
Evaluation of E148Q and Concomitant AA Amyloidosis in Patients with Familial Mediterranean Fever

 , , , and
, , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Patients
2.2. Clinical and Genetic Information
2.3. Statistical Analysis
3. Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Erer, B.; Demirkaya, E.; Ozen, S.; Kallinich, T. What is the best acute phase reactant for familial Mediterranean fever follow-up and its role in the prediction of complications? A systematic review. Rheumatol. Int. 2016, 36, 483–487. [Google Scholar] [CrossRef]
- Abdallah, E.; Waked, E. Incidence and clinical outcome of renal amyloidosis: A retrospective study. Saudi J. Kidney Dis. Transpl. 2013, 24, 950–958. [Google Scholar] [CrossRef]
- Akpolat, T.; Özkaya, O.; Özen, S. Homozygous M694V as a risk factor for amyloidosis in Turkish FMF patients. Gene 2012, 492, 285–289. [Google Scholar] [CrossRef]
- Babaoglu, H.; Armagan, B.; Bodakci, E.; Satis, H.; Atas, N.; Sari, A.; Yasar Bilge, N.S.; Bilici Salman, R.; Yardımcı, G.K.; Avanoglu Guler, A.; et al. Predictors of persistent inflammation in familial Mediterranean fever and association with damage. Rheumatology 2021, 60, 333–339. [Google Scholar] [CrossRef] [PubMed]
- Gershoni-Baruch, R.; Brik, R.; Lidar, M.; Shinawi, M.; Livneh, A. Male sex coupled with articular manifestations cause a 4-fold increase in susceptibility to amyloidosis in patients with familial Mediterranean fever homozygous for the M694V-MEFV mutation. J. Rheumatol. 2003, 30, 308–312. [Google Scholar] [PubMed]
- Kasifoglu, T.; Bilge, S.Y.; Sari, I.; Solmaz, D.; Senel, S.; Emmungil, H.; Kilic, L.; Oner, S.Y.; Yildiz, F.; Yilmaz, S.; et al. Amyloidosis and its related factors in Turkish patients with familial Mediterranean fever: A multicentre study. Rheumatology 2014, 53, 741–745. [Google Scholar] [CrossRef] [Green Version]
- Touitou, I.; Sarkisian, T.; Medlej-Hashim, M.; Tunca, M.; Livneh, A.; Cattan, D.; Yalçinkaya, F.; Ozen, S.; Majeed, H.; Ozdogan, H.; et al. Country as the primary risk factor for renal amyloidosis in familial Mediterranean fever. Arthritis Rheum. 2007, 56, 1706–1712. [Google Scholar] [CrossRef] [PubMed]
- Pras, M.; Bronshpigel, N.; Zemer, D.; Gafni, J. Variable incidence of amyloidosis in familial Mediterranean fever among different ethnic groups. Johns Hopkins Med. J. 1982, 150, 22–26. [Google Scholar] [PubMed]
- Tirosh, I.; Yacobi, Y.; Vivante, A.; Barel, O.; Ben-Moshe, Y.; Erez Granat, O.; Spielman, S.; Semo Oz, R.; Shinar, Y.; Gerstein, M. Clinical significance of E148Q heterozygous variant in paediatric Familial Mediterranean Fever. Rheumatology 2021, keab128. [Google Scholar] [CrossRef]
