
Distinct Mutational Profile of Lynch Syndrome Colorectal Cancers Diagnosed under Regular Colonoscopy Surveillance

 , , ,
, , ,  and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Patients and Tumor Samples
2.2. Histopathology Analysis
2.3. Mutation Analysis
2.4. Immunohistochemical Staining and Quantification of T Cell Density
2.5. Statistical Calculations
3. Results
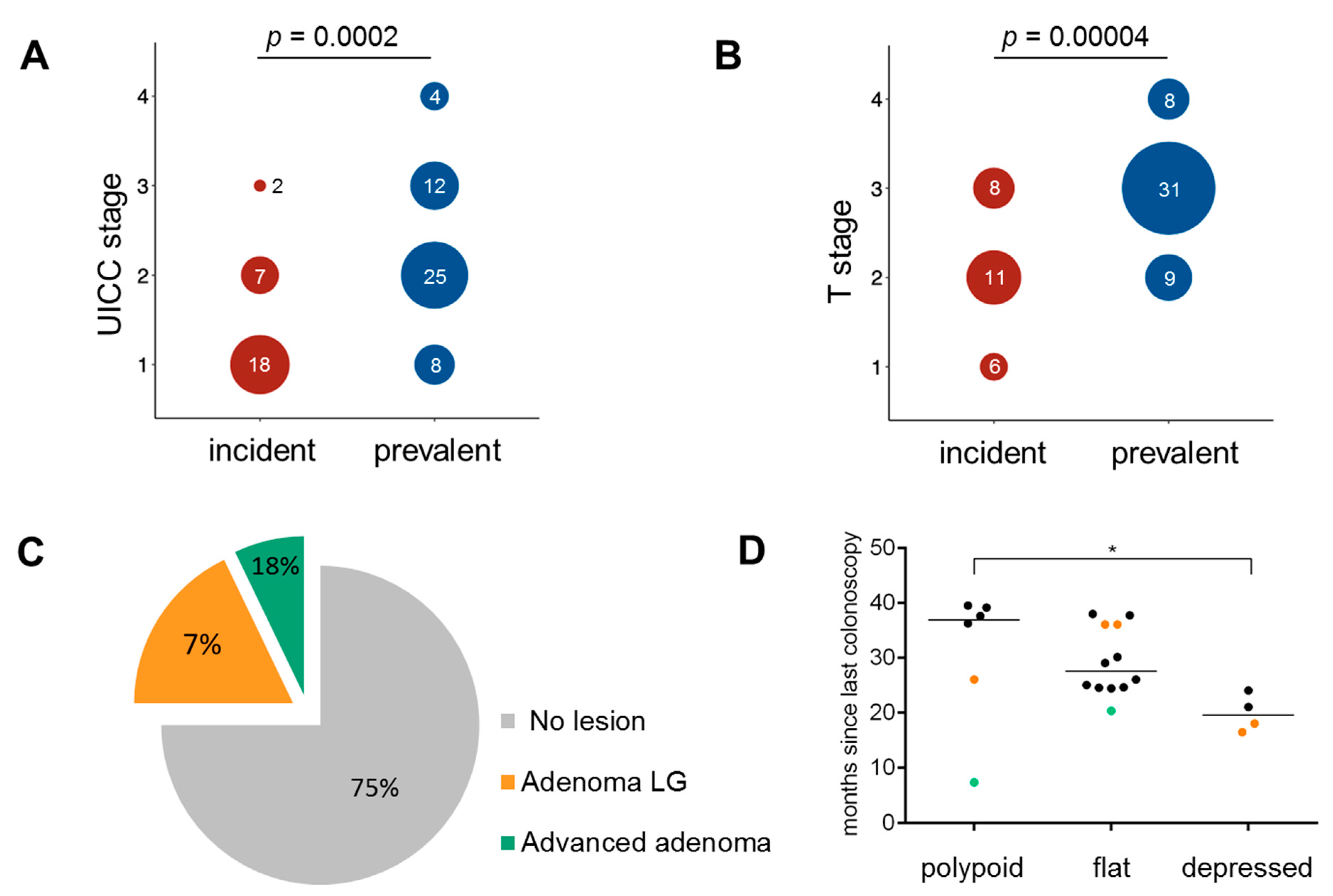
3.1. Clinical Characteristics
3.2. Histopathology of Incident Cancers
3.3. Mutational Profile and MMR Deficiency Signatures in Incident Cancers
3.4. CMS Analysis in Incident Cancers
3.5. Immune Infiltration and Immune Evasion in Incident Cancers
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jasperson, K.W.; Tuohy, T.M.; Neklason, D.W.; Burt, R.W. Hereditary and familial colon cancer. Gastroenterology 2010, 138, 2044–2058. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De la Chapelle, A. Microsatellite instability. N. Engl. J. Med. 2003, 349, 209–210. [Google Scholar] [CrossRef] [Green Version]
- Woerner, S.M.; Kloor, M.; von Knebel Doeberitz, M.; Gebert, J.F. Microsatellite instability in the development of DNA mismatch repair deficient tumors. Cancer Biomark. Sect. A Dis. Mark. 2006, 2, 69–86. [Google Scholar] [CrossRef]
- Linnebacher, M.; Gebert, J.; Rudy, W.; Woerner, S.; Yuan, Y.P.; Bork, P.; von Knebel Doeberitz, M. Frameshift peptide-derived T-cell epitopes: A source of novel tumor-specific antigens. Int. J. Cancer 2001, 93, 6–11. [Google Scholar] [CrossRef]
- Buckowitz, A.; Knaebel, H.P.; Benner, A.; Blaker, H.; Gebert, J.; Kienle, P.; von Knebel Doeberitz, M.; Kloor, M. Microsatellite instability in colorectal cancer is associated with local lymphocyte infiltration and low frequency of distant metastases. Brit. J. Cancer 2005, 92, 1746–1753. [Google Scholar] [CrossRef] [Green Version]
- Popat, S.; Hubner, R.; Houlston, R.S. Systematic review of microsatellite instability and colorectal cancer prognosis. J. Clin. Oncol. 2005, 23, 609–618. [Google Scholar] [CrossRef]
- Le, D.T.; Uram, J.N.; Wang, H.; Bartlett, B.R.; Kemberling, H.; Eyring, A.D.; Skora, A.D.; Luber, B.S.; Azad, N.S.; Laheru, D.; et al. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N. Engl. J. Med. 2015, 372, 2509–2520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le, D.T.; Durham, J.N.; Smith, K.N.; Wang, H.; Bartlett, B.R.; Aulakh, L.K.; Lu, S.; Kemberling, H.; Wilt, C.; Luber, B.S.; et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 2017, 357, 409–413. [Google Scholar] [CrossRef] [Green Version]
- Vasen, H.F.; Blanco, I.; Aktan-Collan, K.; Gopie, J.P.; Alonso, A.; Aretz, S.; Bernstein, I.; Bertario, L.; Burn, J.; Capella, G.; et al. Revised guidelines for the clinical management of Lynch syndrome (HNPCC): Recommendations by a group of European experts. Gut 2013, 62, 812–823. [Google Scholar] [CrossRef] [PubMed]
- Stormorken, A.T.; Clark, N.; Grindedal, E.; Maehle, L.; Moller, P. Prevention of colorectal cancer by colonoscopic surveillance in families with hereditary colorectal cancer. Scand. J. Gastroenterol. 2007, 42, 611–617. [Google Scholar] [CrossRef] [PubMed]
