
Non-Lipid Effects of PCSK9 Monoclonal Antibodies on Vessel Wall


Abstract
:1. Introduction
2. Inflammation
3. Endothelial Dysfunction
4. Haemostasis
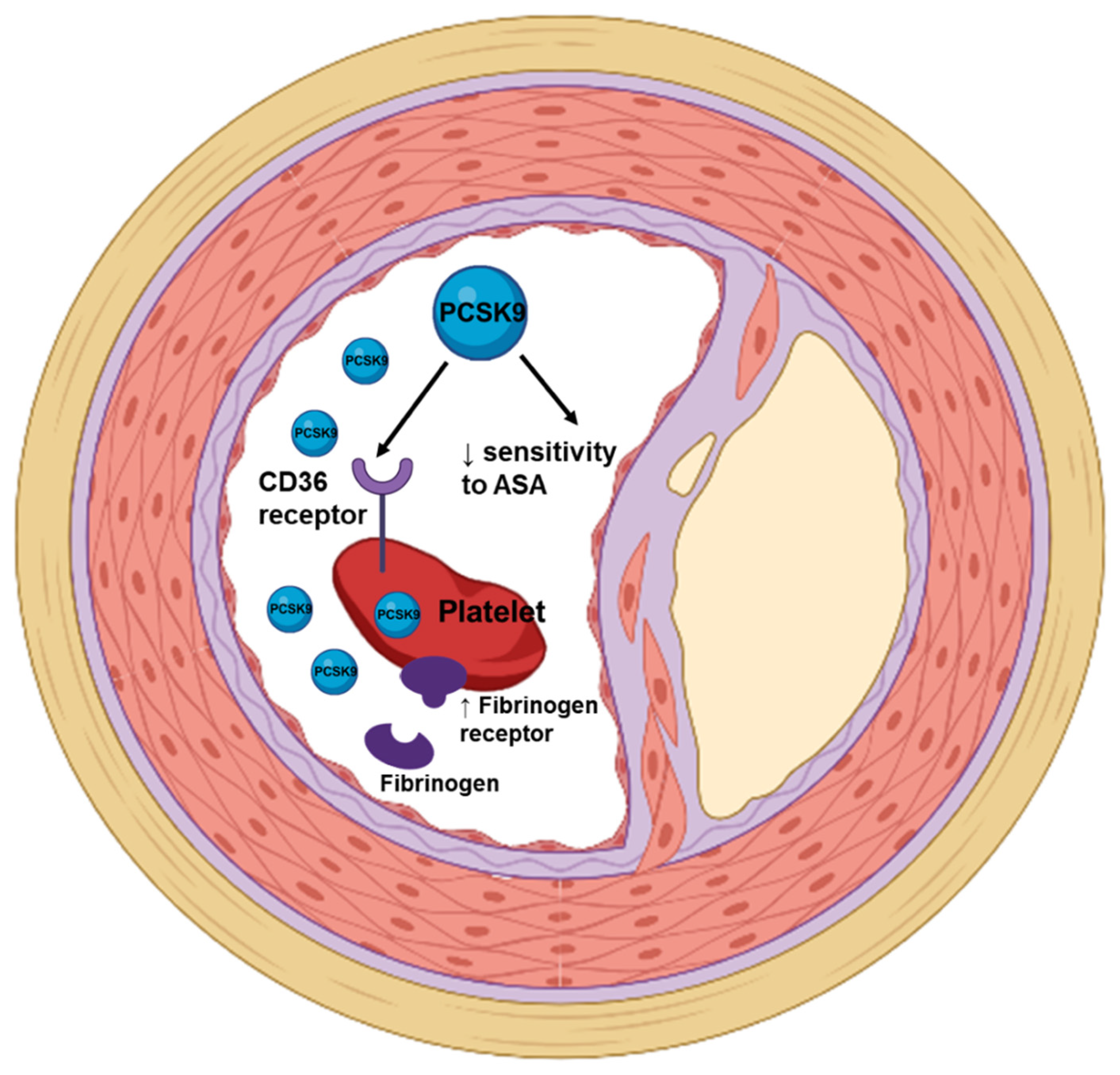
4.1. PCSK9 and Platelets’ Function
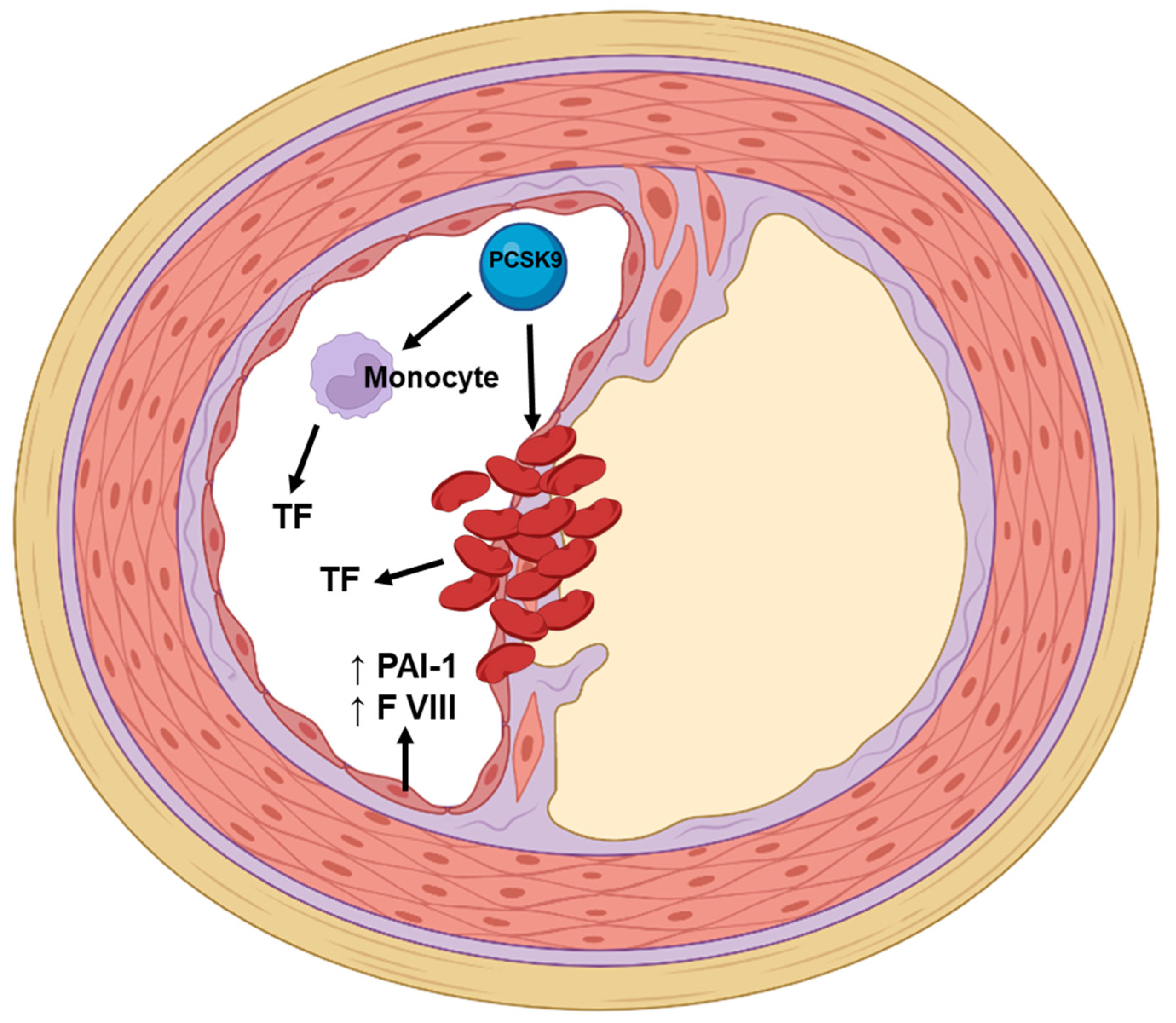
4.2. PCSK9 Monoclonal Antibodies and Coagulation and Fibrinolytic Parameters
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Benjamin, E.J.; Muntner, P.; Alonso, A.; Bittencourt, M.S.; Callaaway, C.W.; Carson, A.P.; Chamberlain, A.M.; Chang, A.R.; Cheng, S.; Das, S.R.; et al. Heart disease and stroke statistics—2019 update: A report from the American Heart Association. Circulation 2019, 139, e1–e473. [Google Scholar] [CrossRef] [PubMed]
- Krychtiuk, K.A.; Kastl, S.P.; Wojta, J.; Speidl, W.S. Inflammation and coagulation in atherosclerosis. Hämostaseologie 2013, 33, 269–282. [Google Scholar] [CrossRef] [PubMed]
- Borén, J.; Chapman, M.J.; Krauss, R.M.; Packard, C.J.; Bentzon, J.F.; Binder, C.J.; Daemen, M.J.; Demer, L.L.; Hegele, R.A.; Nicholls, S.J.; et al. Low-density lipoproteins cause atherosclerotic cardiovascular disease: Pathophysiological, genetic, and therapeutic insights: A consensus statement from the European Atherosclerosis Society Consensus Panel. Eur. Heart J. 2020, 41, 2313–2330. [Google Scholar] [CrossRef] [PubMed]
- Elshourbagy, N.A.; Meyers, H.V.; Abdel-Meguid, S.S. Cholesterol: The Good, the Bad, and the Ugly—Therapeutic Targets for the Treatment of Dyslipidemia. Med. Princ. Pract. 2013, 23, 99–111. [Google Scholar] [CrossRef] [PubMed]
- Krychtiuk, K.A.; Lenz, M.; Hohensinner, P.; Distelmaier, K.; Schrutka, L.; Kastl, S.P.; Huber, K.; Dostal, E.; Oravec, S.; Hengstenberg, C.; et al. Circulating levels of proprotein convertase subtilisin/kexin type 9 (PCSK9) are associated with monocyte subsets in patients with stable coronary artery disease. J. Clin. Lipidol. 2021, 15, 512–521. [Google Scholar] [CrossRef]
- Kosmas, C.E.; Pantou, D.; Sourlas, A.; Papakonstantinou, E.J.; Uceta, R.E.; Guzman, E. New and emerging lipid-modifying drugs to lower LDL cholesterol. Drugs Context 2021, 10, 1–22. [Google Scholar] [CrossRef]
- Ray, K.K.; Molemans, B.; Schoonen, W.M.; Giovas, P.; Bray, S.; Kiru, G.; Murphy, J.; Banach, M.; De Servi, S.; Gaita, D.; et al. EU-Wide Cross-Sectional Observational Study of Lipid-Modifying Therapy Use in Secondary and Primary Care: The DA VINCI study. Eur. J. Prev. Cardiol. 2020, 28, 1279–1289. [Google Scholar] [CrossRef]
- Cybulska, B.; Kłosiewicz-Latoszek, L.; Penson, P.E.; Banach, M. What do we know about the role of lipoprotein(a) in atherogenesis 57 years after its discovery? Prog. Cardiovasc. Dis. 2020, 63, 219–227. [Google Scholar] [CrossRef]
- Nordestgaard, B.G.; Chapman, M.J.; Ray, K.; Borén, J.; Andreotti, F.; Watts, G.; Ginsberg, H.; Amarenco, P.; Catapano, A.L.; Descamps, O.S.; et al. Lipoprotein(a) as a cardiovascular risk factor: Current status. Eur. Heart J. 2010, 31, 2844–2853. [Google Scholar] [CrossRef]
- Kiechl, S.; Willeit, J. The Mysteries of Lipoprotein(a) and Cardiovascular Disease Revisited. J. Am. Coll. Cardiol. 2010, 55, 2168–2170. [Google Scholar] [CrossRef] [Green Version]
- Nordestgaard, B.G.; Nielsen, L.B. Atherosclerosis and arterial influx of lipoproteins. Curr. Opin. Lipidol. 1994, 5, 252–257. [Google Scholar] [CrossRef] [PubMed]
- Tsimikas, S. A Test in Context: Lipoprotein(a): Diagnosis, prognosis, controversies, and emerging therapies. J. Am. Coll. Cardiol. 2017, 69, 692–711. [Google Scholar] [CrossRef] [PubMed]
