self-decision making engineered CAR T-cell. Sci. Rep. 2017, 7, 39833. [Google Scholar] [CrossRef]
- Allen, M.E.; Zhou, W.; Thangaraj, J.; Kyriakakis, P.; Wu, Y.; Huang, Z.; Ho, P.; Pan, Y.; Limsakul, P.; Xu, X.; et al. An AND-Gated Drug and Photoactivatable Cre- loxP System for Spatiotemporal Control in Cell-Based Therapeutics. ACS Synth. Biol. 2019, 8, 2359–2371. [Google Scholar] [CrossRef]
- Huang, Z.; Wu, Y.; Allen, M.E.; Pan, Y.; Kyriakakis, P.; Lu, S.; Chang, Y.-J.; Wang, X.; Chien, S.; Wang, Y. Engineering light-controllable CAR T cells for cancer immunotherapy. Sci. Adv. 2020, 6, eaay9209. [Google Scholar] [CrossRef] [Green Version]
- Hoogi, S.; Eisenberg, V.; Mayer, S.; Shamul, A.; Barliya, T.; Cohen, C.J. A TIGIT-based chimeric co-stimulatory switch receptor improves T-cell anti-tumor function. J. Immunother. Cancer 2019, 7, 243. [Google Scholar] [CrossRef]
- Choi, B.D.; Yu, X.; Castano, A.P.; Bouffard, A.A.; Schmidts, A.; Larson, R.C.; Bailey, S.R.; Boroughs, A.C.; Frigault, M.J.; Leick, M.B.; et al. CAR-T cells secreting BiTEs circumvent antigen escape without detectable toxicity. Nat. Biotechnol. 2019, 37, 1049–1058. [Google Scholar] [CrossRef]
- Wilkie, S.; van Schalkwyk, M.C.I.; Hobbs, S.; Davies, D.M.; van der Stegen, S.J.C.; Pereira, A.C.P.; Burbridge, S.E.; Box, C.; Eccles, S.A.; Maher, J. Dual Targeting of ErbB2 and MUC1 in Breast Cancer Using Chimeric Antigen Receptors Engineered to Provide Complementary Signaling. J. Clin. Immunol. 2012, 32, 1059–1070. [Google Scholar] [CrossRef]
- Abbas, A.K.; Trotta, E.; Simeonov, D.R.; Marson, A.; Bluestone, J.A. Revisiting IL-2: Biology and therapeutic prospects. Sci. Immunol. 2018, 3, eaat1482. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Di, S.; Shi, B.; Zhang, H.; Wang, Y.; Wu, X.; Luo, H.; Wang, H.; Li, Z.; Jiang, H. Armored Inducible Expression of IL-12 Enhances Antitumor Activity of Glypican-3–Targeted Chimeric Antigen Receptor–Engineered T Cells in Hepatocellular Carcinoma. J. Immunol. 2019, 203, 198–207. [Google Scholar] [CrossRef]
- Hurton, L.V.; Singh, H.; Najjar, A.M.; Switzer, K.C.; Mi, T.; Maiti, S.; Olivares, S.; Rabinovich, B.; Huls, H.; Forget, M.-A.; et al. Tethered IL-15 augments antitumor activity and promotes a stem-cell memory subset in tumor-specific T cells. Proc. Natl. Acad. Sci. USA 2016, 113, E7788–E7797. [Google Scholar] [CrossRef] [Green Version]
- Hu, B.; Ren, J.; Luo, Y.; Keith, B.; Young, R.M.; Scholler, J.; Zhao, Y.; June, C.H. Augmentation of Antitumor Immunity by Human and Mouse CAR T Cells Secreting IL-18. Cell Rep. 2017, 20, 3025–3033. [Google Scholar] [CrossRef] [Green Version]
- Ma, X.; Shou, P.; Smith, C.; Chen, Y.; Du, H.; Sun, C.; Porterfield Kren, N.; Michaud, D.; Ahn, S.; Vincent, B.; et al. Interleukin-23 engineering improves CAR T cell function in solid tumors. Nat. Biotechnol. 2020, 38, 448–459. [Google Scholar] [CrossRef]
- Fitzgerald, J.C.; Weiss, S.L.; Maude, S.L.; Barrett, D.M.; Lacey, S.F.; Melenhorst, J.J.; Shaw, P.; Berg, R.A.; June, C.H.; Porter, D.L.; et al. Cytokine Release Syndrome After Chimeric Antigen Receptor T Cell Therapy for Acute Lymphoblastic Leukemia. Crit. Care Med. 2017, 45, e124–e131. [Google Scholar] [CrossRef]
- Schuster, S.J.; Svoboda, J.; Chong, E.A.; Nasta, S.D.; Mato, A.R.; Anak, Ö.; Brogdon, J.L.; Pruteanu-Malinici, I.; Bhoj, V.; Landsburg, D.; et al. Chimeric Antigen Receptor T Cells in Refractory B-Cell Lymphomas. N. Engl. J. Med. 2017, 377, 2545–2554. [Google Scholar] [CrossRef]