- Jarvinen, H.J.; Aarnio, M.; Mustonen, H.; Aktan-Collan, K.; Aaltonen, L.A.; Peltomaki, P.; De La Chapelle, A.; Mecklin, J.P. Controlled 15-year trial on screening for colorectal cancer in families with hereditary nonpolyposis colorectal cancer. Gastroenterology 2000, 118, 829–834. [Google Scholar] [CrossRef]
- Newton, K.; Green, K.; Lalloo, F.; Evans, D.G.; Hill, J. Colonoscopy screening compliance and outcomes in patients with Lynch syndrome. Colorect. Dis. Off. J. Assoc. Coloproctol. G. B. Irel. 2015, 17, 38–46. [Google Scholar] [CrossRef]
- Jarvinen, H.J.; Renkonen-Sinisalo, L.; Aktan-Collan, K.; Peltomaki, P.; Aaltonen, L.A.; Mecklin, J.P. Ten years after mutation testing for Lynch syndrome: Cancer incidence and outcome in mutation-positive and mutation-negative family members. J. Clin. Oncol. 2009, 27, 4793–4797. [Google Scholar] [CrossRef]
- Renkonen-Sinisalo, L.; Aarnio, M.; Mecklin, J.P.; Jarvinen, H.J. Surveillance improves survival of colorectal cancer in patients with hereditary nonpolyposis colorectal cancer. Cancer Detect. Prevent. 2000, 24, 137–142. [Google Scholar]
- Brenner, H.; Chang-Claude, J.; Jansen, L.; Knebel, P.; Stock, C.; Hoffmeister, M. Reduced risk of colorectal cancer up to 10 years after screening, surveillance, or diagnostic colonoscopy. Gastroenterology 2014, 146, 709–717. [Google Scholar] [CrossRef] [Green Version]
- Brenner, H.; Chang-Claude, J.; Seiler, C.M.; Rickert, A.; Hoffmeister, M. Protection from colorectal cancer after colonoscopy: A population-based, case-control study. Ann. Intern. Med. 2011, 154, 22–30. [Google Scholar] [CrossRef]
- Samadder, N.J.; Curtin, K.; Pappas, L.; Boucher, K.; Mineau, G.P.; Smith, K.; Fraser, A.; Wan, Y.; Provenzale, D.; Kinney, A.Y.; et al. Risk of Incident Colorectal Cancer and Death After Colonoscopy: A Population-based Study in Utah. Clin. Gastroenterol. Hepatol. 2016, 14, 279–286.e2. [Google Scholar] [CrossRef] [PubMed]
- Engel, C.; Rahner, N.; Schulmann, K.; Holinski-Feder, E.; Goecke, T.O.; Schackert, H.K.; Kloor, M.; Steinke, V.; Vogelsang, H.; Moslein, G.; et al. Efficacy of annual colonoscopic surveillance in individuals with hereditary nonpolyposis colorectal cancer. Clin. Gastroenterol. Hepatol. 2010, 8, 174–182. [Google Scholar] [CrossRef] [PubMed]
- De Vos tot Nederveen Cappel, W.H.; Nagengast, F.M.; Griffioen, G.; Menko, F.H.; Taal, B.G.; Kleibeuker, J.H.; Vasen, H.F. Surveillance for hereditary nonpolyposis colorectal cancer: A long-term study on 114 families. Dis. Colon Rectum 2002, 45, 1588–1594. [Google Scholar] [CrossRef] [PubMed]
- Vasen, H.F.; Moslein, G.; Alonso, A.; Bernstein, I.; Bertario, L.; Blanco, I.; Burn, J.; Capella, G.; Engel, C.; Frayling, I.; et al. Guidelines for the clinical management of Lynch syndrome (hereditary non-polyposis cancer). J. Med. Genet. 2007, 44, 353–362. [Google Scholar] [CrossRef] [PubMed]
- Mecklin, J.P.; Aarnio, M.; Laara, E.; Kairaluoma, M.V.; Pylvanainen, K.; Peltomaki, P.; Aaltonen, L.A.; Jarvinen, H.J. Development of colorectal tumors in colonoscopic surveillance in Lynch syndrome. Gastroenterology 2007, 133, 1093–1098. [Google Scholar] [CrossRef]
- Engel, C.; Vasen, H.F.; Seppala, T.; Aretz, S.; Bigirwamungu-Bargeman, M.; de Boer, S.Y.; Bucksch, K.; Buttner, R.; Holinski-Feder, E.; Holzapfel, S.; et al. No Difference in Colorectal Cancer Incidence or Stage at Detection by Colonoscopy Among 3 Countries with Different Lynch Syndrome Surveillance Policies. Gastroenterology 2018, 155, 1400–1409. [Google Scholar] [CrossRef]
