
Fibroblast Growth Factor 21 Facilitates the Homeostatic Control of Feeding Behavior


{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Nutritional Context(s) of FGF21 Induction
2.1. Prolonged Fasting and Ketogenic Diet
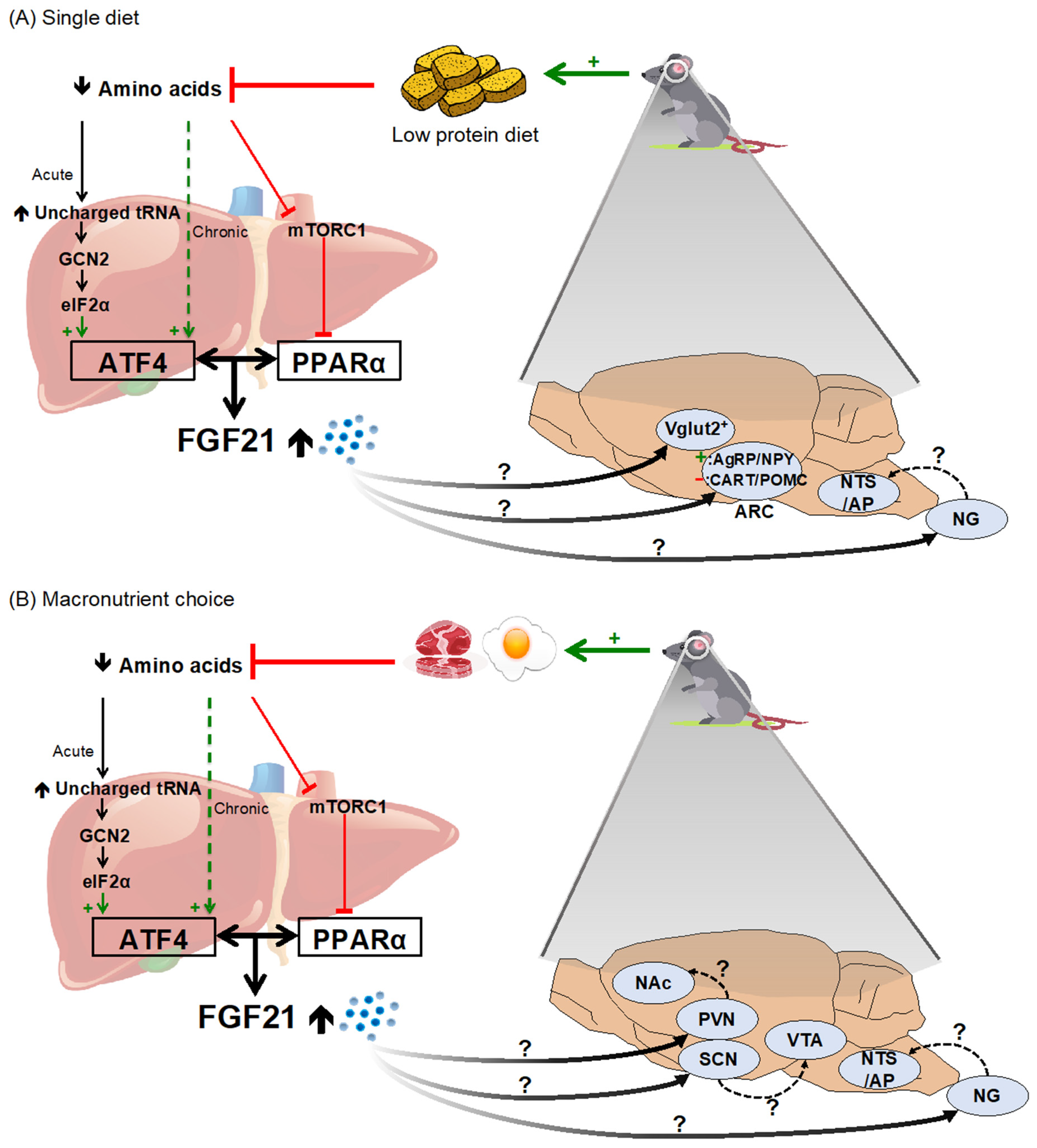
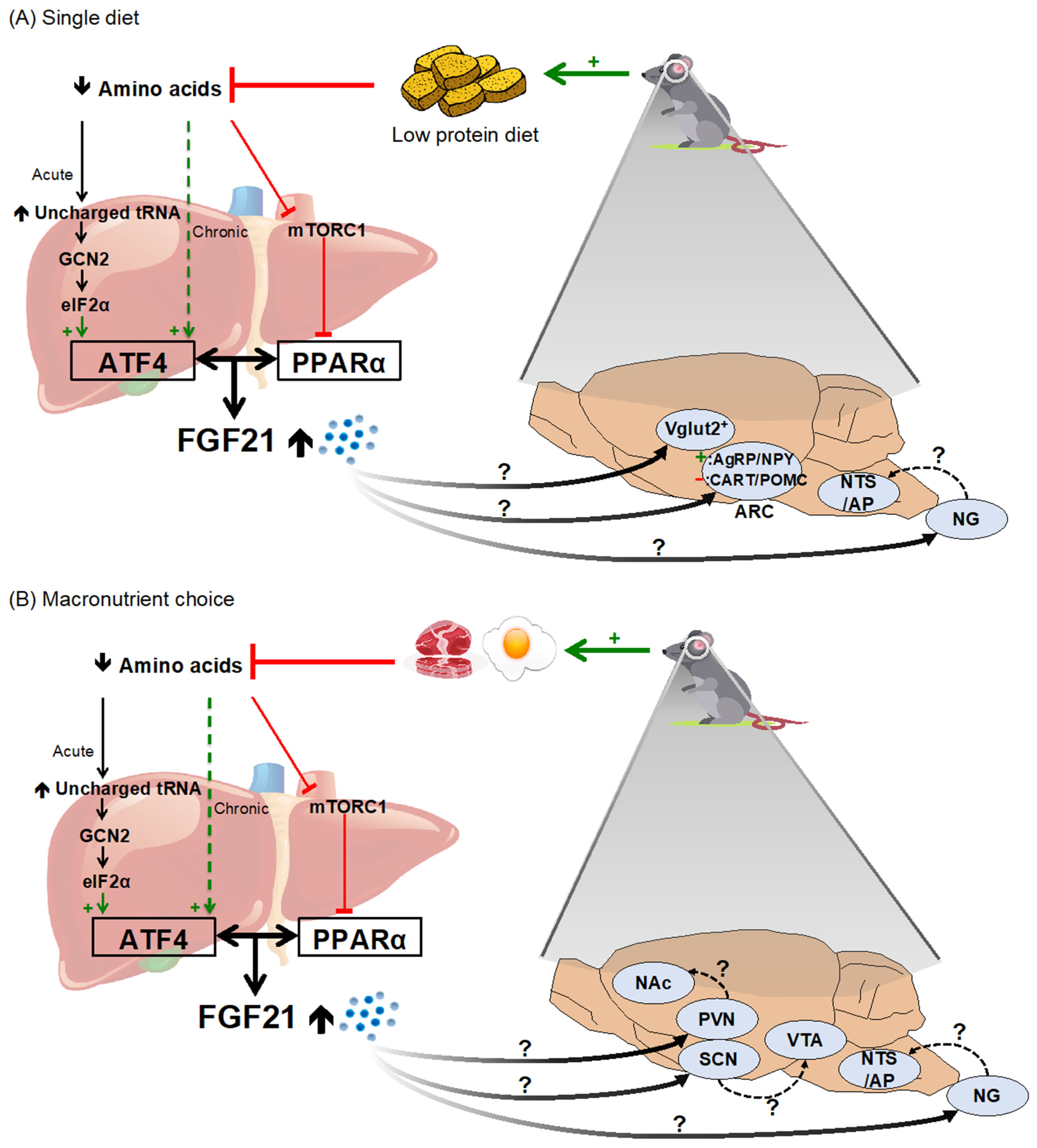
2.2. Dietary Protein and Amino Acid Restriction/Dilution
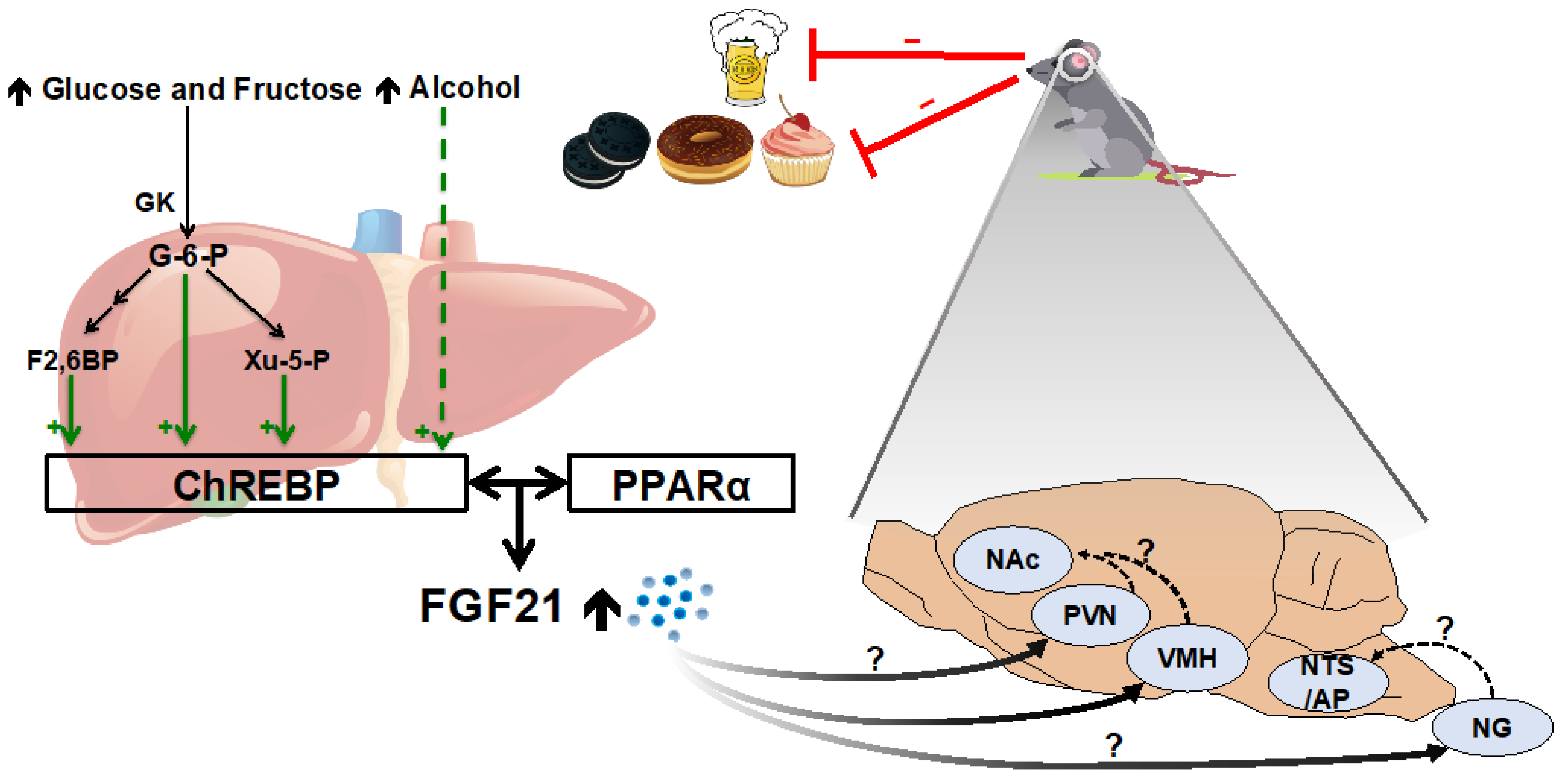
2.3. Simple Sugars and Alcohol
3. FGF21’s Effect on Feeding Behavior
3.1. FGF21 and Caloric Intake
3.2. FGF21 and Macronutrient Selection
3.3. FGF21 Increases Protein Intake
3.4. FGF21 Reduces Sweet and Alcohol Preference
4. Neuroendocrine Mechanisms for the Control of Feeding Behavior by FGF21
4.1. Neuroanatomical Distribution of the FGF21 Receptor Complex
4.2. Mechanistic Basis for FGF21’s Effect on Total Caloric Intake
4.3. Mechanistic Basis for FGF21’s Effect on Macronutrient Intake
5. Future Directions
- (1)
- To what extent are the several behavioral responses to FGF21 unavoidably connected? That is, can the observed reduction in sweet and/or alcohol intake be dissociated from a compensatory increase in protein intake (and vice versa)? This has so far been difficult to disentangle experimentally—for example, in our hands decreased consumption of a sucrose solution following FGF21 injection was balanced by a corresponding increase in the consumption of chow [77]—but this could be addressed with a careful experimental design.
