Abstract
The wide application of next-generation sequencing (NGS) technologies has led to the discovery of multiple genetic alterations in pediatric acute lymphoblastic leukemia (ALL). In this work, we aimed to investigate the mutational spectrum in pediatric ALL. We employed a St. Mary’s customized NGS panel comprising 67 leukemia-related genes. Samples were collected from 139 pediatric ALL patients. Eighty-five patients (61.2%) harbored at least one mutation. In B-cell ALL, the RAS pathway is the most involved pathway, and the three most frequently mutated genes were NRAS (22.4%), KRAS (19.6%), and PTPN11 (8.4%). NRAS and PTPN11 were significantly associated with a high hyperdiploidy karyotype (p = 0.018 and p < 0.001, respectively). In T-cell ALL, the three most frequently mutated genes were NOTCH1 (37.5%), FBXW7 (16.6%), and PTEN (6.2%). Several pairs of co-occurring mutations were found: NRAS with SETD, NRAS with PTPN11 in B-cell ALL (p = 0.024 and p = 0.020, respectively), and NOTCH1 with FBXW7 in T-cell ALL (p < 0.001). The most frequent newly emerged mutation in relapsed ALL was NT5C2. We procured comprehensive genetic information regarding Korean pediatric ALL using NGS technology. Our findings strengthen the current knowledge of recurrent somatic mutations in pediatric ALL.
1. Introduction
The cure rate of childhood acute lymphoblastic leukemia (ALL) has significantly improved owing to contemporary risk-directed treatment, demonstrating a 5-year overall survival (OS) of over 90% in many clinical trials [1,2,3,4]. However, current therapies for ALL are associated with substantial toxic effects, and up to 20% of patients still show relapse after the initial therapy [5]. To further improve outcomes in the upcoming decade, trials need to focus not only on the molecular and immune therapies that might replace toxic chemotherapy but also on the biologic basis of the drug resistance that leads to relapse. In this regard, enhanced knowledge about the genetic background of childhood ALL is gaining importance.
The classification of B-cell precursor (BCP)-ALL and T-cell ALL (T-ALL) has been refined based on gene expression-based subgroups, and recent studies have further identified several novel rearrangements including DUX4-rearranged [6], iAMP21 and MEF2D-rearranged [7,8], ZNF384-rearranged [9], and ETV6-RUNX1-like B-ALL [8]. In addition, the wide application of next-generation sequencing (NGS) technologies has revealed more abnormal molecules and improved the understanding of ALL biology; the pathway most commonly altered in ALL is the transcriptional regulation of lymphoid development (PAX5, IKZF1, and EBF1) [10], and additional mutated pathways include tumor suppression and cell-cycle regulation (TP53, RB1, PTEN, and CDKN2A), RAS signaling (NRAS, KRAS, and PTPN11) [11], and epigenetic modification (CREBBP, EP300, SETD2, and NSD2) [12]. These somatic mutations were found to vary according to the subtype of ALL [13,14] and racial/ethnic disparity [15]. Recently, drug resistance-related mutations, such as those in NT5C2, PRPS1, NR3C1, CREBBP, and TP53, which facilitated relapse of pediatric ALL, were reported [16].
There are few reports analyzing the comprehensive and detailed mutation data of Korean pediatric patients with ALL using NGS. In this study, we aimed to investigate the mutational spectrum and frequency of pediatric ALL using a panel-based NGS with 67 leukemia-related genes at a single Korean institution and to reveal its clinical impact on disease relapse and survival outcomes.
2. Materials and Methods
2.1. Study Cohort
A total of 139 pediatric patients (≤18 years) who were diagnosed with either BCP-ALL (n = 123) or T-ALL (n = 16) and treated at our institution were enrolled in this study. The patients’ samples and clinical information between April 2018 and October 2020 were collected. We excluded cases of Philadelphia chromosome-positive ALL and Down syndrome and included cases of infant ALL (n = 6) in the study cohort. A diagnosis of ALL was confirmed via bone marrow (BM) pathology, immunophenotyping, cytogenetics, and molecular genetics. The presence of recurrent genetic abnormalities, including ETV6-RUNX1, TCF3-PBX1, KMT2A-rearrangement, high hyperdiploidy, and hypodiploidy, was diagnosed using reverse transcription polymerase chain reaction (RT-PCR), fluorescence in situ hybridization (FISH), and Giemsa band karyotyping. The cytogenetic and molecular genetic abnormalities were analyzed using BM samples at the time of diagnosis. All patients were risk-classified and treated according to an institutional protocol, which has been reported previously [17]. Clinical characteristics including age at diagnosis and initial white blood count (WBC), National Cancer Institute/Rome (NCI) risk group [18], as well as institutional risk group [17], were reported. The study received institutional review board approval from Seoul St. Mary’s Hospital, The Catholic University of Korea (IRB No. KC21RISI0481).