- Yilmaz, M.I.; Demirkaya, E.; Acikel, C.; Saldir, M.; Akar, S.; Cayci, T.; Saglam, M.; Unal, H.U.; Gok, M.; Polat, A.; et al. Endothelial function in patients with familial Mediterranean fever-related amyloidosis and association with cardiovascular events. Rheumatology 2014, 53, 2002–2008. [Google Scholar] [CrossRef] [Green Version]
- Sawyer, S.M.; McNeil, R.; Francis, K.L.; Matskarofski, J.Z.; Patton, G.C.; Bhutta, Z.A.; Esangbedo, D.O.; Klein, J.D. The age of paediatrics. Lancet Child. Adolesc. Health 2019, 3, 822–830. [Google Scholar] [CrossRef]
- Daher, R.T.; Khalik, R.N.; Hoteit, R.M.; Sarieddine, D.S.; Charafeddine, K.M.; Cortas, N.K.; Mahfouz, R.A. The use of a reverse hybridization strip assay for the study of hemochromatosis-associated gene mutations in Lebanon. Genet. Test. Mol. Biomark. 2011, 15, 909–911. [Google Scholar] [CrossRef]
- Tunca, M.; Akar, S.; Onen, F.; Ozdogan, H.; Kasapcopur, O.; Yalcinkaya, F.; Tutar, E.; Ozen, S.; Topaloglu, R.; Yilmaz, E.; et al. Familial Mediterranean fever (FMF) in Turkey: Results of a nationwide multicenter study. Medicine 2005, 84, 1–11. [Google Scholar] [CrossRef]
- Ben-Chetrit, E.; Lerer, I.; Malamud, E.; Domingo, C.; Abeliovich, D. The E148Q mutation in the MEFV gene: Is it a disease-causing mutation or a sequence variant? Hum. Mutat. 2000, 15, 385–386. [Google Scholar] [CrossRef]
- Yilmaz, E.; Ozen, S.; Balci, B.; Duzova, A.; Topaloglu, R.; Besbas, N.; Saatci, U.; Bakkaloglu, A.; Ozguc, M. Mutation frequency of Familial Mediterranean Fever and evidence for a high carrier rate in the Turkish population. Eur. J. Hum. Genet. 2001, 9, 553–555. [Google Scholar] [CrossRef] [PubMed]
- Livneh, A.; Langevitz, P.; Shinar, Y.; Zaks, N.; Kastner, D.L.; Pras, M.; Pras, E. MEFV mutation analysis in patients suffering from amyloidosis of familial Mediterranean fever. Amyloid 1999, 6, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Topaloglu, R.; Batu, E.D.; Yıldız, Ç.; Korkmaz, E.; Özen, S.; Beşbaş, N.; Özaltın, F. Familial Mediterranean fever patients homozygous for E148Q variant may have milder disease. Int. J. Rheum. Dis. 2018, 21, 1857–1862. [Google Scholar] [CrossRef]
- Topaloglu, R.; Ozaltin, F.; Yilmaz, E.; Ozen, S.; Balci, B.; Besbas, N.; Bakkaloglu, A. E148Q is a disease-causing MEFV mutation: A phenotypic evaluation in patients with familial Mediterranean fever. Ann. Rheum. Dis. 2005, 64, 750–752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nursal, A.F.; Tekcan, A.; Kaya, S.U.; Turkmen, E.; Yigit, S. Mutational Spectrum of the MEFV Gene in AA Amyloidosis Associated With Familial Mediterranean Fever. Iran. J. Kidney Dis. 2016, 10, 107–112. [Google Scholar]