- Lee, D.W.; Santomasso, B.D.; Locke, F.L.; Ghobadi, A.; Turtle, C.J.; Brudno, J.N.; Maus, M.V.; Park, J.H.; Mead, E.; Pavletic, S.; et al. ASTCT Consensus Grading for Cytokine Release Syndrome and Neurologic Toxicity Associated with Immune Effector Cells. Biol. Blood Marrow Transplant. 2019, 25, 625–638. [Google Scholar] [CrossRef] [Green Version]
- Maus, M.V.; Haas, A.R.; Beatty, G.L.; Albelda, S.M.; Levine, B.L.; Liu, X.; Zhao, Y.; Kalos, M.; June, C.H. T Cells Expressing Chimeric Antigen Receptors Can Cause Anaphylaxis in Humans. Cancer Immunol. Res. 2013, 1, 26–31. [Google Scholar] [CrossRef] [Green Version]
- Griffioen, M.; van Egmond, E.H.M.; Kester, M.G.D.; Willemze, R.; Falkenburg, J.H.F.; Heemskerk, M.H.M. Retroviral transfer of human CD20 as a suicide gene for adoptive T-cell therapy. Haematologica 2009, 94, 1316–1320. [Google Scholar] [CrossRef] [Green Version]
- Serafini, M.; Manganini, M.; Borleri, G.; Bonamino, M.; Imberti, L.; Biondi, A.; Golay, J.; Rambaldi, A.; Introna, M. Characterization of CD20-Transduced T Lymphocytes as an Alternative Suicide Gene Therapy Approach for the Treatment of Graft-Versus-Host Disease. Hum. Gene Ther. 2004, 15, 63–76. [Google Scholar] [CrossRef]
- Wang, X.; Chang, W.-C.; Wong, C.W.; Colcher, D.; Sherman, M.; Ostberg, J.R.; Forman, S.J.; Riddell, S.R.; Jensen, M.C. A transgene-encoded cell surface polypeptide for selection, in vivo tracking, and ablation of engineered cells. Blood 2011, 118, 1255–1263. [Google Scholar] [CrossRef]
- Zhao, B.; Wang, Y.; Tan, X.; Zheng, X.; Wang, F.; Ke, K.; Zhang, C.; Liao, N.; Dang, Y.; Shi, Y.; et al. An Optogenetic Controllable T Cell System for Hepatocellular Carcinoma Immunotherapy. Theranostics 2019, 9, 1837–1850. [Google Scholar] [CrossRef]
- Wherry, E.J.; Kurachi, M. Molecular and cellular insights into T cell exhaustion. Nat. Rev. Immunol. 2015, 15, 486–499. [Google Scholar] [CrossRef]
- Gautam, S.; Fioravanti, J.; Zhu, W.; Le Gall, J.B.; Brohawn, P.; Lacey, N.E.; Hu, J.; Hocker, J.D.; Hawk, N.V.; Kapoor, V.; et al. The transcription factor c-Myb regulates CD8+ T cell stemness and antitumor immunity. Nat. Immunol. 2019, 20, 337–349. [Google Scholar] [CrossRef]
- Lynn, R.C.; Weber, E.W.; Sotillo, E.; Gennert, D.; Xu, P.; Good, Z.; Anbunathan, H.; Lattin, J.; Jones, R.; Tieu, V.; et al. c-Jun overexpression in CAR T cells induces exhaustion resistance. Nature 2019, 576, 293–300. [Google Scholar] [CrossRef]
- Chen, J.; López-Moyado, I.F.; Seo, H.; Lio, C.W.J.; Hempleman, L.J.; Sekiya, T.; Yoshimura, A.; Scott-Browne, J.P.; Rao, A. NR4A transcription factors limit CAR T cell function in solid tumours. Nature 2019, 567, 530–534. [Google Scholar] [CrossRef]
- Seo, H.; Chen, J.; González-Avalos, E.; Samaniego-Castruita, D.; Das, A.; Wang, Y.H.; López-Moyado, I.F.; Georges, R.O.; Zhang, W.; Onodera, A.; et al. TOX and TOX2 transcription factors cooperate with NR4A transcription factors to impose CD8 + T cell exhaustion. Proc. Natl. Acad. Sci. USA 2019, 116, 12410–12415. [Google Scholar] [CrossRef] [Green Version]
- Fraietta, J.A.; Nobles, C.L.; Sammons, M.A.; Lundh, S.; Carty, S.A.; Reich, T.J.; Cogdill, A.P.; Morrissette, J.J.D.; DeNizio, J.E.; Reddy, S.; et al. Disruption of TET2 promotes the therapeutic efficacy of CD19-targeted T cells. Nature 2018, 558, 307–312. [Google Scholar] [CrossRef]