2.2. Conventional Cytogenetics and Fluorescence In Situ Hybridization
Chromosomal analyses were performed by examining short-term cultures of BM specimens according to standard conventional cytogenetic protocols. At least 20 cells in metaphase were analyzed in each case. Clonal abnormalities were classified according to the 2020 International System for Human Cytogenetic Nomenclature guidelines [19].
FISH analyses were performed to identify gene fusions, using a TEL/AML1 (ETV6/RUNX1) translocation dual-fusion probe (Cytocell, Banbury, UK), an E2A (TCF3) break-apart probe (Cytocell, Banbury, UK), and an MLL (KMT2A) break-apart probe (Cytocell, Banbury, UK). These FISH analyses used pellets of cells remaining after conventional cytogenetic studies. Slides for FISH were prepared by using cells harvested for conventional cytogenetics and by processing them for FISH according to the manufacturer’s guidelines (Abbott Vysis, Des Plaines, IL, USA). Analyses were performed on cells in either interphase or metaphase.
2.3. Detection of Mutations Using Next-Generation Sequencing
NGS was performed using a St. Mary’s customized NGS panel for acute leukemia, i.e., the “SM Acute leukemia panel”. Ion AmpliSeq Technology was used to amplify 67 genes (Supplementary Table S1) using an Ion Chef™ system and an Ion S5 XL Sequencer (all from Thermo Fisher Scientific, Waltham, MA, USA) [20]. Sequenced reads were mapped to the human reference genome (hg19, Genome Reference Consortium, February 2009). Annotated variants were classified into four tiers according to the Standards and Guidelines of the Association for Molecular Pathology [21]. Bioinformatics analysis was carried out using both customized and manufacturer-provided pipelines. Variants were selected and annotated using analytics algorithms and public databases [22]. All mutations were manually verified using the Integrative Genomic Viewer [23]. Among the 139 patients, 106 patients and 19 patients were analyzed with samples at diagnosis and at relapse, respectively, while 14 patients were analyzed with both samples.
2.4. Multiplex Ligation-Dependent Probe Amplification
The targeted copy number of Ikaros family zinc finger 1 (IKZF1) was analyzed using a SALSA MLPA Probemix P335-C1 ALL-IKZF1 kit (MRC-Holland, Amsterdam, The Netherlands) according to the manufacturer’s instructions. The MLPA results were analyzed using GeneMarker software (Softgenetics, State College, PA, USA). Peak heights were normalized, and deletions or duplications were defined as recommended by the manufacturer. Direct sequencing of the probe-binding and ligation sites was performed for the relevant exons to detect nearby variants, which can lead to a false decrease in peak signal [24].
2.5. Statistical Analysis
A complex karyotype is defined as more than or equal to three chromosomal aberrations, including at least one structural aberration. Clinical characteristics, including median age and WBC count at diagnosis, and NCI and overall risk groups between non-relapsed and relapsed cases of ALL were compared using the Mann–Whitney test and Pearson’s chi-square test. The spectrum and number of genetic mutations for BCP-ALL and T-ALL were compared using Fisher’s exact test and the Kruskal–Wallis test. Event-free survival (EFS) and OS were determined via the Kaplan–Meier method, with comparisons of survival rate performed using a log-lank test. The date of the last follow-up was 28 February 2021. p < 0.05 was considered statistically significant. SPSS version 24.0 (SPSS, Chicago, IL, USA) was used for all statistical analyses.
3. Results
3.1. Genetic Characteristics of the Study Cohort
Among 139 pediatric patients with ALL, seventy-eight (56.1%) patients had recurrent genetic abnormalities (ETV6-RUNX1 in 34, high hyperdiploidy in 29, TCF3-PBX1 in 7, KMT2A-rearrangement in 7, and hypodiploidy in 1), and nine (6.5%) patients had complex karyotypes. When comparing the two phenotypes, recurrent genetic abnormalities were more frequently found in BCP-ALL (p < 0.001), whereas a complex karyotype was more dominant in T-ALL (p = 0.001). Eighty-five patients (61.2%) harbored at least one genetic mutation that seemed either pathogenic (P) or likely pathogenic (LP), and the median number of these mutations was 1 (range, 1–10). The number of mutations in patients with BCP-ALL was significantly lower than that in patients with T-ALL (0.86 ± 1.081 vs. 2.75 ± 2.206) (p < 0.001). The main pathways detected via the NGS panel of this study were RAS and NOTCH signaling pathways and transcriptional regulation. The five most frequently mutated genes in the total cohort were NRAS, KRAS, NOTCH1, FBXW7, and PTPN11, in that order (Figure 1). We also analyzed the co-occurrence of mutations and complex karyotypes. Several pairs of co-occurring mutations were found, such as NRAS with SETD, NRAS with PTPN11, and NOTCH1 with FBXW7 (p = 0.024, p = 0.020, and p < 0.001, respectively) (Figure 2) [25].