- Altunoğlu, A.; Erten, Ş.; Canoz, M.B.; Yuksel, A.; Ceylan, G.G.; Balci, S.; Dogan, H.T. Phenotype 2 familial mediterranean fever: Evaluation of 22 case series and review of the literature on phenotype 2 FMF. Ren. Fail. 2013, 35, 226–230. [Google Scholar] [CrossRef] [Green Version]
- Atoyan, S.; Hayrapetyan, H.; Yeghiazaryan, A.; Ben-Chetrit, E.; Sarkisian, T. Is the country of living important in the phenotypic expression of E148Q mutation? The Armenian experience. Clin. Exp. Rheumatol. 2020, 38 (Suppl. 127), 124–125. [Google Scholar] [PubMed]
- Ozen, S.; Demirkaya, E.; Amaryan, G.; Koné-Paut, I.; Polat, A.; Woo, P.; Uziel, Y.; Modesto, C.; Finetti, M.; Quartier, P.; et al. Results from a multicentre international registry of familial Mediterranean fever: Impact of environment on the expression of a monogenic disease in children. Ann. Rheum. Dis. 2014, 73, 662–667. [Google Scholar] [CrossRef] [PubMed]
- Twig, G.; Livneh, A.; Vivante, A.; Afek, A.; Shamiss, A.; Derazne, E.; Tzur, D.; Ben-Zvi, I.; Tirosh, A.; Barchana, M.; et al. Mortality risk factors associated with familial Mediterranean fever among a cohort of 1.25 million adolescents. Ann. Rheum. Dis. 2014, 73, 704–709. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Age of Diagnosis < 18 (n = 101) | Age of Diagnosis ≥ 18 (n = 68) | Total (n = 169) | p | |
|---|---|---|---|---|
| Age Median (min-max) (years) | 35 (20–49) | 38 (19–45) | 36 (19–49) | 0.08 |
| Age of diagnosis for FMF Median (min-max) (years) | 13.9 (5–17) | 20 (18–25) | 16 (5–25) | <0.001 |
| Age of diagnosis for amyloidosis Median (min-max) (years) | 21 (13–31) | 20 (18–25) | 20 (13–31) | 0.86 |
| Follow-up duration Median (min-max) (years) | 13 (4–17) | 16 (4–17) | 15 (4–17) | 0.17 |
| Male n (%) | 65 (64.4) | 39 (57.4) | 104 (61.5) | 0.35 |
| History of FMF n (%) | 45 (44.6) | 11 (16.2) | 56 (33.1) | <0.001 |
| History of Amyloidosis n (%) | 34 (33.7) | 7 (10.3) | 41 (24.3) | 0.001 |
| Death n (%) | 11 (10.9) | 4 (5.6) | 15 (8.9) | 0.26 |
| Clinical Findings n (%) | ||||
| Fever | 87 (86.1) | 56 (82.4) | 143 (84.6) | 0.52 |
| Abdominal pain | 72 (71.3) | 49 (72.1) | 121 (71.6) | 0.91 |
| Arthritis | 70 (69.3) | 43 (63.2) | 113 (66.9) | 0.41 |
| Chest pain | 65 (64.4) | 36 (52.9) | 101 (59.8) | 0.13 |
| Arthralgia | 45 (44.6) | 36 (52.9) | 81(47.9) | 0.28 |