- Moller, P.; Seppala, T.; Bernstein, I.; Holinski-Feder, E.; Sala, P.; Evans, D.G.; Lindblom, A.; Macrae, F.; Blanco, I.; Sijmons, R.; et al. Cancer incidence and survival in Lynch syndrome patients receiving colonoscopic and gynaecological surveillance: First report from the prospective Lynch syndrome database. Gut 2017, 66, 464–472. [Google Scholar] [CrossRef]
- Rutter, M.D.; Beintaris, I.; Valori, R.; Chiu, H.M.; Corley, D.A.; Cuatrecasas, M.; Dekker, E.; Forsberg, A.; Gore-Booth, J.; Haug, U.; et al. World Endoscopy Organization Consensus Statements on Post-Colonoscopy and Post-Imaging Colorectal Cancer. Gastroenterology 2018, 155, 909–925. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moller, P.; Seppala, T.T.; Bernstein, I.; Holinski-Feder, E.; Sala, P.; Gareth Evans, D.; Lindblom, A.; Macrae, F.; Blanco, I.; Sijmons, R.H.; et al. Cancer risk and survival in path_MMR carriers by gene and gender up to 75 years of age: A report from the Prospective Lynch Syndrome Database. Gut 2018, 67, 1306–1316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonadona, V.; Bonaiti, B.; Olschwang, S.; Grandjouan, S.; Huiart, L.; Longy, M.; Guimbaud, R.; Buecher, B.; Bignon, Y.J.; Caron, O.; et al. Cancer risks associated with germline mutations in MLH1, MSH2, and MSH6 genes in Lynch syndrome. JAMA 2011, 305, 2304–2310. [Google Scholar] [CrossRef] [PubMed]
- Dowty, J.G.; Win, A.K.; Buchanan, D.D.; Lindor, N.M.; Macrae, F.A.; Clendenning, M.; Antill, Y.C.; Thibodeau, S.N.; Casey, G.; Gallinger, S.; et al. Cancer risks for MLH1 and MSH2 mutation carriers. Hum. Mutat. 2013, 34, 490–497. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ten Broeke, S.W.; van der Klift, H.M.; Tops, C.M.J.; Aretz, S.; Bernstein, I.; Buchanan, D.D.; de la Chapelle, A.; Capella, G.; Clendenning, M.; Engel, C.; et al. Cancer Risks for PMS2-Associated Lynch Syndrome. J. Clin. Oncol. 2018, 36, 2961–2968. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahadova, A.; Seppala, T.T.; Engel, C.; Gallon, R.; Burn, J.; Holinski-Feder, E.; Steinke-Lange, V.; Moslein, G.; Nielsen, M.; Ten Broeke, S.W.; et al. The "unnatural" history of colorectal cancer in Lynch syndrome: Lessons from colonoscopy surveillance. Int. J. Cancer 2021, 148, 800–811. [Google Scholar] [CrossRef] [PubMed]
- Latchford, A. How Should Colonoscopy Surveillance in Lynch Syndrome Be Performed? Gastroenterology 2020, 158, 818–819. [Google Scholar] [CrossRef]
- Sawhney, M.S.; Farrar, W.D.; Gudiseva, S.; Nelson, D.B.; Lederle, F.A.; Rector, T.S.; Bond, J.H. Microsatellite instability in interval colon cancers. Gastroenterology 2006, 131, 1700–1705. [Google Scholar] [CrossRef] [PubMed]
- Ahadova, A.; von Knebel Doeberitz, M.; Blaker, H.; Kloor, M. CTNNB1-mutant colorectal carcinomas with immediate invasive growth: A model of interval cancers in Lynch syndrome. Fam. Cancer 2016, 15, 579–586. [Google Scholar] [CrossRef] [PubMed]
- Kloor, M.; Huth, C.; Voigt, A.Y.; Benner, A.; Schirmacher, P.; von Knebel Doeberitz, M.; Blaker, H. Prevalence of mismatch repair-deficient crypt foci in Lynch syndrome: A pathological study. Lancet Oncol. 2012, 13, 598–606. [Google Scholar] [CrossRef]