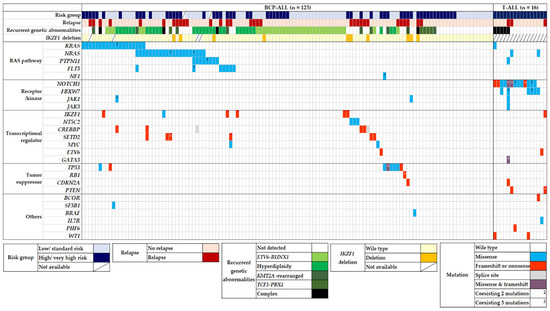
Figure 1.
Mutational landscape of 139 pediatric ALL patients. Heatmap diagram showing genomic data of recurrent translocations, IKZF1 deletion, and somatic mutations in pediatric ALL. Abbreviation: BCP-ALL, B-cell precursor acute lymphoblastic leukemia.
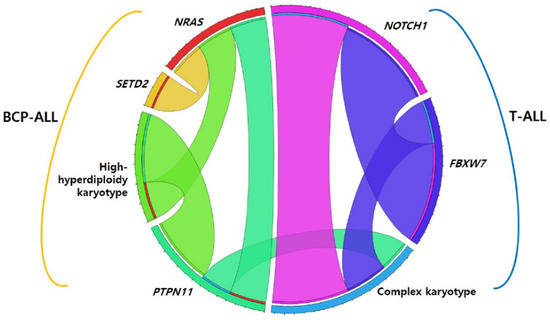
Figure 2.
Co-occurring mutations in pediatric acute lymphoblastic leukemia. Abbreviation: BCP-ALL, B-cell precursor acute lymphoblastic leukemia.
3.2. Mutational Spectrum of Pediatric ALL According to Disease Category
3.2.1. BCP-ALL: Mutations in the RAS Pathway Were More Abundant
In BCP-ALL (n = 123), 68 patients (55.3%) harbored mutations, and the median number of mutations was 1 (range, 0–3). The five most frequently mutated genes in BCP-ALL were NRAS (22.4%), KRAS (19.6%), PTPN11 (8.4%), TP53 (8.4%), and FLT3 (7.4%), in that order (Figure 3A). The number of somatic mutations in patient groups with ETV6-RUNX1 (0.38 ± 0.70) was significantly lower than that in patient groups with no recurrent genetic abnormality (1.11 ± 1.18), high hyperdiploidy (1.17 ± 0.90), and KMT2A rearrangement (1.17 ± 0.98) (p < 0.001, p < 0.001, and p = 0.037, respectively). A total of 47 (38.2%) out of 123 patients showed mutations in the RAS pathway (NRAS, KRAS, PTPN11, FLT3, and NF1 in order), which is the most commonly involved pathway in BCP-ALL. All RAS pathway mutations found in this study were missense mutations (Figure 1). RAS pathway mutation had a particularly high frequency in patients with high hyperdiploidy (n = 34); 19 patients (67.8%) with high hyperdiploidy carried the RAS pathway mutation. NRAS and PTPN11 were significantly associated with a high hyperdiploidy karyotype (p = 0.018 and p < 0.001, respectively). IKZF1 deletions were detected in 10 patients (8.1%) with BCP-ALL, and three and seven patients showed whole and partial gene deletions, respectively. The two main detected focal deletions were exons 4–7 and exons 4–8.
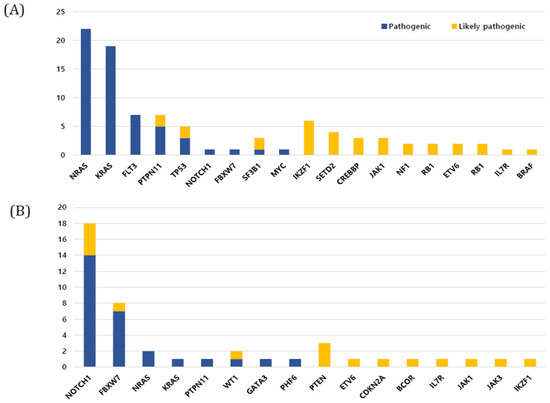
Figure 3.
Frequency of mutations in study cohort. (A) B-cell precursor acute lymphoblastic leukemia (ALL) (B) T-cell ALL.
3.2.2. T-ALL: Mutation in the Notch Pathway Were More Common
All patients with T-ALL (n = 16) harbored at least one mutation and the median number of mutations was 3 (range, 1–10), which was significantly higher than that in BCP-ALL (p < 0.01). The three most frequently mutated genes in T-ALL were NOTCH1 (37.5%), FBXW7 (16.6%), and PTEN (6.2%), in that order. In particular, NOTCH1, FBXW7, PTEN, and WT1 mutations were found more frequently in T-ALL than in BCP-ALL (p < 0.001, p < 0.001, p = 0.013, and p = 0.013, respectively) (Figure 3B). Among T-ALL patients, a complex karyotype was significantly associated with NOTCH1, PTPN11, and FBXW7 mutations (p < 0.001, p = 0.048, and p = 0.006, respectively) (Figure 2). Five (31.3%) patients harbored both NOTCH1 and FBXW7 mutations, a significant co-occurring mutation in T-ALL.