| Vomiting | 30 (29.7) | 22 (32.4) | 52 (30.8) | 0.73 |
| Constipation | 21 (20.8) | 16 (23.5) | 37 (21.9) | 0.67 |
| Diarrhea | 23 (22.8) | 9 (13.2) | 32 (18.9) | 0.12 |
| Mood disorder | 26 (25.7) | 14 (20.6) | 40 (23.7) | 0.43 |
| Myalgia/myositis | 27 (26.7) | 19 (27.9) | 46 (27.2) | 0.86 |
| Protracted-febrile-myalgia | 17 (16.8) | 9 (13.2) | 26 (15.4) | 0.52 |
| Fatigue | 12 (11.9) | 10 (14.7) | 22 (13.0) | 0.59 |
| Headache | 10 (9.9) | 9 (13.2) | 19 (11.2) | 0.50 |
| Erysipeloid erythema | 7 (6.9) | 3 (4.4) | 10 (5.9) | 0.74 * |
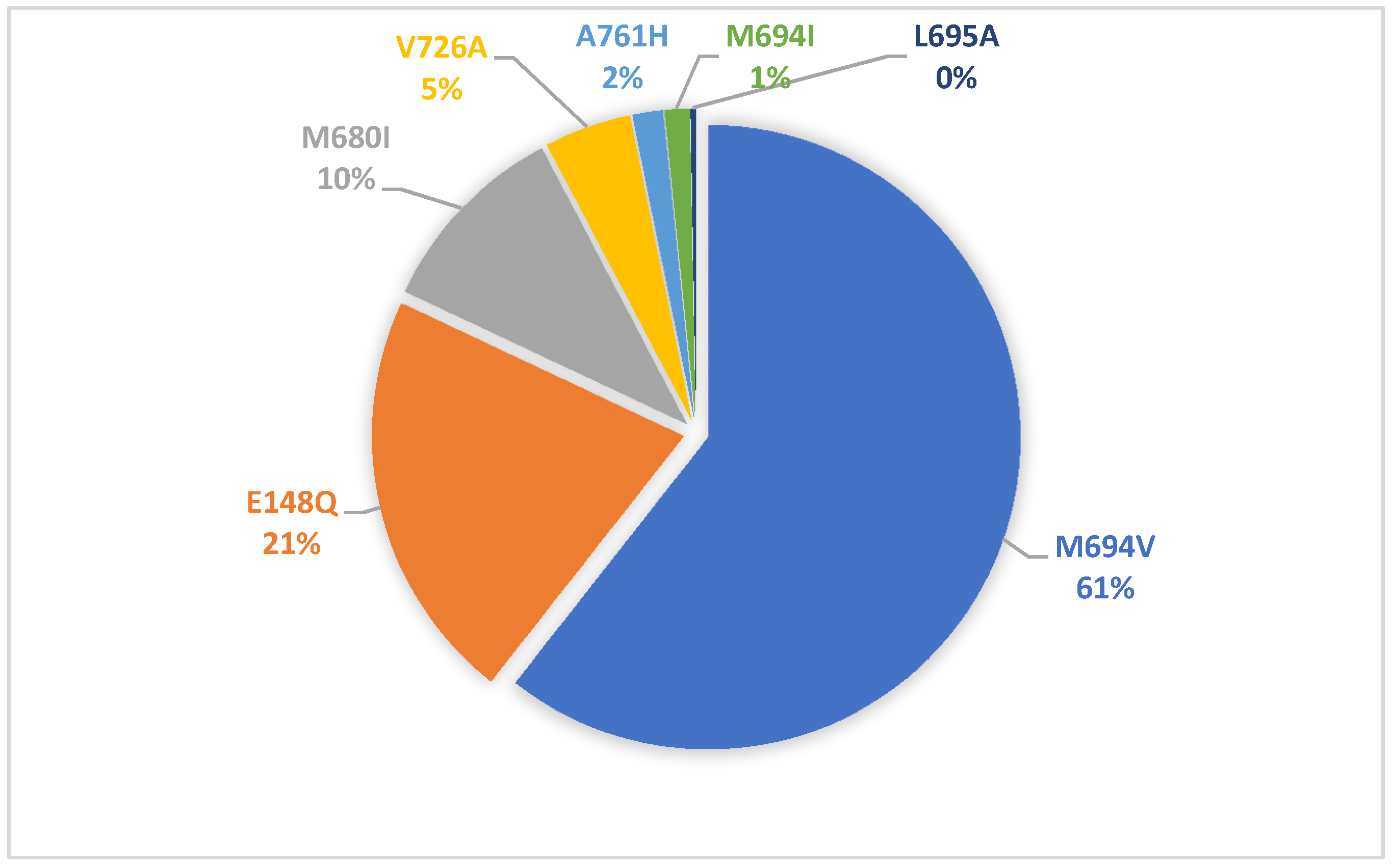
| Genotypes in MEFV n (%) | ||||
| M694V/M694V | 48 (47.5) | 28 (41.2) | 76 (45.0) | 0.46 |
| M694V/E148Q | 13 (12.9) | 12 (17.6) | 25 (14.8) | |
| E148Q/E148Q | 9 (8.9) | 10 (14.7) | 19 (11.2) | |
| E148Q/other variants | 1 (1.0) | 2 (2.9) | 3 (1.8) | |
| Other | 30 (29.7) | 16 (23.6) | 46 (27.2) | |
| E148Q/E148Q (n = 19) | E148Q/Other Variants (n = 28) | M694V/M694V (n = 76) | Total (n = 123) | p | |
|---|---|---|---|---|---|
| Age Median (min-max) (years) | 37 (22–44) | 36 (25–49) | 36.5 (19–49) | 36 (19–49) | 0.65 |
| Age of diagnosis for FMF Median (min-max) (years) | 18 (8–24) | 17.5 (7–22) | 16 (5–25) | 16 (5–25) | 0.79 |
| Age of diagnosis for amyloidosis Median (min-max) (years) | 19 (16–26) | 19.5 (16–25) | 20 (13–31) | 20 (13–31) | 0.76 |
| Follow up duration Median (min-max) (years) | 16 (4–17) | 14.5 (4–17) | 16 (4–17) | 16 (4–17) | 0.96 |
| Gender n (%) | |||||
| Male | 9 (47.4) | 17 (60.7) | 50 (65.8) | 76 (61.8) | 0.33 |
| Female | 10 (52.6) | 11 (39.3) | 26 (34.2) | 47 (38.2) | |
| History of FMF n (%) | 7 (36.8) | 8 (28.6) | 30 (39.5) | 45 (36.6) | 0.59 |
| History of Amyloidosis n (%) | 5 (26.3) | 7 (25.0) | 19 (25.0) | 31 (25.2) | 0.99 |
| Death | 1 (5.3) | - | 14 (18.4) | 15 (12.2) | 0.016 |
| Clinical Findings n (%) | |||||
| Fever | 16 (84.2) | 25 (89.3) | 65 (85.5) | 106 (86.2) | 0.86 |
| Abdominal pain | 15 (78.9) | 21 (75.0) | 55 (72.4) | 91 (74.0) | 0.83 |
| Arthritis | 15 (78.9) | 13 (46.4) | 58 (76.3) | 86 (69.9) | 0.008 |
| Chest pain | 13 (68.4) | 17 (60.7) | 47 (61.8) | 77 (62.6) | 0.84 |
| Arthralgia | 8 (42.1) | 12 (42.9) | 39 (51.3) | 59 (48.0) | 0.63 |
| Vomiting | 4 (21.1) | 4 (14.3) | 26 (34.2) | 34 (27.6) | 0.10 |
| Constipation | 3 (15.8) | 5 (17.9) | 15 (19.7) | 23 (18.7) | 0.91 |
| Diarrhea | 2 (10.5) | 4 (14.3) | 17 (22.4) | 23 (18.7) | 0.39 |
| Mood disorder | 3 (15.8) | 3 (10.7) | 18 (23.7) | 24 (19.5) | 0.30 |
| Myalgia/myositis | 7 (36.8) | 2 (7.1) | 20 (26.3) | 29 (23.6) | 0.04 |