- Tsimikas, S.; Fazio, S.; Ferdinand, K.C.; Ginsberg, H.N.; Koschinsky, M.; Marcovina, S.M.; Moriarty, P.M.; Rader, D.J.; Remaley, A.T.; Reyes-Soffer, G.; et al. NHLBI Working Group Recommendations to Reduce Lipoprotein(a)-Mediated Risk of Cardiovascular Disease and Aortic Stenosis. J. Am. Coll. Cardiol. 2018, 71, 177–192. [Google Scholar] [CrossRef] [PubMed]
- Stulnig, T.M.; Morozzi, C.; Reindl-Schwaighofer, R.; Stefanutti, C. Looking at Lp(a) and Related Cardiovascular Risk: From Scientific Evidence and Clinical Practice. Curr. Atheroscler. Rep. 2019, 21, 37. [Google Scholar] [CrossRef] [PubMed]
- Ferretti, G.; Bacchetti, T.; Johnston, T.P.; Banach, M.; Pirro, M.; Sahebkar, A. Lipoprotein(a): A missing culprit in the management of athero-thrombosis? J. Cell. Physiol. 2017, 233, 2966–2981. [Google Scholar] [CrossRef] [PubMed]
- Yurtseven, E.; Ural, D.; Baysal, K.; Tokgözoğlu, L. An Update on the Role of PCSK9 in Atherosclerosis. J. Atheroscler. Thromb. 2020, 27, 909–918. [Google Scholar] [CrossRef]
- Sabatine, M.S.; Giugliano, R.P.; Keech, A.C.; Honarpour, N.; Wiviott, S.D.; Murphy, S.A.; Kuder, J.F.; Wang, H.; Liu, T.; Wasserman, S.M.; et al. Evolocumab and Clinical Outcomes in Patients with Cardiovascular Disease. N. Engl. J. Med. 2017, 376, 1713–1722. [Google Scholar] [CrossRef]
- Robinson, J.G.; Farnier, M.; Krempf, M.; Bergeron, J.; Luc, G.; Averna, M.; Stroes, E.S.; Langslet, G.; Raal, F.J.; El Shahawy, M.; et al. Efficacy and Safety of Alirocumab in Reducing Lipids and Cardiovascular Events. N. Engl. J. Med. 2015, 372, 1489–1499. [Google Scholar] [CrossRef]
- Stoekenbroek, R.M.; Kallend, D.; Wijngaard, P.L.; Kastelein, J.J. Inclisiran for the treatment of cardiovascular disease: The ORION clinical development program. Futur. Cardiol. 2018, 14, 433–442. [Google Scholar] [CrossRef]
- Ross, R. Atherosclerosis as an inflammatory disease. N. Engl. J. Med. 1999, 340, 115–126. [Google Scholar] [CrossRef]
- Aday, A.W.; Ridker, P.M. Targeting Residual Inflammatory Risk: A Shifting Paradigm for Atherosclerotic Disease. Front. Cardiovasc. Med. 2019, 6, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gabay, C.; Kushner, I. Acute-Phase Proteins and Other Systemic Responses to Inflammation. N. Engl. J. Med. 1999, 340, 448–454. [Google Scholar] [CrossRef] [PubMed]
- Levstek, T.; Podkrajšek, N.; Likozar, A.R.; Šebeštjen, M.; Podkrajšek, K.T. The Influence of Treatment with PCSK9 Inhibitors and Variants in the CRP (rs1800947), TNFA (rs1800629), and IL6 (rs1800795) Genes on the Corresponding Inflammatory Markers in Patients with Very High Lipoprotein(a) Levels. J. Cardiovasc. Dev. Dis. 2022, 9, 127. [Google Scholar] [CrossRef] [PubMed]
- Jeppesen, J.; Hansen, T.; Olsen, M.H.; Rasmussen, S.; Lbsen, H.; Torp-Pedersen, C.; Hildebrandt, P.R.; Madsbad, S. C-reactive protein, insulin resistance and risk of cardiovascular disease: A population-based study. Eur. J. Cardiovasc. Prev. Rehabil. 2008, 15, 594–598. [Google Scholar] [CrossRef] [PubMed]
- Held, C.; White, H.D.; Stewart, R.A.H.; Budaj, A.; Cannon, C.P.; Hochman, J.S.; Koenig, W.; Siegbahn, A.; Steg, P.G.; Soffer, J.; et al. Inflammatory Biomarkers Interleukin-6 and C-Reactive Protein and Outcomes in Stable Coronary Heart Disease: Experiences from the STABILITY (Stabilization of Atherosclerotic Plaque by Initiation of Darapladib Therapy) Trial. J. Am. Heart Assoc. 2017, 6, e005077. [Google Scholar] [CrossRef] [Green Version]
- Ridker, P.M.; Danielson, E.; Fonseca, F.A.; Genest, J.; Gotto, A.M., Jr.; Kastelein, J.J.; Koenig, W.; Libby, P.; Lorenzatti, A.J.; MacFadyen, J.G.; et al. Rosuvastatin to Prevent Vascular Events in Men and Women with Elevated C-Reactive Protein. N. Engl. J. Med. 2008, 359, 2195–2207. [Google Scholar] [CrossRef] [Green Version]