- Staffa, L.; Echterdiek, F.; Nelius, N.; Benner, A.; Werft, W.; Lahrmann, B.; Grabe, N.; Schneider, M.; Tariverdian, M.; von Knebel Doeberitz, M.; et al. Mismatch repair-deficient crypt foci in Lynch syndrome—Molecular alterations and association with clinical parameters. PLoS ONE 2015, 10, e0121980. [Google Scholar] [CrossRef] [Green Version]
- Pai, R.K.; Dudley, B.; Karloski, E.; Brand, R.E.; O’Callaghan, N.; Rosty, C.; Buchanan, D.D.; Jenkins, M.A.; Thibodeau, S.N.; French, A.J.; et al. DNA mismatch repair protein deficient non-neoplastic colonic crypts: A novel indicator of Lynch syndrome. Mod. Pathol. 2018, 31, 1608–1618. [Google Scholar] [CrossRef]
- Argillander, T.E.; Koornstra, J.J.; van Kouwen, M.; Langers, A.M.; Nagengast, F.M.; Vecht, J.; de Vos Tot Nederveen Cappel, W.H.; Dekker, E.; van Duijvendijk, P.; Vasen, H.F. Features of incident colorectal cancer in Lynch syndrome. United Eur. Gastroenterol. J. 2018, 6, 1215–1222. [Google Scholar] [CrossRef] [Green Version]
- Lambert, R. The Paris endoscopic classification of superficial neoplastic lesions: Esophagus, stomach, and colon: November 30 to December 1, 2002. Gastrointest. Endosc. 2003, 58, S3–S43. [Google Scholar] [CrossRef]
- Ahadova, A.; Gallon, R.; Gebert, J.; Ballhausen, A.; Endris, V.; Kirchner, M.; Stenzinger, A.; Burn, J.; von Knebel Doeberitz, M.; Blaker, H.; et al. Three molecular pathways model colorectal carcinogenesis in Lynch syndrome. Int. J. Cancer 2018, 143, 139–150. [Google Scholar] [CrossRef] [Green Version]
- Jesinghaus, M.; Pfarr, N.; Endris, V.; Kloor, M.; Volckmar, A.L.; Brandt, R.; Herpel, E.; Muckenhuber, A.; Lasitschka, F.; Schirmacher, P.; et al. Genotyping of colorectal cancer for cancer precision medicine: Results from the IPH Center for Molecular Pathology. Genes Chromosom. Cancer 2016, 55, 505–521. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef] [PubMed]
- Robinson, J.T.; Thorvaldsdottir, H.; Winckler, W.; Guttman, M.; Lander, E.S.; Getz, G.; Mesirov, J.P. Integrative genomics viewer. Nat. Biotechnol. 2011, 29, 24–26. [Google Scholar] [CrossRef] [Green Version]
- Findeisen, P.; Kloor, M.; Merx, S.; Sutter, C.; Woerner, S.M.; Dostmann, N.; Benner, A.; Dondog, B.; Pawlita, M.; Dippold, W.; et al. T25 repeat in the 3’ untranslated region of the CASP2 gene: A sensitive and specific marker for microsatellite instability in colorectal cancer. Cancer Res. 2005, 65, 8072–8078. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woerner, S.M.; Yuan, Y.P.; Benner, A.; Korff, S.; von Knebel Doeberitz, M.; Bork, P. SelTarbase, a database of human mononucleotide-microsatellite mutations and their potential impact to tumorigenesis and immunology. Nucleic Acids Res. 2010, 38, D682–D689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jurtz, V.; Paul, S.; Andreatta, M.; Marcatili, P.; Peters, B.; Nielsen, M. NetMHCpan-4.0: Improved Peptide-MHC Class I Interaction Predictions Integrating Eluted Ligand and Peptide Binding Affinity Data. J. Immunol. 2017, 199, 3360–3368. [Google Scholar] [CrossRef]
- Ballhausen, A.; Przybilla, M.J.; Jendrusch, M.; Haupt, S.; Pfaffendorf, E.; Seidler, F.; Witt, J.; Hernandez Sanchez, A.; Urban, K.; Draxlbauer, M.; et al. The shared frameshift mutation landscape of microsatellite-unstable cancers suggests immunoediting during tumor evolution. Nat. Commun. 2020, 11, 4740. [Google Scholar] [CrossRef] [PubMed]