3.2.3. Relapsed ALL: IKZF1 Deletion and NT5C2 Mutation Seemed to Associate with Disease Relapse
The changes in mutations between the time of diagnosis and the time of relapse are shown in Figure 4. A higher number of mutations was observed in patients who experienced relapse (n = 34) than in patients who did not experience relapse (p < 0.01). We compared the mutation changes in 14 patients for whom both diagnostic and relapse samples were available: eight patients showed the same mutations, five patients had newly emerged mutations, and one patient lost the initial mutation. The most frequent newly emerged mutation was NT5C2 (42%), which was found in three of three cases of relapse. In addition to the NT5C2 gene, ETV6, SETD2, and IL7R were newly observed genes in relapsed samples. Two of two patients with PTEN and CDKN2A, four out of eight patients with TP53, and three out of six patients with CREBBP relapsed, respectively. There was no difference in clinical outcomes according to presence of TP53 mutation in our cohort. The proportion of patients who showed IKZF1 deletion was higher in the relapsed ALL group (17.6%) than in the non-relapsed ALL group (3.8%) (p < 0.001) (Table 1).
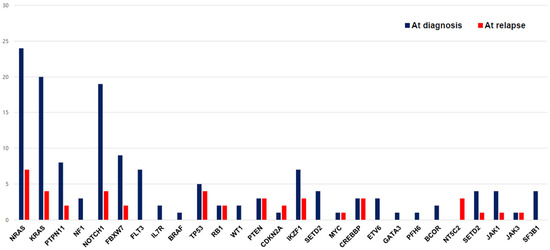
Figure 4.
The change in mutational distribution of the study cohort at the time of diagnosis and relapse.

Table 1.
Comparison of clinical characteristics between non-relapsed and relapsed ALL.
3.2.4. Comparison of Clinical Characteristics Based on Relapse
The median age at diagnosis was 5.0 years (range, 0.1–18.0) in the study cohort. In T-ALL cases, the median age at diagnosis and the median value of initial WBC counts were significantly higher than those in BCP-ALL cases (p < 0.01). When comparing the clinical characteristics between non-relapsed and relapsed cases of ALL, we found that the median value of initial WBC count was significantly higher in relapsed ALL (25.5 × 109/L, range 0.9–630) than in non-relapsed ALL (10.5 × 109/L, range 0.6–339) (p < 0.001). In addition, more patients in the NCI high-risk (HR) group experienced relapse than those in the NCI standard-risk (SR) group (NCI HR 67.7% vs. NCI SR 40.9%, p = 0.016).
The 3-year EFS and OS of the total cohort were 65.8 ± 8.5% and 87.1 ± 5.3%, respectively. There were no significant differences in survival outcomes between BCP-ALL and T-ALL (EFS: BCP-ALL 66.5% ± 8.6% vs. T-ALL 80.8 ± 10.0%, p = 0.881; OS: BCP-ALL 89.0 ± 5.3% vs. T-ALL 79.1% ± 10.8%, p = 0.160). When compared with patients in the NCI SR group, patients in the NCI HR group showed an inferior EFS (NCI HR 62.2 ± 8.8% vs. NCI SR 66.8 ± 14.7%, p = 0.011) and comparable OS (NCI HR 87.0 ± 5.2% vs. NCI SR 85.0 ± 10.7%, p = 0.082). The 3-year OS of patients who experienced relapse was 76.5 ± 7.8%, while no patient died of disease among those who did not experience relapse. No significant differences were found in the EFS and OS according to the response in the corticosteroid window phase, institution risk group, presence of RAS pathway mutation, IKZF1 deletion, and presence of recurrent genetic abnormalities.
4. Discussion
To assess the mutational spectrum in Korean pediatric ALL, we performed a panel based NGS analysis that involved various signaling pathways and showed comprehensive genetic profiles among these patients. This study showed a distinctive mutational spectrum and frequency between BCP-ALL and T-ALL; in BCP-ALL, the dominant mutations were enriched in the RAS pathway, while in T-ALL, the dominant ones were those in the NOTCH pathway. Though no significant mutations predicting disease prognosis were found in our cohort, IKZF1 deletion and NT5C2 mutation seemed to be associated with disease relapse.
As previous studies reported, RAS pathway mutations were well-known recurrent somatic variants in pediatric BCP-ALL, and the vast majority of mutations occurred in KRAS, NRAS, PTPN11, FLT3, and NF1 [26,27]. We reported similar results that the most common somatic mutations were in the RAS signaling pathway, and all these mutations were missense mutations. RAS pathway mutations have been associated with disease relapse [28] and chemotherapy resistance [29] in pediatric BCP-ALL. However, the prognostic significance of clonal and subclonal mutations remains unknown. Although RAS pathway mutations were not correlated with relapse in our cohort, all RAS pathway mutations observed at the time of relapse were clonal mutations, except in one patient. This finding deserves further analysis with a larger cohort in future studies.