- (2)
- What are the key neural circuit mechanisms mediating the effect of FGF21 on feeding behavior? Recent findings have identified critical neuronal phenotypes for sweet taste preferences (e.g., glutamatergic neurons of the VMH [97]), but some discrepancies remain unresolved, and downstream neural circuit mediators must be identified. The key first-order neurons for changing caloric and protein intake remain unknown, as is the extent to which circuits controlling caloric, sweet, and protein intake are intertwined.
- (3)
- How does the nervous system integrate information conveyed by FGF21 with other well-characterized signals of energy status? For example, does FGF21 influence leptin and insulin signaling in the arcuate and elsewhere [125,126,127,128]; glucose sensing via glucokinase in neurons of the VMH [97] and elsewhere [129,130]; and/or amino acid sensing via mTOR or GCN2 in the mediobasal hypothalamus, hindbrain [131,132,133], and anterior piriform cortex [44]?
- (4)
- What is the role of FGF21 in influencing the circadian control of feeding behavior? Plasma FGF21 follows a circadian rhythm that peaks early in the light phase and falls throughout the dark phase [134,135], and FGF21-transgenic mice have a dysregulated circadian pattern of locomotor behavior [103]. Since macronutrient intake is known to follow a circadian rhythm, with rodents favoring carbohydrate intake at the onset of dark and protein and fat intake at the onset of light [136,137], it is interesting to speculate about the potential role of FGF21 in facilitating this pattern.
- (5)
- What is the therapeutic potential of these new findings? Because pharmacologic administration of recombinant FGF21 elicits multiple metabolic benefits in animal models, including decreased body weight, improved insulin and leptin sensitivity, and decreased hepatic steatosis, as reviewed by [9,10,11,138], several pharmaceutical companies have now developed FGF21 analogues for clinical use in metabolic disease [11,139,140,141]. One exciting possibility is that these drugs may also be useful to modify behavior. Potential applications include the treatment of alcoholism [142] or combating protein malnutrition and sarcopenia in aging [143].