- Sahebkar, A.; Simental-Mendía, L.E.; Guerrero-Romero, F.; Golledge, J.; Watts, G.F. Effect of statin therapy on plasma proprotein convertase subtilisin kexin 9 (PCSK9) concentrations: A systematic review and meta-analysis of clinical trials. Diabetes Obes. Metab. 2015, 17, 1042–1055. [Google Scholar] [CrossRef]
- Roth, E.M.; Davidson, M.H. PCSK9 Inhibitors: Mechanism of Action, Efficacy, and Safety. Rev. Cardiovasc. Med. 2018, 19, 31–46. [Google Scholar] [CrossRef]
- Barale, C.; Melchionda, E.; Morotti, A.; Russo, I. PCSK9 Biology and Its Role in Atherothrombosis. Int. J. Mol. Sci. 2021, 22, 5880. [Google Scholar] [CrossRef]
- Gencer, B.; Montecucco, F.; Nanchen, D.; Carbone, F.; Klingenberg, R.; Vuilleumier, N.; Aghlmandi, S.; Heg, D.; Räber, L.; Auer, R.; et al. Prognostic value of PCSK9 levels in patients with acute coronary syndromes. Eur. Heart J. 2016, 37, 546–553. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Zhang, Y.; Xu, R.-X.; Guo, Y.-L.; Zhu, C.-G.; Wu, N.-Q.; Qing, P.; Liu, G.; Dong, Q.; Li, J.-J. Proprotein convertase subtilisin-kexin type 9 as a biomarker for the severity of coronary artery disease. Ann. Med. 2015, 47, 386–393. [Google Scholar] [CrossRef] [PubMed]
- Navarese, E.P.; Kołodziejczak, M.; Winter, M.-P.; Alimohammadi, A.; Lang, I.M.; Buffon, A.; Lip, G.Y.; Siller-Matula, J.M. Association of PCSK9 with platelet reactivity in patients with acute coronary syndrome treated with prasugrel or ticagrelor: The PCSK9-REACT study. Int. J. Cardiol. 2017, 227, 644–649. [Google Scholar] [CrossRef] [PubMed]
- Punch, E.; Klein, J.; Diaba-Nuhoho, P.; Morawietz, H.; Garelnabi, M. Effects of PCSK9 Targeting: Alleviating Oxidation, Inflammation, and Atherosclerosis. J. Am. Heart Assoc. 2022, 11, e023328. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.-X.; Li, S.; Liu, H.-H.; Li, J.-J. Impact of PCSK9 monoclonal antibodies on circulating hs-CRP levels: A systematic review and meta-analysis of randomised controlled trials. BMJ Open 2018, 8, e022348. [Google Scholar] [CrossRef] [Green Version]
- Stiekema, L.C.A.; Stroes, E.S.G.; Verweij, S.L.; Kassahun, H.; Chen, L.; Wasserman, S.M.; Sabatine, M.S.; Mani, V.; Fayad, Z.A. Persistent arterial wall inflammation in patients with elevated lipoprotein(a) despite strong low-density lipoprotein cholesterol reduction by proprotein convertase subtilisin/kexin type 9 antibody treatment. Eur. Heart J. 2018, 40, 2775–2781. [Google Scholar] [CrossRef] [Green Version]
- Hoogeveen, R.M.; Opstal, T.S.; Kaiser, Y.; Stiekema, L.C.; Kroon, J.; Knol, R.J.; Bax, W.A.; Verberne, H.J.; Cornel, J.; Stroes, E.S. PCSK9 Antibody Alirocumab Attenuates Arterial Wall Inflammation Without Changes in Circulating Inflammatory Markers. JACC Cardiovasc. Imaging 2019, 12, 2571–2573. [Google Scholar] [CrossRef]
- Stiekema, L.C.A.; Prange, K.H.M.; Hoogeveen, R.M.; Verweij, S.L.; Kroon, J.; Schnitzler, J.G.; Dzobo, K.E.; Cupido, A.J.; Tsimikas, S.; Stroes, E.S.G.; et al. Potent lipoprotein(a) lowering following apolipoprotein(a) antisense treatment reduces the pro-inflammatory activation of circulating monocytes in patients with elevated lipoprotein(a). Eur. Heart J. 2020, 41, 2262–2271. [Google Scholar] [CrossRef]
- Nicholls, S.J.; Puri, R.; Anderson, T.; Ballantyne, C.M.; Cho, L.; Kastelein, J.J.P.; Koenig, W.; Somaratne, R.; Kassahun, H.; Yang, J.; et al. Effect of Evolocumab on Progression of Coronary Disease in Statin-Treated Patients: The GLAGOV randomized clinical trial. JAMA J. Am. Med. Assoc. 2016, 316, 2373–2384. [Google Scholar] [CrossRef]