Although ALL with high hyperdiploidy (51–67 chromosomes) generally represents a favorable prognostic subtype, less than 20% of these patients experienced disease relapse [30]. Studies have reported a high incidence of RAS pathway mutations in patients with high hyperdiploidy and have linked it to disease recurrence [27,31]. We also observed that RAS pathway mutations occurred in more than half of ALL patients with high hyperdiploidy (67.8%) with a significant association, particularly between a high hyperdiploidy karyotype and NRAS-PTPN11 mutation. In this study, ALL patients with high hyperdiploidy harbored more somatic mutations than patients with other recurrent translocations, such as ETV6-RUNX1 and TCF3-PBX1. Among them, ETV6-RUNX1-positive ALL patients harbored the least number of somatic variants.
A deletion of IKZF1 is known to be associated with adverse outcomes and has been described as a high-risk marker in pediatric BCP-ALL [32]. Most of the published data on IKZF1 deletions in pediatric ALL have been generated via MLPA analysis and have revealed its reliability to detect all deletions targeting the coding sequence [33,34]. We reported a IKZF1 deletion frequency of 8.1%, which is slightly lower than previous findings of an overall frequency around 15% [35]. The presence of IKZF1 deletions was linked to features indicating poor prognosis, including an older age at diagnosis, higher initial WBC count, and higher levels of minimal residual disease after induction chemotherapy [34,35]. As expected, 6 out of 10 patients with IKZF1 deletion were risk-stratified into a ‘very high risk’ group because of either high WBC count or older age at presentation, and significantly more patients with IKZF1 deletion experienced disease relapse compared to those with wild-type IKZF1 in this study. These findings reiterate the urgency of introducing new specific therapeutic approaches for this patient group.
Recently, several studies on pediatric ALL reported that mutations in NT5C2 caused disease relapse, especially during maintenance chemotherapy, by conferring purine analog resistance [36,37,38]. In this study, NT5C2 mutations were found to have newly emerged in three patients with relapsed ALL. Among them, two patients with subclonal mutation died after relapse while one patient with clonal mutation remained alive throughout the study period. Barz et al. reported that subclonal NT5C2 mutations independently predict an inferior outcome of disease relapse [37]. In addition to alterations in NT5C2, relapse-specific somatic alterations, such as those in TP53 and CREBBP, were also more frequently found in patients who experienced relapse in our cohort.
Nevertheless, this study has some limitations. Primarily, the mutational analysis was performed in a disease-only setting. We could not obtain germline samples, such as skin fibroblasts, and did not perform a familial study to search for germline mutations. In addition, samples from the time of diagnosis were not available for all patients who experienced relapse; therefore, the mutational difference between the time of diagnosis and relapse could not be fully elucidated. Third, we used a custom panel for NGS that only including 67 leukemia-related genes. The use of a wider panel (e.g., commonly used 409-gene panel) in future study could provide more comprehensive results. Finally, although our study employed one of the largest cohorts of Korean pediatric patients, a relatively smaller number of cases were enrolled than in multicenter studies.
5. Conclusions
We found a difference in recurrent somatic mutations between BCP-ALL and T-ALL in Korean pediatric patients. Our findings strengthen the current knowledge of genetic profiles in pediatric ALL. Furthermore, we demonstrate the usefulness of panel-based NGS techniques in providing comprehensive genetic information. We expect that these data will serve as a foundation to develop a mutational roadmap for Korean pediatric patients for nation-wide studies in the future.
Supplementary Materials
The following supporting information can be downloaded at https://www.mdpi.com/article/10.3390/jcm11216298/s1, Table S1: Gene targets of SM acute leukemia panel.
Author Contributions
Conceptualization and design: J.W.Y., M.K. and N.-G.C. Patient data and samples: J.-M.L., S.J., S.K., J.W.L. and B.C. Experiments, collection, and assembly of data: A.A., Y.K. and M.K. Data analysis and interpretation: A.A., J.W.Y., M.K. and N.-G.C. Manuscript writing and editing: J.W.Y., A.A., M.K. and N.-G.C. All authors have read and agreed to the published version of the manuscript.
Funding
This research was supported by a grant (ZC21SISI0440) from the Catholic University of Korea in 2021.
Institutional Review Board Statement
The study received institutional review board approval from Seoul St. Mary’s Hospital, The Catholic University of Korea (IRB No. KC21RISI0481).