- Wei, J.; Long, L.; Zheng, W.; Dhungana, Y.; Lim, S.A.; Guy, C.; Wang, Y.; Wang, Y.-D.; Qian, C.; Xu, B.; et al. Targeting REGNASE-1 programs long-lived effector T cells for cancer therapy. Nature 2019, 576, 471–476. [Google Scholar] [CrossRef]
- LaFleur, M.W.; Nguyen, T.H.; Coxe, M.A.; Miller, B.C.; Yates, K.B.; Gillis, J.E.; Sen, D.R.; Gaudiano, E.F.; Al Abosy, R.; Freeman, G.J.; et al. PTPN2 regulates the generation of exhausted CD8+ T cell subpopulations and restrains tumor immunity. Nat. Immunol. 2019, 20, 1335–1347. [Google Scholar] [CrossRef]
- Palmer, D.C.; Guittard, G.C.; Franco, Z.; Crompton, J.G.; Eil, R.L.; Patel, S.J.; Ji, Y.; Van Panhuys, N.; Klebanoff, C.A.; Sukumar, M.; et al. Cish actively silences TCR signaling in CD8+ T cells to maintain tumor tolerance. J. Exp. Med. 2015, 212, 2095–2113. [Google Scholar] [CrossRef] [Green Version]
- Dudda, J.C.; Salaun, B.; Ji, Y.; Palmer, D.C.; Monnot, G.C.; Merck, E.; Boudousquie, C.; Utzschneider, D.T.; Escobar, T.M.; Perret, R.; et al. MicroRNA-155 Is Required for Effector CD8+ T Cell Responses to Virus Infection and Cancer. Immunity 2013, 38, 742–753. [Google Scholar] [CrossRef] [Green Version]
- Chikuma, S.; Kanamori, M.; Mise-Omata, S.; Yoshimura, A. Suppressors of cytokine signaling: Potential immune checkpoint molecules for cancer immunotherapy. Cancer Sci. 2017, 108, 574–580. [Google Scholar] [CrossRef] [Green Version]
- Jiang, M.; Zhang, W.; Liu, P.; Yu, W.; Liu, T.; Yu, J. Dysregulation of SOCS-Mediated Negative Feedback of Cytokine Signaling in Carcinogenesis and Its Significance in Cancer Treatment. Front. Immunol. 2017, 8. [Google Scholar] [CrossRef] [Green Version]
- Stromnes, I.M.; Fowler, C.; Casamina, C.C.; Georgopolos, C.M.; McAfee, M.S.; Schmitt, T.M.; Tan, X.; Kim, T.-D.; Choi, I.; Blattman, J.N.; et al. Abrogation of Src Homology Region 2 Domain-Containing Phosphatase 1 in Tumor-Specific T Cells Improves Efficacy of Adoptive Immunotherapy by Enhancing the Effector Function and Accumulation of Short-Lived Effector T Cells In Vivo. J. Immunol. 2012, 189, 1812–1825. [Google Scholar] [CrossRef]
- Riese, M.J.; Wang, L.-C.S.; Moon, E.K.; Joshi, R.P.; Ranganathan, A.; June, C.H.; Koretzky, G.A.; Albelda, S.M. Enhanced Effector Responses in Activated CD8+ T Cells Deficient in Diacylglycerol Kinases. Cancer Res. 2013, 73, 3566–3577. [Google Scholar] [CrossRef] [Green Version]
- Brownlie, R.J.; Wright, D.; Zamoyska, R.; Salmond, R.J. Deletion of PTPN22 improves effector and memory CD8+ T cell responses to tumors. JCI Insight 2019, 4. [Google Scholar] [CrossRef]
- Thaker, Y.R.; Raab, M.; Strebhardt, K.; Rudd, C.E. GTPase-activating protein Rasal1 associates with ZAP-70 of the TCR and negatively regulates T-cell tumor immunity. Nat. Commun. 2019, 10, 4804. [Google Scholar] [CrossRef] [Green Version]
- Stromnes, I.M.; Blattman, J.N.; Tan, X.; Jeevanjee, S.; Gu, H.; Greenberg, P.D. Abrogating Cbl-b in effector CD8+ T cells improves the efficacy of adoptive therapy of leukemia in mice. J. Clin. Investig. 2010, 120, 3722–3734. [Google Scholar] [CrossRef] [Green Version]
- Hinterleitner, R.; Gruber, T.; Pfeifhofer-Obermair, C.; Lutz-Nicoladoni, C.; Tzankov, A.; Schuster, M.; Penninger, J.M.; Loibner, H.; Lametschwandtner, G.; Wolf, D.; et al. Adoptive Transfer of siRNA Cblb-Silenced CD8+ T Lymphocytes Augments Tumor Vaccine Efficacy in a B16 Melanoma Model. PLoS ONE 2012, 7, e44295. [Google Scholar] [CrossRef] [Green Version]