- Nelson, A.J.; Puri, R.; Brennan, D.M.; Anderson, T.J.; Cho, L.; Ballantyne, C.M.; Kastelein, J.J.; Koenig, W.; Kassahun, H.; Somaratne, R.M.; et al. C-reactive protein levels and plaque regression with evolocumab: Insights from GLAGOV. Am. J. Prev. Cardiol. 2020, 3, 100091. [Google Scholar] [CrossRef]
- Nicholls, S.J.; Kataoka, Y.; Nissen, S.E.; Prati, F.; Windecker, S.; Puri, R.; Hucko, T.; Aradi, D.; Herrman, J.-P.R.; Hermanides, R.S.; et al. Effect of Evolocumab on Coronary Plaque Phenotype and Burden in Statin-Treated Patients Following Myocardial Infarction. JACC Cardiovasc. Imaging 2022, in press. [Google Scholar] [CrossRef]
- Park, K.-H.; Park, W.J. Endothelial Dysfunction: Clinical Implications in Cardiovascular Disease and Therapeutic Approaches. J. Korean Med. Sci. 2015, 30, 1213–1225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonetti, P.O.; Lerman, L.O.; Lerman, A. Endothelial Dysfunction: A marker of atherosclerotic risk. Arter. Thromb. Vasc. Biol. 2003, 23, 168–175. [Google Scholar] [CrossRef] [PubMed]
- Heitzer, T.; Schlinzig, T.; Krohn, K.; Meinertz, T.; Münzel, T. Cardiovascular events in patients with coronary artery disease. Circulation 2001, 104, 673–2678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maulucci, G.; Cipriani, F.; Russo, D.; Casavecchia, G.; Di Staso, C.; Di Martino, L.F.M.; Ruggiero, A.; Di Biase, M.; Brunetti, N.D. Improved endothelial function after short-term therapy with evolocumab. J. Clin. Lipidol. 2018, 12, 669–673. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.D.; Berglund, L.; Dimayuga, C.; Jones, J.; Sciacca, R.R.; Di Tullio, M.R.; Homma, S. High lipoprotein(a) levels and small apolipoprotein(a) sizes are associated with endothelial dysfunction in a multiethnic cohort. J. Am. Coll. Cardiol. 2004, 43, 1828–1833. [Google Scholar] [CrossRef] [Green Version]
- Di Minno, A.; Gentile, M.; Iannuzzo, G.; Calcaterra, I.; Tripaldella, M.; Porro, B.; Cavalca, V.; Di Taranto, M.D.; Tremoli, E.; Fortunato, G.; et al. Endothelial function improvement in patients with familial hypercholesterolemia receiving PCSK-9 inhibitors on top of maximally tolerated lipid lowering therapy. Thromb. Res. 2020, 194, 229–236. [Google Scholar] [CrossRef]
- Metzner, T.; Leitner, D.R.; Dimsity, G.; Gunzer, F.; Opriessnig, P.; Mellitzer, K.; Beck, A.; Sourij, H.; Stojakovic, T.; Deutschmann, H.; et al. Short-Term Treatment with Alirocumab, Flow-Dependent Dilatation of the Brachial Artery and Use of Magnetic Resonance Diffusion Tensor Imaging to Evaluate Vascular Structure: An Exploratory Pilot Study. Biomedicines 2022, 10, 152. [Google Scholar] [CrossRef]
- Bittner, V.A.; Szarek, M.; Aylward, P.E.; Bhatt, D.L.; Diaz, R.; Edelberg, J.M.; Fras, Z.; Goodman, S.G.; Halvorsen, S.; Hanotin, C.; et al. Effect of Alirocumab on Lipoprotein(a) and Cardiovascular Risk After Acute Coronary Syndrome. J. Am. Coll. Cardiol. 2020, 75, 133–144. [Google Scholar] [CrossRef]
- Leucker, T.M.; Gerstenblith, G.; Schär, M.; Brown, T.T.; Jones, S.R.; Afework, Y.; Weiss, R.G.; Hays, A.G. Evolocumab, a PCSK9-Monoclonal Antibody, Rapidly Reverses Coronary Artery Endothelial Dysfunction in People Living with HIV and People with Dyslipidemia. J. Am. Heart Assoc. 2020, 9, e016263. [Google Scholar] [CrossRef]
- Luquero, A.; Badimon, L.; Borrell-Pages, M. PCSK9 Functions in Atherosclerosis Are Not Limited to Plasmatic LDL-Cholesterol Regulation. Front. Cardiovasc. Med. 2021, 8, 639727. [Google Scholar] [CrossRef]
- Luchetti, F.; Crinelli, R.; Nasoni, M.G.; Benedetti, S.; Palma, F.; Fraternale, A.; Iuliano, L. LDL receptors, caveolae and cholesterol in endothelial dysfunction: OxLDLs accomplices or victims? J. Cereb. Blood Flow Metab. 2020, 178, 3104–3114. [Google Scholar] [CrossRef] [PubMed]