Informed Consent Statement
Informed consent was obtained from all subjects involved in the study.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Vrooman, L.M.; Blonquist, T.M.; Harris, M.H.; Stevenson, K.E.; Place, A.E.; Hunt, S.K.; O’Brien, J.E.; Asselin, B.L.; Athale, U.H.; Clavell, L.A.; et al. Refining risk classification in childhood B acute lymphoblastic leukemia: Results of DFCI ALL Consortium Protocol 05-001. Blood Adv. 2018, 2, 1449–1458. [Google Scholar] [CrossRef] [PubMed]
- Toft, N.; Birgens, H.; Abrahamsson, J.; Griskevicius, L.; Hallbook, H.; Heyman, M.; Klausen, T.W.; Jonsson, O.G.; Palk, K.; Pruunsild, K.; et al. Results of NOPHO ALL2008 treatment for patients aged 1–45 years with acute lymphoblastic leukemia. Leukemia 2018, 32, 606–615. [Google Scholar] [CrossRef]
- Jeha, S.; Pei, D.; Choi, J.; Cheng, C.; Sandlund, J.T.; Coustan-Smith, E.; Campana, D.; Inaba, H.; Rubnitz, J.E.; Ribeiro, R.C.; et al. Improved CNS Control of Childhood Acute Lymphoblastic Leukemia Without Cranial Irradiation: St Jude Total Therapy Study 16. J. Clin. Oncol. 2019, 37, 3377–3391. [Google Scholar] [CrossRef] [PubMed]
- Angiolillo, A.L.; Schore, R.J.; Kairalla, J.A.; Devidas, M.; Rabin, K.R.; Zweidler-McKay, P.; Borowitz, M.J.; Wood, B.; Carroll, A.J.; Heerema, N.A.; et al. Excellent Outcomes With Reduced Frequency of Vincristine and Dexamethasone Pulses in Standard-Risk B-Lymphoblastic Leukemia: Results From Children’s Oncology Group AALL0932. J. Clin. Oncol. 2021, 39, 1437–1447. [Google Scholar] [CrossRef]
- Pui, C.H.; Robison, L.L.; Look, A.T. Acute lymphoblastic leukaemia. Lancet 2008, 371, 1030–1043. [Google Scholar] [CrossRef]
- Yasuda, T.; Tsuzuki, S.; Kawazu, M.; Hayakawa, F.; Kojima, S.; Ueno, T.; Imoto, N.; Kohsaka, S.; Kunita, A.; Doi, K.; et al. Recurrent DUX4 fusions in B cell acute lymphoblastic leukemia of adolescents and young adults. Nat. Genet. 2016, 48, 569–574. [Google Scholar] [CrossRef] [PubMed]
- Gu, Z.; Churchman, M.; Roberts, K.; Li, Y.; Liu, Y.; Harvey, R.C.; McCastlain, K.; Reshmi, S.C.; Payne-Turner, D.; Iacobucci, I.; et al. Genomic analyses identify recurrent MEF2D fusions in acute lymphoblastic leukaemia. Nat. Commun. 2016, 7, 13331. [Google Scholar] [CrossRef] [PubMed]
- Zaliova, M.; Kotrova, M.; Bresolin, S.; Stuchly, J.; Stary, J.; Hrusak, O.; Te Kronnie, G.; Trka, J.; Zuna, J.; Vaskova, M. ETV6/RUNX1-like acute lymphoblastic leukemia: A novel B-cell precursor leukemia subtype associated with the CD27/CD44 immunophenotype. Genes Chromosomes Cancer 2017, 56, 608–616. [Google Scholar] [CrossRef]
- Hirabayashi, S.; Ohki, K.; Nakabayashi, K.; Ichikawa, H.; Momozawa, Y.; Okamura, K.; Yaguchi, A.; Terada, K.; Saito, Y.; Yoshimi, A.; et al. ZNF384-related fusion genes define a subgroup of childhood B-cell precursor acute lymphoblastic leukemia with a characteristic immunotype. Haematologica 2017, 102, 118–129. [Google Scholar] [CrossRef]
- Roberts, K.G.; Mullighan, C.G. Genomics in acute lymphoblastic leukaemia: Insights and treatment implications. Nat. Rev. Clin. Oncol. 2015, 12, 344–357. [Google Scholar] [CrossRef]
- Lee, J.W.; Kim, Y.; Cho, B.; Kim, S.; Jang, P.S.; Lee, J.; Cho, H.; Lee, G.D.; Chung, N.G.; Kim, M. High incidence of RAS pathway mutations among sentinel genetic lesions of Korean pediatric BCR-ABL1-like acute lymphoblastic leukemia. Cancer Med. 2020, 9, 4632–4639. [Google Scholar] [CrossRef] [PubMed]
- Pui, C.H.; Nichols, K.E.; Yang, J.J. Somatic and germline genomics in paediatric acute lymphoblastic leukaemia. Nat. Rev. Clin. Oncol. 2019, 16, 227–240. [Google Scholar] [CrossRef] [PubMed]