6. Summary
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Ryan, K.K.; Woods, S.C.; Seeley, R.J. Central Nervous System Mechanisms Linking the Consumption of Palatable High-Fat Diets to the Defense of Greater Adiposity. Cell Metab. 2012, 15, 137–149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morton, G.J.; Meek, T.H.; Schwartz, M.W. Neurobiology of Food Intake in Health and Disease. Nat. Rev. Neurosci. 2014, 15, 367–378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, K.-S.; Seeley, R.J.; Sandoval, D.A. Signalling from the Periphery to the Brain That Regulates Energy Homeostasis. Nat. Rev. Neurosci. 2018, 19, 185–196. [Google Scholar] [CrossRef]
- Raubenheimer, D.; Simpson, S.J. Integrative Models of Nutrient Balancing: Application to Insects and Vertebrates. Nutr. Res. Rev. 1997, 10, 151–179. [Google Scholar] [CrossRef] [Green Version]
- Berthoud, H.-R.; Münzberg, H.; Richards, B.K.; Morrison, C.D. Neural and Metabolic Regulation of Macronutrient Intake and Selection. Proc. Nutr. Soc. 2012, 71, 390–400. [Google Scholar] [CrossRef] [PubMed]
- Berthoud, H.-R.; Seeley, R.J. Neural and Metabolic Control of Macronutrient Intake; CRC PRESS: Boca Raton, FL, USA, 2019; ISBN 978-0-367-39935-1. [Google Scholar]
- Hill, C.M.; Qualls-Creekmore, E.; Berthoud, H.-R.; Soto, P.; Yu, S.; McDougal, D.H.; Münzberg, H.; Morrison, C.D. FGF21 and the Physiological Regulation of Macronutrient Preference. Endocrinology 2020, 161, bqaa019. [Google Scholar] [CrossRef] [Green Version]
- Nishimura, T.; Nakatake, Y.; Konishi, M.; Itoh, N. Identification of a Novel FGF, FGF-21, Preferentially Expressed in the Liver. Biochim. Biophys. Acta 2000, 1492, 203–206. [Google Scholar] [CrossRef]
- Fisher, F.M.; Maratos-Flier, E. Understanding the Physiology of FGF21. Annu. Rev. Physiol. 2016, 78, 223–241. [Google Scholar] [CrossRef] [Green Version]
- BonDurant, L.D.; Potthoff, M.J. Fibroblast Growth Factor 21: A Versatile Regulator of Metabolic Homeostasis. Annu. Rev. Nutr. 2018, 38, 173–196. [Google Scholar] [CrossRef]
- Geng, L.; Lam, K.S.L.; Xu, A. The Therapeutic Potential of FGF21 in Metabolic Diseases: From Bench to Clinic. Nat. Rev. Endocrinol. 2020, 16, 654–667. [Google Scholar] [CrossRef]
- Fisher, F.M.; Kleiner, S.; Douris, N.; Fox, E.C.; Mepani, R.J.; Verdeguer, F.; Wu, J.; Kharitonenkov, A.; Flier, J.S.; Maratos-Flier, E.; et al. FGF21 Regulates PGC-1α and Browning of White Adipose Tissues in Adaptive Thermogenesis. Genes Dev. 2012, 26, 271–281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coate, K.C.; Hernandez, G.; Thorne, C.A.; Sun, S.; Le, T.D.V.; Vale, K.; Kliewer, S.A.; Mangelsdorf, D.J. FGF21 Is an Exocrine Pancreas Secretagogue. Cell Metab. 2017, 25, 472–480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, C.L.; Weston, J.Y.; Chadi, S.A.; Fazio, E.N.; Huff, M.W.; Kharitonenkov, A.; Köester, A.; Pin, C.L. Fibroblast Growth Factor 21 Reduces the Severity of Cerulein-Induced Pancreatitis in Mice. Gastroenterology 2009, 137, 1795–1804. [Google Scholar] [CrossRef] [PubMed]
- Markan, K.R.; Naber, M.C.; Ameka, M.K.; Anderegg, M.D.; Mangelsdorf, D.J.; Kliewer, S.A.; Mohammadi, M.; Potthoff, M.J. Circulating FGF21 Is Liver Derived and Enhances Glucose Uptake During Refeeding and Overfeeding. Diabetes 2014, 63, 4057–4063. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inagaki, T.; Dutchak, P.; Zhao, G.; Ding, X.; Gautron, L.; Parameswara, V.; Li, Y.; Goetz, R.; Mohammadi, M.; Esser, V.; et al. Endocrine Regulation of the Fasting Response by PPARα-Mediated Induction of Fibroblast Growth Factor 21. Cell Metab. 2007, 5, 415–425. [Google Scholar] [CrossRef] [Green Version]