- Ricci, C.; Ruscica, M.; Camera, M.; Rossetti, L.; Macchi, C.; Colciago, A.; Zanotti, I.; Lupo, M.G.; Adorni, M.P.; Cicero, A.F.G.; et al. PCSK9 induces a pro-inflammatory response in macrophages. Sci. Rep. 2018, 8, 2267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moens, S.J.B.; Neele, A.E.; Kroon, J.; Van Der Valk, F.M.; Bossche, J.V.D.; Hoeksema, M.A.; Hoogeveen, R.M.; Schnitzler, J.G.; Baccara-Dinet, M.T.; Manvelian, G.; et al. PCSK9 monoclonal antibodies reverse the pro-inflammatory profile of monocytes in familial hypercholesterolaemia. Eur. Heart J. 2017, 38, 1584–1593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marques, P.; Domingo, E.; Rubio, A.; Martinez-Hervás, S.; Ascaso, J.F.; Piqueras, L.; Real, J.T.; Sanz, M.-J. Beneficial effects of PCSK9 inhibition with alirocumab in familial hypercholesterolemia involve modulation of new immune players. Biomed. Pharmacother. 2021, 145, 112460. [Google Scholar] [CrossRef] [PubMed]
- Karagiannis, A.D.; Liu, M.; Toth, P.P.; Zhao, S.; Agrawal, D.K.; Libby, P.; Chatzizisis, Y.S. Pleiotropic Anti-atherosclerotic Effects of PCSK9 Inhibitors from Molecular Biology to Clinical Translation. Curr. Atheroscler. Rep. 2018, 20, 20. [Google Scholar] [CrossRef] [PubMed]
- Ben Zadok, O.I.; Mager, A.; Leshem-Lev, D.; Lev, E.; Kornowski, R.; Eisen, A. The Effect of Proprotein Convertase Subtilisin Kexin Type 9 Inhibitors on Circulating Endothelial Progenitor Cells in Patients with Cardiovascular Disease. Cardiovasc. Drugs Ther. 2021, 36, 85–92. [Google Scholar] [CrossRef]
- Hill, J.M.; Zalos, G.; Halcox, J.P.J.; Schenke, W.H.; Waclawiw, M.A.; Quyyumi, A.A.; Finkel, T. Circulating Endothelial Progenitor Cells, Vascular Function, and Cardiovascular Risk. N. Engl. J. Med. 2003, 348, 593–600. [Google Scholar] [CrossRef]
- Barale, C.; Bonomo, K.; Frascaroli, C.; Morotti, A.; Guerrasio, A.; Cavalot, F.; Russo, I. Platelet function and activation markers in primary hypercholesterolemia treated with anti-PCSK9 monoclonal antibody: A 12-month follow-up. Nutr. Metab. Cardiovasc. Dis. 2019, 30, 282–291. [Google Scholar] [CrossRef]
- Pawelczyk, M.; Kaczorowska, B.; Baj, Z. The impact of hyperglycemia and hyperlipidemia on plasma P-selectin and platelet markers after ischemic stroke. Arch. Med. Sci. 2017, 13, 1049–1056. [Google Scholar] [CrossRef] [Green Version]
- Cipollone, F.; Mezzetti, A.; Porreca, E.; Di Febbo, C.; Nutini, M.; Fazia, M.; Falco, A.; Cuccurullo, F.; Davì, G. Association Between Enhanced Soluble CD40L and Prothrombotic State in Hypercholesterolemia: Effects of statin therapy. Circulation 2002, 106, 399–402. [Google Scholar] [CrossRef] [Green Version]
- Camera, M.; Rossetti, L.; Barbieri, S.S.; Zanotti, I.; Canciani, B.; Trabattoni, D.; Ruscica, M.; Tremoli, E.; Ferri, N. PCSK9 as a Positive Modulator of Platelet Activation. J. Am. Coll. Cardiol. 2018, 71, 952–954. [Google Scholar] [CrossRef] [PubMed]
- Hachem, A.; Hariri, E.; Saoud, P.; Lteif, C.; Lteif, L.; Welty, F. The Role of Proprotein Convertase Subtilisin/Kexin Type 9 (PCSK9) in Cardiovascular Homeostasis: A Non-Systematic Literature Review. Curr. Cardiol. Rev. 2017, 13, 274–282. [Google Scholar] [CrossRef] [PubMed]
- Momtazi, A.A.; Sabouri-Rad, S.; Gotto, A.M.; Pirro, M.; Banach, M.; Awan, Z.; Barreto, G.E.; Sahebkar, A. PCSK9 and inflammation: A review of experimental and clinical evidence. Eur. Heart J. Cardiovasc. Pharmacother. 2019, 5, 237–245. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Zhu, C.-G.; Guo, Y.-L.; Xu, R.-X.; Zhang, Y.; Sun, J.; Li, J.-J. The Relationship between the Plasma PCSK9 Levels and Platelet Indices in Patients with Stable Coronary Artery Disease. J. Atheroscler. Thromb. 2015, 22, 76–84. [Google Scholar] [CrossRef] [Green Version]