- Pfuderer, P.L.; Ballhausen, A.; Seidler, F.; Stark, H.J.; Grabe, N.; Frayling, I.M.; Ager, A.; von Knebel Doeberitz, M.; Kloor, M.; Ahadova, A. High endothelial venules are associated with microsatellite instability, hereditary background and immune evasion in colorectal cancer. Brit. J. Cancer 2019, 121, 395–404. [Google Scholar] [CrossRef]
- Kloor, M.; Sutter, C.; Wentzensen, N.; Cremer, F.W.; Buckowitz, A.; Keller, M.; von Knebel Doeberitz, M.; Gebert, J. A large MSH2 Alu insertion mutation causes HNPCC in a German kindred. Hum. Genet. 2004, 115, 432–438. [Google Scholar] [CrossRef] [PubMed]
- Janikovits, J.; Muller, M.; Krzykalla, J.; Korner, S.; Echterdiek, F.; Lahrmann, B.; Grabe, N.; Schneider, M.; Benner, A.; Doeberitz, M.V.K.; et al. High numbers of PDCD1 (PD-1)-positive T cells and B2M mutations in microsatellite-unstable colorectal cancer. Oncoimmunology 2018, 7, e1390640. [Google Scholar] [CrossRef] [Green Version]
- Team, R.C. R: A Language and Environment for Statistical Computing; R Version 3.6.0.R; R Foundation for Statistical Computing: Vienna, Austria, 2019. [Google Scholar]
- Team, R. RStudio: Integrated Development for R; RStudio: Boston, MA, USA, 2018. [Google Scholar]
- Engel, C.; Ahadova, A.; Seppala, T.; Aretz, S.; Bigirwamungu-Bargeman, M.; Blaker, H.; Bucksch, K.; Buttner, R.; de Vos Tot Nederveen Cappel, W.; Endris, V.; et al. Associations of Pathogenic Variants in MLH1, MSH2, and MSH6 with Risk of Colorectal Adenomas and Tumors and with Somatic Mutations in Patients with Lynch Syndrome. Gastroenterology 2020. [Google Scholar] [CrossRef]
- Binder, H.; Hopp, L.; Schweiger, M.R.; Hoffmann, S.; Juhling, F.; Kerick, M.; Timmermann, B.; Siebert, S.; Grimm, C.; Nersisyan, L.; et al. Genomic and transcriptomic heterogeneity of colorectal tumours arising in Lynch syndrome. J. Pathol. 2017, 243, 242–254. [Google Scholar] [CrossRef]
- Kloor, M.; Michel, S.; Buckowitz, B.; Ruschoff, J.; Buttner, R.; Holinski-Feder, E.; Dippold, W.; Wagner, R.; Tariverdian, M.; Benner, A.; et al. Beta2-microglobulin mutations in microsatellite unstable colorectal tumors. Int. J. Cancer 2007, 121, 454–458. [Google Scholar] [CrossRef] [PubMed]
- Seppala, T.T.; Ahadova, A.; Dominguez-Valentin, M.; Macrae, F.; Evans, D.G.; Therkildsen, C.; Sampson, J.; Scott, R.; Burn, J.; Moslein, G.; et al. Lack of association between screening interval and cancer stage in Lynch syndrome may be accounted for by over-diagnosis; a prospective Lynch syndrome database report. Hered. Cancer Clin. Pract. 2019, 17, 8. [Google Scholar] [CrossRef] [PubMed]
- Seppala, T.; Pylvanainen, K.; Evans, D.G.; Jarvinen, H.; Renkonen-Sinisalo, L.; Bernstein, I.; Holinski-Feder, E.; Sala, P.; Lindblom, A.; Macrae, F.; et al. Colorectal cancer incidence in path_MLH1 carriers subjected to different follow-up protocols: A Prospective Lynch Syndrome Database report. Hered. Cancer Clin. Pract. 2017, 15, 18. [Google Scholar] [CrossRef] [Green Version]
- Hartman, D.J.; Nikiforova, M.N.; Chang, D.T.; Chu, E.; Bahary, N.; Brand, R.E.; Zureikat, A.H.; Zeh, H.J.; Choudry, H.; Pai, R.K. Signet ring cell colorectal carcinoma: A distinct subset of mucin-poor microsatellite-stable signet ring cell carcinoma associated with dismal prognosis. Am. J. Surg. Pathol. 2013, 37, 969–977. [Google Scholar] [CrossRef]