- Frei, T.; Cella, F.; Tedeschi, F.; Gutiérrez, J.; Stan, G.-B.; Khammash, M.; Siciliano, V. Characterization and mitigation of gene expression burden in mammalian cells. Nat. Commun. 2020, 11, 4641. [Google Scholar] [CrossRef]
- Nissim, L.; Wu, M.R.; Pery, E.; Binder-Nissim, A.; Suzuki, H.I.; Stupp, D.; Wehrspaun, C.; Tabach, Y.; Sharp, P.A.; Lu, T.K. Synthetic RNA-Based Immunomodulatory Gene Circuits for Cancer Immunotherapy. Cell 2017, 171, 1138–1150.e15. [Google Scholar] [CrossRef] [Green Version]
- Rijal, G.; Li, W. Native-mimicking in vitro microenvironment: An elusive and seductive future for tumor modeling and tissue engineering. J. Biol. Eng. 2018, 12, 20. [Google Scholar] [CrossRef] [Green Version]
- Pennacchio, F.A.; Caliendo, F.; Iaccarino, G.; Langella, A.; Siciliano, V.; Santoro, F. Three-dimensionally Patterned Scaffolds Modulate the Biointerface at the Nanoscale. Nano Lett. 2019, 19, 5118–5123. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| Synthetic Biology Approach | Control Module | Purpose | References |
|---|---|---|---|
| Tandem CAR | Logic Gate OR | Avoid OFF-target | [45,46,47] |
| sCAR | Logic Gate AND | Increase specificity | [48,49,50,51] |
| SUPRA CAR | Docking system | Tune T cell activation | [52] |
| Engineered TGF-β-RII | Chimeric receptors/Rewiring | Overcome TME immunosuppression | [53,54] |
| Engineered IL-4-R | Chimeric receptors/Rewiring | Overcome TME immunosuppression | [55,56] |
| Engineered IRs | Chimeric receptors/Rewiring | Overcome TME immunosuppression | [57,58] |
| CCR a-CAR | Logic Gate AND | Increase specificity | [59] |
| SynNotch CAR | Logic Gate AND | Increase specificity | [60] |
| COVER-T | Logic Gate AND-like | Avoid OFF-target | [61] |
| iCAR | Logic Gate AND NOT | Avoid OFF-target | [62] |
| Tmod2 | Logic Gate AND NOT | Target LOH | [63] |
| Engineered IL-2R/IL-7R | Cytokine signaling | Reduce cytokine cytotoxicity | [64,65] |
| Engineered IL-12/IL-18 | Cytokine signaling | Reduce cytokine cytotoxicity | [66,67,68,69] |
| iCasp9 | Safety switches | Control CAR-T cell viability | [70,71] |
| TET-ON/TET-OFF | Safety switches | Reduce side effects | [72,73] |
| HIF-CAR | Safety switches | Activate T cell only in the tumor core | [74] |
| Blue light-activated CAR | Safety switches | Tune T cell activation | [75,76] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bonfá, G.; Blazquez-Roman, J.; Tarnai, R.; Siciliano, V. Precision Tools in Immuno-Oncology: Synthetic Gene Circuits for Cancer Immunotherapy. Vaccines 2020, 8, 732. https://doi.org/10.3390/vaccines8040732
Bonfá G, Blazquez-Roman J, Tarnai R, Siciliano V. Precision Tools in Immuno-Oncology: Synthetic Gene Circuits for Cancer Immunotherapy. Vaccines. 2020; 8(4):732. https://doi.org/10.3390/vaccines8040732
Chicago/Turabian StyleBonfá, Giuliano, Juan Blazquez-Roman, Rita Tarnai, and Velia Siciliano. 2020. "Precision Tools in Immuno-Oncology: Synthetic Gene Circuits for Cancer Immunotherapy" Vaccines 8, no. 4: 732. https://doi.org/10.3390/vaccines8040732
APA StyleBonfá, G., Blazquez-Roman, J., Tarnai, R., & Siciliano, V. (2020). Precision Tools in Immuno-Oncology: Synthetic Gene Circuits for Cancer Immunotherapy. Vaccines, 8(4), 732. https://doi.org/10.3390/vaccines8040732