- Pastori, D.; Nocella, C.; Farcomeni, A.; Bartimoccia, S.; Santulli, M.; Vasaturo, F.; Carnevale, R.; Menichelli, D.; Violi, F.; Pignatelli, P.; et al. Relationship of PCSK9 and Urinary Thromboxane Excretion to Cardiovascular Events in Patients with Atrial Fibrillation. J. Am. Coll. Cardiol. 2017, 70, 1455–1462. [Google Scholar] [CrossRef]
- Puteri, M.U.; Azmi, N.U.; Kato, M.; Saputri, F.C. PCSK9 Promotes Cardiovascular Diseases: Recent Evidence about Its Association with Platelet Activation-Induced Myocardial Infarction. Life 2022, 12, 190. [Google Scholar] [CrossRef]
- Qi, Z.; Hu, L.; Zhang, J.; Yang, W.; Liu, X.; Jia, D.; Yao, Z.; Chang, L.; Pan, G.; Zhong, H.; et al. PCSK9 (Proprotein Convertase Subtilisin/Kexin 9) Enhances Platelet Activation, Thrombosis, and Myocardial Infarct Expansion by Binding to Platelet CD36. Circulation 2021, 143, 45–61. [Google Scholar] [CrossRef]
- Petersen-Uribe, A.; Kremser, M.; Rohlfing, A.-K.; Castor, T.; Kolb, K.; Dicenta, V.; Emschermann, F.; Li, B.; Borst, O.; Rath, D.; et al. Platelet-Derived PCSK9 Is Associated with LDL Metabolism and Modulates Atherothrombotic Mechanisms in Coronary Artery Disease. Int. J. Mol. Sci. 2021, 22, 11179. [Google Scholar] [CrossRef]
- Morofuji, Y.; Nakagawa, S.; Ujifuku, K.; Fujimoto, T.; Otsuka, K.; Niwa, M.; Tsutsumi, K. Beyond Lipid-Lowering: Effects of Statins on Cardiovascular and Cerebrovascular Diseases and Cancer. Pharmaceuticals 2022, 15, 151. [Google Scholar] [CrossRef]
- Pęczek, P.; Leśniewski, M.; Mazurek, T.; Szarpak, L.; Filipiak, K.; Gąsecka, A. Antiplatelet Effects of PCSK9 Inhibitors in Primary Hypercholesterolemia. Life 2021, 11, 466. [Google Scholar] [CrossRef]
- Luzak, B.; Boncler, M.; Rywaniak, J.; Wilk, R.; Stanczyk, L.; Czyz, M.; Rysz, J.; Watala, C. The effect of a platelet cholesterol modulation on the acetylsalicylic acid-mediated blood platelet inhibition in hypercholesterolemic patients. Eur. J. Pharmacol. 2011, 658, 91–97. [Google Scholar] [CrossRef] [PubMed]
- Tirnaksiz, E.; Pamukcu, B.; Oflaz, H.; Nisanci, Y. Effect of high dose statin therapy on platelet function; statins reduce aspirin resistant platelet aggregation in patients with coronary heart disease. J. Thromb. Thrombolysis 2007, 27, 24–28. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Tall, A.R. Cholesterol in platelet biogenesis and activation. Blood 2016, 127, 1949–1953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paciullo, F.; Momi, S.; Gresele, P. PCSK9 in Haemostasis and Thrombosis: Possible Pleiotropic Effects of PCSK9 Inhibitors in Cardiovascular Prevention. Thromb. Haemost. 2019, 119, 359–367. [Google Scholar] [CrossRef] [Green Version]
- Ugovšek, S.; Šebeštjen, M. Lipoprotein(a)—The Crossroads of Atherosclerosis, Atherothrombosis and Inflammation. Biomolecules 2021, 12, 26. [Google Scholar] [CrossRef]
- Boffa, M.; Koschinsky, M.L. Thematic review series: Lipoprotein (a): Coming of age at last: Lipoprotein (a): Truly a direct prothrombotic factor in cardiovascular disease? J. Lipid Res. 2016, 57, 745–757. [Google Scholar] [CrossRef] [Green Version]
- Schwartz, G.G.; Steg, P.G.; Szarek, M.; Bittner, V.A.; Diaz, R.; Goodman, S.G.; Kim, Y.-U.; Jukema, J.W.; Pordy, R.; Roe, M.T.; et al. Peripheral Artery Disease and Venous Thromboembolic Events After Acute Coronary Syndrome: Role of lipoprotein(a) and modification by alirocumab: Prespecified analysis of the ODYSSEY OUTCOMES randomized clinical trial. Circulation 2020, 141, 1608–1617. [Google Scholar] [CrossRef]