- Dominguez-Valentin, M.; Seppala, T.T.; Sampson, J.R.; Macrae, F.; Winship, I.; Evans, D.G.; Scott, R.J.; Burn, J.; Moslein, G.; Bernstein, I.; et al. Survival by colon cancer stage and screening interval in Lynch syndrome: A prospective Lynch syndrome database report. Hered. Cancer Clin. Pract. 2019, 17, 28. [Google Scholar] [CrossRef] [Green Version]
- Shia, J.; Schultz, N.; Kuk, D.; Vakiani, E.; Middha, S.; Segal, N.H.; Hechtman, J.F.; Berger, M.F.; Stadler, Z.K.; Weiser, M.R.; et al. Morphological characterization of colorectal cancers in The Cancer Genome Atlas reveals distinct morphology-molecular associations: Clinical and biological implications. Mod. Pathol. 2017, 30, 599–609. [Google Scholar] [CrossRef] [PubMed]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Borresen-Dale, A.L.; et al. Signatures of mutational processes in human cancer. Nature 2013, 500, 415–421. [Google Scholar] [CrossRef] [Green Version]
- Oliveira, C.; Westra, J.L.; Arango, D.; Ollikainen, M.; Domingo, E.; Ferreira, A.; Velho, S.; Niessen, R.; Lagerstedt, K.; Alhopuro, P.; et al. Distinct patterns of KRAS mutations in colorectal carcinomas according to germline mismatch repair defects and hMLH1 methylation status. Hum. Mol. Genet. 2004, 13, 2303–2311. [Google Scholar] [CrossRef]
- Zauber, P.; Marotta, S.; Sabbath-Solitare, M. KRAS gene mutations are more common in colorectal villous adenomas and in situ carcinomas than in carcinomas. Int. J. Mol. Epidemiol. Genet. 2013, 4, 1–10. [Google Scholar]
- Juarez, M.; Egoavil, C.; Rodriguez-Soler, M.; Hernandez-Illan, E.; Guarinos, C.; Garcia-Martinez, A.; Alenda, C.; Giner-Calabuig, M.; Murcia, O.; Mangas, C.; et al. KRAS and BRAF somatic mutations in colonic polyps and the risk of metachronous neoplasia. PLoS ONE 2017, 12, e0184937. [Google Scholar] [CrossRef] [Green Version]
- Hoffmeister, M.; Blaker, H.; Jansen, L.; Alwers, E.; Amitay, E.L.; Carr, P.R.; Kloor, M.; Herpel, E.; Roth, W.; Chang-Claude, J.; et al. Colonoscopy and Reduction of Colorectal Cancer Risk by Molecular Tumor Subtypes: A Population-Based Case-Control Study. Am. J. Gastroenterol. 2020, 115, 2007–2016. [Google Scholar] [CrossRef]
- Shaukat, A.; Arain, M.; Anway, R.; Manaktala, S.; Pohlman, L.; Thyagarajan, B. Is KRAS mutation associated with interval colorectal cancers? Digest. Dis. Sci. 2012, 57, 913–917. [Google Scholar] [CrossRef]
- Fearon, E.R.; Vogelstein, B. A genetic model for colorectal tumorigenesis. Cell 1990, 61, 759–767. [Google Scholar] [CrossRef]
- Arnold, A.; Tronser, M.; Sers, C.; Ahadova, A.; Endris, V.; Mamlouk, S.; Horst, D.; Mobs, M.; Bischoff, P.; Kloor, M.; et al. Correction to: The majority of beta-catenin mutations in colorectal cancer is homozygous. BMC Cancer 2020, 20, 1151. [Google Scholar] [CrossRef]
- Aaltonen, L.A.; Salovaara, R.; Kristo, P.; Canzian, F.; Hemminki, A.; Peltomaki, P.; Chadwick, R.B.; Kaariainen, H.; Eskelinen, M.; Jarvinen, H.; et al. Incidence of hereditary nonpolyposis colorectal cancer and the feasibility of molecular screening for the disease. N. Engl. J. Med. 1998, 338, 1481–1487. [Google Scholar] [CrossRef] [PubMed]