| Protracted-febrile-myalgia | 4 (21.1) | 4 (14.8) | 10 (13.2) | 18 (14.6) | 0.65 |
| Fatigue | 4 (21.1) | 2 (7.1) | 10 (13.2) | 16 (13.0) | 0.36 |
| Headache | 3 (15.8) | 3 (10.7) | 7 (9.2) | 13 (10.6) | 0.65 |
| Erysipeloid erythema | - | 2 (7.1) | 7 (9.2) | 9 (7.3) | 0.53 |
| Number | Sex | Age of FMF Diagnosis (Years) | Age of Amyloidosis Diagnosis (Years) | Age of Death (Years) | Genotype | Disease Duration (Years) | Cause of Death |
|---|---|---|---|---|---|---|---|
| 1 | Female | 13 | 21 | 36 | M694V/M694V | 23 | Arrhythmia |
| 2 | Male | 11 | 23 | 36 | M694V/M694V | 25 | MI * |
| 3 | Male | 9 | 18 | 36 | M694V/M694V | 27 | Arrhythmia |
| 4 | Male | 18 | 18 | 37 | M694V/M694V | 19 | MI |
| 5 | Male | 11 | 21 | 39 | M694V/M694V | 28 | MI |
| 6 | Female | 13 | 22 | 39 | M694V/M694V | 26 | MI |
| 7 | Female | 14 | 18 | 40 | E148Q/E148Q | 26 | MI |
| 8 | Male | 7 | 21 | 41 | M694V/M694V | 34 | MI |
| 9 | Male | 11 | 22 | 43 | M694V/M694V | 32 | MI |
| 10 | Male | 21 | 22 | 44 | M694V/M694V | 23 | MI |
| 11 | Female | 18 | 23 | 44 | M694V/M694V | 26 | MI |
| 12 | Male | 14 | 19 | 44 | M694V/M694V | 30 | Arrhythmia |
| 13 | Male | 17 | 17 | 45 | M694V/M694V | 28 | MI |
| 14 | Male | 20 | 20 | 47 | M694V/M694V | 27 | MI |
| 15 | Female | 15 | 19 | 49 | M694V/M694V | 35 | MI |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Arici, Z.S.; Romano, M.; Piskin, D.; Guzel, F.; Sahin, S.; Berard, R.A.; Yilmaz, M.I.; Demirkaya, E. Evaluation of E148Q and Concomitant AA Amyloidosis in Patients with Familial Mediterranean Fever. J. Clin. Med. 2021, 10, 3511. https://doi.org/10.3390/jcm10163511
Arici ZS, Romano M, Piskin D, Guzel F, Sahin S, Berard RA, Yilmaz MI, Demirkaya E. Evaluation of E148Q and Concomitant AA Amyloidosis in Patients with Familial Mediterranean Fever. Journal of Clinical Medicine. 2021; 10(16):3511. https://doi.org/10.3390/jcm10163511
Chicago/Turabian StyleArici, Zehra Serap, Micol Romano, David Piskin, Ferhat Guzel, Sezgin Sahin, Roberta A. Berard, Mahmut I. Yilmaz, and Erkan Demirkaya. 2021. "Evaluation of E148Q and Concomitant AA Amyloidosis in Patients with Familial Mediterranean Fever" Journal of Clinical Medicine 10, no. 16: 3511. https://doi.org/10.3390/jcm10163511
APA StyleArici, Z. S., Romano, M., Piskin, D., Guzel, F., Sahin, S., Berard, R. A., Yilmaz, M. I., & Demirkaya, E. (2021). Evaluation of E148Q and Concomitant AA Amyloidosis in Patients with Familial Mediterranean Fever. Journal of Clinical Medicine, 10(16), 3511. https://doi.org/10.3390/jcm10163511