- Zaliova, M.; Stuchly, J.; Winkowska, L.; Musilova, A.; Fiser, K.; Slamova, M.; Starkova, J.; Vaskova, M.; Hrusak, O.; Sramkova, L.; et al. Genomic landscape of pediatric B-other acute lymphoblastic leukemia in a consecutive European cohort. Haematologica 2019, 104, 1396–1406. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Easton, J.; Shao, Y.; Maciaszek, J.; Wang, Z.; Wilkinson, M.R.; McCastlain, K.; Edmonson, M.; Pounds, S.B.; Shi, L.; et al. The genomic landscape of pediatric and young adult T-lineage acute lymphoblastic leukemia. Nat. Genet. 2017, 49, 1211–1218. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.Y.; Bhatia, S.; Robison, L.L.; Yang, J.J. Genomics of racial and ethnic disparities in childhood acute lymphoblastic leukemia. Cancer 2014, 120, 955–962. [Google Scholar] [CrossRef]
- Li, B.; Brady, S.W.; Ma, X.; Shen, S.; Zhang, Y.; Li, Y.; Szlachta, K.; Dong, L.; Liu, Y.; Yang, F.; et al. Therapy-induced mutations drive the genomic landscape of relapsed acute lymphoblastic leukemia. Blood 2020, 135, 41–55. [Google Scholar] [CrossRef]
- Lee, J.W.; Kim, S.K.; Jang, P.S.; Jeong, D.C.; Chung, N.G.; Cho, B.; Kim, H.K. Treatment of children with acute lymphoblastic leukemia with risk group based intensification and omission of cranial irradiation: A Korean study of 295 patients. Pediatr. Blood Cancer 2016, 63, 1966–1973. [Google Scholar] [CrossRef]
- Smith, M.; Bleyer, A.; Crist, W.; Murphy, S.; Sallan, S.E. Uniform criteria for childhood acute lymphoblastic leukemia risk classification. J. Clin. Oncol 1996, 14, 680–681. [Google Scholar] [CrossRef]
- McGowan-Jordan, J.; Hastings, R.J.; Moore, S. ISCN2020: An International System for Human Cytogenomic Nomenclature (2020); Karger: Basel, Switzerland, 2020. [Google Scholar]
- Kim, H.J.; Kim, Y.; Kang, D.; Kim, H.S.; Lee, J.M.; Kim, M.; Cho, B.S. Prognostic value of measurable residual disease monitoring by next-generation sequencing before and after allogeneic hematopoietic cell transplantation in acute myeloid leukemia. Blood Cancer J. 2021, 11, 109. [Google Scholar] [CrossRef]
- Li, M.M.; Datto, M.; Duncavage, E.J.; Kulkarni, S.; Lindeman, N.I.; Roy, S.; Tsimberidou, A.M.; Vnencak-Jones, C.L.; Wolff, D.J.; Younes, A.; et al. Standards and Guidelines for the Interpretation and Reporting of Sequence Variants in Cancer: A Joint Consensus Recommendation of the Association for Molecular Pathology, American Society of Clinical Oncology, and College of American Pathologists. J. Mol. Diagn. 2017, 19, 4–23. [Google Scholar] [CrossRef]
- Lee, J.; Cho, S.; Hong, S.E.; Kang, D.; Choi, H.; Lee, J.M.; Yoon, J.H.; Cho, B.S.; Lee, S.; Kim, H.J.; et al. Integrative Analysis of Gene Expression Data by RNA Sequencing for Differential Diagnosis of Acute Leukemia: Potential Application of Machine Learning. Front. Oncol. 2021, 11, 717616. [Google Scholar] [CrossRef] [PubMed]
- Robinson, J.T.; Thorvaldsdottir, H.; Wenger, A.M.; Zehir, A.; Mesirov, J.P. Variant Review with the Integrative Genomics Viewer. Cancer Res. 2017, 77, e31–e34. [Google Scholar] [CrossRef] [PubMed]
- De Luca, A.; Bottillo, I.; Dasdia, M.C.; Morella, A.; Lanari, V.; Bernardini, L.; Divona, L.; Giustini, S.; Sinibaldi, L.; Novelli, A.; et al. Deletions of NF1 gene and exons detected by multiplex ligation-dependent probe amplification. J. Med. Genet. 2007, 44, 800–808. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Krzywinski, M.; Schein, J.; Birol, I.; Connors, J.; Gascoyne, R.; Horsman, D.; Jones, S.J.; Marra, M.A. Circos: An information aesthetic for comparative genomics. Genome Res. 2009, 19, 1639–1645. [Google Scholar] [CrossRef] [PubMed]
- Case, M.; Matheson, E.; Minto, L.; Hassan, R.; Harrison, C.J.; Bown, N.; Bailey, S.; Vormoor, J.; Hall, A.G.; Irving, J.A. Mutation of genes affecting the RAS pathway is common in childhood acute lymphoblastic leukemia. Cancer Res. 2008, 68, 6803–6809. [Google Scholar] [CrossRef] [PubMed]