- Maruvka, Y.E.; Mouw, K.W.; Karlic, R.; Parasuraman, P.; Kamburov, A.; Polak, P.; Haradhvala, N.J.; Hess, J.M.; Rheinbay, E.; Brody, Y.; et al. Analysis of somatic microsatellite indels identifies driver events in human tumors. Nat. Biotechnol. 2017, 35, 951–959. [Google Scholar] [CrossRef] [PubMed]
- Roeckel, N.; Woerner, S.M.; Kloor, M.; Yuan, Y.P.; Patsos, G.; Gromes, R.; Kopitz, J.; Gebert, J. High frequency of LMAN1 abnormalities in colorectal tumors with microsatellite instability. Cancer Res. 2009, 69, 292–299. [Google Scholar] [CrossRef] [Green Version]
- Lockwood, W.W.; Thu, K.L.; Lin, L.; Pikor, L.A.; Chari, R.; Lam, W.L.; Beer, D.G. Integrative genomics identified RFC3 as an amplified candidate oncogene in esophageal adenocarcinoma. Clin. Cancer Res. 2012, 18, 1936–1946. [Google Scholar] [CrossRef] [Green Version]
- Woerner, S.M.; Benner, A.; Sutter, C.; Schiller, M.; Yuan, Y.P.; Keller, G.; Bork, P.; Doeberitz, M.; Gebert, J.F. Pathogenesis of DNA repair-deficient cancers: A statistical meta-analysis of putative Real Common Target genes. Oncogene 2003, 22, 2226–2235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lai, E.J.; Calderwood, A.H.; Doros, G.; Fix, O.K.; Jacobson, B.C. The Boston bowel preparation scale: A valid and reliable instrument for colonoscopy-oriented research. Gastrointest. Endosc. 2009, 69, 620–625. [Google Scholar] [CrossRef] [Green Version]
- Lappalainen, J.; Holmstrom, D.; Lepisto, A.; Saarnio, J.; Mecklin, J.P.; Seppala, T. Incident colorectal cancer in Lynch syndrome is usually not preceded by compromised quality of colonoscopy. Scand. J. Gastroenterol. 2019, 54, 1473–1480. [Google Scholar] [CrossRef] [PubMed]
- Haanstra, J.F.; Vasen, H.F.; Sanduleanu, S.; van der Wouden, E.J.; Koornstra, J.J.; Kleibeuker, J.H.; de Vos Tot Nederveen Cappel, W.H. Quality colonoscopy and risk of interval cancer in Lynch syndrome. Int. J. Colorectal Dis. 2013, 28, 1643–1649. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient | Age at Diagnosis | Gender | Location | TNM Stage | UICC Stage | Gene | Age at Last FU | Age at Death | Cause of Death | Months Since Last Colonoscopy | Reason of Examination | Finding at Last Colonoscopy * |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 54.6 | M | splenic flexure | T2N0M0 | I | MLH1 | 62.9 | 21.0 | symptoms | 0 | ||
| 2 | 61.7 | F | ascendens | T1N0M0 | I | MLH1 | 76.5 | 20.3 | follow-up | advanced adenoma | ||
| 3 | 44.2 | M | descendens | Dukes A | I | MLH1 | 55.1 | 36.0 | follow-up | adenoma with LG dysplasia | ||
| 4 | 69.7 | F | sigmoid | T1N0M0 | I | MSH2 | 74.9 | 75.1 | cardiac insufficiency | 7.3 | follow-up | advanced adenoma |
| 5 | 63.1 | F | tranverse | T3N0M0 | II | MSH2 | 72.6 | 72.6 | pancreatic cancer | 25.0 | follow-up | 0 |
| 6 | 70.5 | M | sigmoid | TisNxM0 | 0 | MLH1 | 78.4 | 24.5 | follow-up | 0 | ||
| 7 | 35.5 | M | caecum | T3N1M0 | III | MLH1 | 44.3 | 44.3 | gastric cancer | 28.0 | symptoms | 0 |
| 8 | 57.3 | F | sigmoid | T3N0M0 | II | MLH1 | 63.3 | 30.0 | follow-up | 0 | ||
| 9 | 54.5 | F | caecum | T2N0M0 | I | MLH1 | 63.4 | 63.4 | CRC | 31.2 | follow-up | 0 |
| 10 | 71.6 | F | rectum | T2N0M0 | I | MLH1 | 75.1 | 75.1 | biliary tract cancer | 24.0 | follow-up | 0 |
| 11 | 41.7 | F | descendens | T3N2M0 | III | MLH1 | 44.7 | 44.7 | CRC | 23.0 | symptoms | 0 |