- Marston, N.A. The effect of PCSK9 inhibition on the risk of venous thromboembolism. Circulation 2020, 141, 1600–1607. [Google Scholar] [CrossRef]
- von Depka, M.; Nowak-Gottl, U.; Eisert, R.; Dieterich, C.; Barthels, M.; Scharrer, I.; Ganser, A.; Ehrenforth, S. Increased lipo-protein (a) levels as an independent risk factor for venous thromboembolism. Blood 2000, 96, 3364–3368. [Google Scholar] [CrossRef]
- Marcucci, R.; Liotta, A.A.; Cellai, A.P.; Rogolino, A.; Gori, A.M.; Giusti, B.; Poli, D.; Fedi, S.; Abbate, R.; Prisco, D. Increased plasma levels of lipoprotein(a) and the risk of idiopathic and recurrent venous thromboembolism. Am. J. Med. 2003, 115, 601–605. [Google Scholar] [CrossRef]
- Grifoni, E.; Marcucci, R.; Ciuti, G.; Cenci, C.; Poli, D.; Mannini, L.; Liotta, A.A.; Miniati, M.; Abbate, R.; Prisco, D. The Thrombophilic Pattern of Different Clinical Manifestations of Venous Thromboembolism: A Survey of 443 Cases of Venous Thromboembolism. Semin. Thromb. Hemost. 2012, 38, 230–234. [Google Scholar] [CrossRef] [PubMed]
- Kunutsor, S.K.; Mäkikallio, T.H.; Kauhanen, J.; Voutilainen, A.; Laukkanen, J.A. Lipoprotein(a) is not associated with venous thromboembolism risk. Scand. Cardiovasc. J. 2019, 53, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Kamstrup, P.R.; Tybjærg-Hansen, A.; Nordestgaard, B.G. Genetic Evidence That Lipoprotein(a) Associates with Atherosclerotic Stenosis Rather Than Venous Thrombosis. Arter. Thromb. Vasc. Biol. 2012, 32, 1732–1741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsai, A.W.; Cushman, M.; Rosamond, W.D.; Heckbert, S.R.; Polak, J.F.; Folsom, A.R. Cardiovascular Risk Factors and Venous Thromboembolism Incidence: The longitudinal investigation of thromboembolism etiology. Arch. Intern. Med. 2002, 162, 1182–1189. [Google Scholar] [CrossRef] [PubMed]
- Danik, J.S.; Buring, J.E.; Chasman, D.I.; Zee, R.Y.L.; Ridker, P.M.; Glynn, R.J. Lipoprotein(a), polymorphisms in the LPA gene, and incident venous thromboembolism among 21 483 women. J. Thromb. Haemost. 2012, 11, 205–208. [Google Scholar] [CrossRef] [Green Version]
- Mahmoodi, B.K.; Gansevoort, R.T.; Muntinghe, F.L.; Dullaart, R.P.; Kluin-Nelemans, H.C.; Veeger, N.J.; van Schouwenburg, I.M.; Meijer, K. Lipid levels do not influence the risk of venous thromboembolism. Results of a population-based cohort study. Thromb. Haemost. 2012, 108, 923–929. [Google Scholar] [CrossRef] [Green Version]
- Boffa, M.B.; Marar, T.T.; Yeang, C.; Viney, N.J.; Xia, S.; Witztum, J.L.; Koschinsky, M.; Tsimikas, S. Potent reduction of plasma lipoprotein (a) with an antisense oligonucleotide in human subjects does not affect ex vivo fibrinolysis. J. Lipid Res. 2019, 60, 2082–2089. [Google Scholar] [CrossRef]
- Schol-Gelok, S.; Galema-Boers, J.M.; van Gelder, T.; Kruip, M.J.; van Lennep, J.E.R.; Versmissen, J. No effect of PCSK9 inhibitors on D-dimer and fibrinogen levels in patients with familial hypercholesterolemia. Biomed. Pharmacother. 2018, 108, 1412–1414. [Google Scholar] [CrossRef]
- Scalise, V.; Sanguinetti, C.; Neri, T.; Cianchetti, S.; Lai, M.; Carnicelli, V.; Celi, A.; Pedrinelli, R. PCSK9 Induces Tissue Factor Expression by Activation of TLR4/NFkB Signaling. Int. J. Mol. Sci. 2021, 22, 12640. [Google Scholar] [CrossRef]
- Ragusa, R.; Basta, G.; Neglia, D.; De Caterina, R.; Del Turco, S.; Caselli, C. PCSK9 and atherosclerosis: Looking beyond LDL regulation. Eur. J. Clin. Investig. 2020, 51, e13459. [Google Scholar] [CrossRef]
- The Official ORION-4 Study Website. Available online: https://www.orion4trial.org/homepage-uk (accessed on 10 May 2022).