- Jerchel, I.S.; Hoogkamer, A.Q.; Aries, I.M.; Steeghs, E.M.P.; Boer, J.M.; Besselink, N.J.M.; Boeree, A.; van de Ven, C.; de Groot-Kruseman, H.A.; de Haas, V.; et al. RAS pathway mutations as a predictive biomarker for treatment adaptation in pediatric B-cell precursor acute lymphoblastic leukemia. Leukemia 2018, 32, 931–940. [Google Scholar] [CrossRef] [PubMed]
- Irving, J.; Matheson, E.; Minto, L.; Blair, H.; Case, M.; Halsey, C.; Swidenbank, I.; Ponthan, F.; Kirschner-Schwabe, R.; Groeneveld-Krentz, S.; et al. Ras pathway mutations are prevalent in relapsed childhood acute lymphoblastic leukemia and confer sensitivity to MEK inhibition. Blood 2014, 124, 3420–3430. [Google Scholar] [CrossRef] [PubMed]
- Aries, I.M.; van den Dungen, R.E.; Koudijs, M.J.; Cuppen, E.; Voest, E.; Molenaar, J.J.; Caron, H.N.; Pieters, R.; den Boer, M.L. Towards personalized therapy in pediatric acute lymphoblastic leukemia: RAS mutations and prednisolone resistance. Haematologica 2015, 100, e132–e136. [Google Scholar] [CrossRef] [PubMed]
- Moorman, A.V.; Richards, S.M.; Martineau, M.; Cheung, K.L.; Robinson, H.M.; Jalali, G.R.; Broadfield, Z.J.; Harris, R.L.; Taylor, K.E.; Gibson, B.E.; et al. Outcome heterogeneity in childhood high-hyperdiploid acute lymphoblastic leukemia. Blood 2003, 102, 2756–2762. [Google Scholar] [CrossRef]
- Davidsson, J.; Paulsson, K.; Lindgren, D.; Lilljebjorn, H.; Chaplin, T.; Forestier, E.; Andersen, M.K.; Nordgren, A.; Rosenquist, R.; Fioretos, T.; et al. Relapsed childhood high hyperdiploid acute lymphoblastic leukemia: Presence of preleukemic ancestral clones and the secondary nature of microdeletions and RTK-RAS mutations. Leukemia 2010, 24, 924–931. [Google Scholar] [CrossRef]
- Mullighan, C.G.; Su, X.; Zhang, J.; Radtke, I.; Phillips, L.A.; Miller, C.B.; Ma, J.; Liu, W.; Cheng, C.; Schulman, B.A.; et al. Deletion of IKZF1 and prognosis in acute lymphoblastic leukemia. N. Engl. J. Med. 2009, 360, 470–480. [Google Scholar] [CrossRef] [PubMed]
- Kuiper, R.P.; Waanders, E.; van der Velden, V.H.; van Reijmersdal, S.V.; Venkatachalam, R.; Scheijen, B.; Sonneveld, E.; van Dongen, J.J.; Veerman, A.J.; van Leeuwen, F.N.; et al. IKZF1 deletions predict relapse in uniformly treated pediatric precursor B-ALL. Leukemia 2010, 24, 1258–1264. [Google Scholar] [CrossRef] [PubMed]
- Stanulla, M.; Dagdan, E.; Zaliova, M.; Moricke, A.; Palmi, C.; Cazzaniga, G.; Eckert, C.; Te Kronnie, G.; Bourquin, J.P.; Bornhauser, B.; et al. IKZF1(plus) Defines a New Minimal Residual Disease-Dependent Very-Poor Prognostic Profile in Pediatric B-Cell Precursor Acute Lymphoblastic Leukemia. J. Clin. Oncol. 2018, 36, 1240–1249. [Google Scholar] [CrossRef] [PubMed]
- Stanulla, M.; Cave, H.; Moorman, A.V. IKZF1 deletions in pediatric acute lymphoblastic leukemia: Still a poor prognostic marker? Blood 2020, 135, 252–260. [Google Scholar] [CrossRef] [PubMed]
- Dieck, C.L.; Tzoneva, G.; Forouhar, F.; Carpenter, Z.; Ambesi-Impiombato, A.; Sanchez-Martin, M.; Kirschner-Schwabe, R.; Lew, S.; Seetharaman, J.; Tong, L.; et al. Structure and Mechanisms of NT5C2 Mutations Driving Thiopurine Resistance in Relapsed Lymphoblastic Leukemia. Cancer Cell 2018, 34, 136–147.e136. [Google Scholar] [CrossRef]
- Barz, M.J.; Hof, J.; Groeneveld-Krentz, S.; Loh, J.W.; Szymansky, A.; Astrahantseff, K.; von Stackelberg, A.; Khiabanian, H.; Ferrando, A.A.; Eckert, C.; et al. Subclonal NT5C2 mutations are associated with poor outcomes after relapse of pediatric acute lymphoblastic leukemia. Blood 2020, 135, 921–933. [Google Scholar] [CrossRef]
- Dieck, C.L.; Ferrando, A. Genetics and mechanisms of NT5C2-driven chemotherapy resistance in relapsed ALL. Blood 2019, 133, 2263–2268. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).