| 12 | 43.6 | F | ascendens | T1N0M0 | I | MLH1 | 57.5 | 57.5 | breast cancer | 26.0 | follow-up | 0 |
| 13 | 41.7 | F | tranverse | T1N0M0 | I | MLH1 | 50.1 | 26.0 | follow-up | adenoma with LG dysplasia | ||
| 14 | 42.4 | F | tranverse | T3N0M0 | II | MLH1 | 48.8 | 48.8 | pancreatic cancer | 24.4 | follow-up | 0 |
| 15 | 71.5 | M | ascendens | T2N0M0 | I | MSH2 | 84.1 | 36.0 | follow-up | adenoma with LG dysplasia | ||
| 16 | 43.6 | M | tranverse | T3N0M0 | II | MLH1 | 53.9 | 53.9 | another CRC | 26.0 | follow-up | 0 |
| 17 | 71.9 | F | caecum | T2N0M0 | I | MLH1 | 81.4 | 81.4 | pneumonia | 29.0 | follow-up | 0 |
| 18 | 69.0 | F | caecum | T2N0M0 | I | MSH2 | 69.0 | 69.0 | postoperative complication | 37.7 | symptoms | 0 |
| 19 | 42.0 | F | caecum | T1N0M0 | I | MLH1 | 58.2 | 39.5 | follow-up | 0 | ||
| 20 | 35.1 | M | caecum | Dukes B | II | MLH1 | 64.4 | 37.6 | follow-up | 0 | ||
| 21.a | 54.2 | F | ascendens | T2N0M0 | I | MLH1 | 63.3 | 30.1 | follow-up | 0 | ||
| 21.b | 56.8 | F | sigmoid | T3N0M0 | II | MLH1 | 63.3 | 28.8 | follow-up | 0 | ||
| 22 | 54.2 | M | ascendens | T3N0M0 | II | MLH1 | 63.4 | 65.0 | CUP (brain. lung) | 18.0 | follow-up | adenoma with LG dysplasia |
| 23 | 53.8 | M | ascendens | T2N0M0 | I | MLH1 | 58.8 | 16.4 | follow-up | adenoma with LG dysplasia | ||
| 24 | 82.8 | M | descendens | T2N0M0 | I | MLH1 | 85.1 | 24.6 | follow-up | 0 | ||
| 25 | 55.0 | F | caecum | T1N0M0 | I | MLH1 | 66.4 | 39.1 | follow-up | 0 | ||
| 26 | 43.5 | M | caecum | T2N0M0 | I | MLH1 | 52.5 | 36.2 | follow-up | 0 | ||
| 27 | 27.2 | M | caecum | T2N0M0 | I | MLH1 | 42.9 | 37.9 | follow-up | 0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ahadova, A.; Pfuderer, P.L.; Ahtiainen, M.; Ballhausen, A.; Bohaumilitzky, L.; Kösegi, S.; Müller, N.; Tang, Y.L.; Kosmalla, K.; Witt, J.; et al. Distinct Mutational Profile of Lynch Syndrome Colorectal Cancers Diagnosed under Regular Colonoscopy Surveillance. J. Clin. Med. 2021, 10, 2458. https://doi.org/10.3390/jcm10112458
Ahadova A, Pfuderer PL, Ahtiainen M, Ballhausen A, Bohaumilitzky L, Kösegi S, Müller N, Tang YL, Kosmalla K, Witt J, et al. Distinct Mutational Profile of Lynch Syndrome Colorectal Cancers Diagnosed under Regular Colonoscopy Surveillance. Journal of Clinical Medicine. 2021; 10(11):2458. https://doi.org/10.3390/jcm10112458
Chicago/Turabian StyleAhadova, Aysel, Pauline Luise Pfuderer, Maarit Ahtiainen, Alexej Ballhausen, Lena Bohaumilitzky, Svenja Kösegi, Nico Müller, Yee Lin Tang, Kosima Kosmalla, Johannes Witt, and et al. 2021. "Distinct Mutational Profile of Lynch Syndrome Colorectal Cancers Diagnosed under Regular Colonoscopy Surveillance" Journal of Clinical Medicine 10, no. 11: 2458. https://doi.org/10.3390/jcm10112458
APA StyleAhadova, A., Pfuderer, P. L., Ahtiainen, M., Ballhausen, A., Bohaumilitzky, L., Kösegi, S., Müller, N., Tang, Y. L., Kosmalla, K., Witt, J., Endris, V., Stenzinger, A., von Knebel Doeberitz, M., Bläker, H., Renkonen-Sinisalo, L., Lepistö, A., Böhm, J., Mecklin, J.-P., Seppälä, T. T., & Kloor, M. (2021). Distinct Mutational Profile of Lynch Syndrome Colorectal Cancers Diagnosed under Regular Colonoscopy Surveillance. Journal of Clinical Medicine, 10(11), 2458. https://doi.org/10.3390/jcm10112458