{kind=link}
{kind=link}
{kind=link}
| Study | Study Population | Treatment | Primary Endpoint | Outcome | |
|---|---|---|---|---|---|
| (n) | Characteristics | ||||
| Cao et al. (2018) [34] | 4198 | FH or non-FH | A or E or LY3015014 or RG7652 | Change in hsCRP | No beneficial changes in hsCRP |
| Stiekema et al. (2019) [35] | 129 | Elevated Lp(a) | E vs. placebo | Change in arterial wall inflammation | No beneficial changes in arterial wall inflammation, assessed as MDS TBR of the index vessel |
| Hoogeveen et al. (2019) [36] | 50 | Atherosclerotic disease or FH | A vs. placebo | Change in arterial wall inflammation | Reduced arterial wall inflammation assessed as MDS TBR of the index carotid (−6.1%) |
| Stiekema et al. (2020) [37] | 18 | Elevated Lp(a) | E | Change in gene expression and function of monocytes | No beneficial changes in pro-inflammatory state of monocytes |
| 14 | CVD and elevated Lp(a) | AKCEA-APO(a)-LRX | Reduced pro-inflammatory state of monocytes (−17%) | ||
| GLAGOV [38] | 968 | Angiographic coronary disease | E vs. placebo | Change in percent atheroma volume | Reduced percent atheroma volume (−0.95%), total atheroma volume |
| HUYGENS [40] | 161 | Non-ST-segment elevation myocardial infarction | E vs. placebo | Changes in plaque composition | Increased fibrous cap thickness, decreased atheroma volume, lipid arc and macrophage index |
| Maulucci et al. (2018) [44] | 14 | Myocardial infarction | E | Changes in endothelial function | Increased FMD (+40%), brachial artery diameter and velocity time integral |
| Di Minno et al. (2020) [46] | 25 | FH | E | Changes in endothelial function, lipid profile and oxidation markers | Increased FMD and RHI, reduced 11-dehydro-thromboxane (−18%) and 8-iso-prostaglandin F2α (−17%) |
| ALIROCKS [47] | 24 | Indication for treatment with PCSK9 antibodies | A | Changes in endothelial function | No beneficial changes in FMD carotid intima-media thickness, fractional anisotropy of carotid artery, P-selectin and VEGF |
| Leucker et al. (2020) [49] | 19 | Patients with HIV | E | Changes in coronary endothelial function at rest and during isometric handgrip exercise | Increased coronary CSA and CBF, no beneficial changes in CRP, IL-6, INFγ, TNFα and CD163 |
| 11 | Dyslipidemia | ||||
| Marques et al. (2022) [54] | 14 | FH | A | Changes in inflammatory state, endothelial function and cardiovascular outcomes | Reduced activation of platelets and leukocytes, increased IL-10, reduced INFγ and soluble PCSK9 |
| Itzhaki et al. (2020) [56] | 26 | CVD | A or E | Change in cEPC | Increased CD34+/CD133+ (+0.98%), VEGF receptor-2+ (0.66%) and PCSK9 |
| Barale et al. (2019)[58] | 24 | Hyper- cholesterolemia | A or E | Change in platelet function | Reduced platelet aggregation and expression of CD62P, soluble CD40 ligand, platelet factor-4 and soluble P-selectin |
| Schwartz et al. (2020) [77] | 18,924 | ACS | A vs. placebo | Peripheral artery disease event or venous thromboembolism | Reduced risk of peripheral artery disease (hazard ratio 0.69), no beneficial changes in reducing the risk of venous thromboembolism |
| Marston (2020) [78] | 27,564 | Stable atherosclerosis, hyperlipidemia | E | Venous thromboembolism | Reduced risk of venous thromboembolism (hazard ratio 0.54) |
| Schol-Gelok et al. (2018) [88] | 30 | Statin-intolerant patients with FH | A or E | Change in D-dimer and fibrinogen | No beneficial changes in D-dimer and fibrinogen |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ugovšek, S.; Šebeštjen, M. Non-Lipid Effects of PCSK9 Monoclonal Antibodies on Vessel Wall. J. Clin. Med. 2022, 11, 3625. https://doi.org/10.3390/jcm11133625
Ugovšek S, Šebeštjen M. Non-Lipid Effects of PCSK9 Monoclonal Antibodies on Vessel Wall. Journal of Clinical Medicine. 2022; 11(13):3625. https://doi.org/10.3390/jcm11133625
Chicago/Turabian StyleUgovšek, Sabina, and Miran Šebeštjen. 2022. "Non-Lipid Effects of PCSK9 Monoclonal Antibodies on Vessel Wall" Journal of Clinical Medicine 11, no. 13: 3625. https://doi.org/10.3390/jcm11133625