- Badman, M.K.; Pissios, P.; Kennedy, A.R.; Koukos, G.; Flier, J.S.; Maratos-Flier, E. Hepatic Fibroblast Growth Factor 21 Is Regulated by PPARα and Is a Key Mediator of Hepatic Lipid Metabolism in Ketotic States. Cell Metab. 2007, 5, 426–437. [Google Scholar] [CrossRef] [Green Version]
- Reitman, M.L. FGF21: A Missing Link in the Biology of Fasting. Cell Metab. 2007, 5, 405–407. [Google Scholar] [CrossRef] [Green Version]
- Kersten, S.; Seydoux, J.; Peters, J.M.; Gonzalez, F.J.; Desvergne, B.; Wahli, W. Peroxisome Proliferator–Activated Receptor α Mediates the Adaptive Response to Fasting. J. Clin. Investig. 1999, 103, 1489–1498. [Google Scholar] [CrossRef] [Green Version]
- Potthoff, M.J.; Inagaki, T.; Satapati, S.; Ding, X.; He, T.; Goetz, R.; Mohammadi, M.; Finck, B.N.; Mangelsdorf, D.J.; Kliewer, S.A.; et al. FGF21 Induces PGC-1alpha and Regulates Carbohydrate and Fatty Acid Metabolism during the Adaptive Starvation Response. Proc. Natl. Acad. Sci. USA 2009, 106, 10853–10858. [Google Scholar] [CrossRef] [Green Version]
- Lundåsen, T.; Hunt, M.C.; Nilsson, L.-M.; Sanyal, S.; Angelin, B.; Alexson, S.E.H.; Rudling, M. PPARα Is a Key Regulator of Hepatic FGF21. Biochem. Biophys. Res. Commun. 2007, 360, 437–440. [Google Scholar] [CrossRef] [Green Version]
- Araki, M.; Nakagawa, Y.; Oishi, A.; Han, S.; Wang, Y.; Kumagai, K.; Ohno, H.; Mizunoe, Y.; Iwasaki, H.; Sekiya, M.; et al. The Peroxisome Proliferator-Activated Receptor α (PPARα) Agonist Pemafibrate Protects against Diet-Induced Obesity in Mice. Int. J. Mol. Sci. 2018, 19, 2148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eto, K.; Tumenbayar, B.; Nagashima, S.; Tazoe, F.; Miyamoto, M.; Takahashi, M.; Ando, A.; Okada, K.; Yagyu, H.; Ishibashi, S. Distinct Association of Serum FGF21 or Adiponectin Levels with Clinical Parameters in Patients with Type 2 Diabetes. Diabetes Res. Clin. Pract. 2010, 89, 52–57. [Google Scholar] [CrossRef] [PubMed]
- Ong, K.L.; Rye, K.-A.; O’Connell, R.; Jenkins, A.J.; Brown, C.; Xu, A.; Sullivan, D.R.; Barter, P.J.; Keech, A.C. FIELD study investigators Long-Term Fenofibrate Therapy Increases Fibroblast Growth Factor 21 and Retinol-Binding Protein 4 in Subjects with Type 2 Diabetes. J. Clin. Endocrinol. Metab. 2012, 97, 4701–4708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Xu, Y.; Hu, Y.; Wang, G. The Role of Fibroblast Growth Factor 21 in the Pathogenesis of Non-Alcoholic Fatty Liver Disease and Implications for Therapy. Metabolism 2015, 64, 380–390. [Google Scholar] [CrossRef] [PubMed]
- Dushay, J.; Chui, P.C.; Gopalakrishnan, G.S.; Varela-Rey, M.; Crawley, M.; Fisher, F.M.; Badman, M.K.; Chantar, M.L.M.; Maratos-Flier, E. Increased Fibroblast Growth Factor 21 in Obesity and Nonalcoholic Fatty Liver Disease. Gastroenterology 2010, 139, 456–463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fazeli, P.K.; Lun, M.; Kim, S.M.; Bredella, M.A.; Wright, S.; Zhang, Y.; Lee, H.; Catana, C.; Klibanski, A.; Patwari, P.; et al. FGF21 and the Late Adaptive Response to Starvation in Humans. J. Clin. Investig. 2015, 125, 4601–4611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gälman, C.; Lundåsen, T.; Kharitonenkov, A.; Bina, H.A.; Eriksson, M.; Hafström, I.; Dahlin, M.; Åmark, P.; Angelin, B.; Rudling, M. The Circulating Metabolic Regulator FGF21 Is Induced by Prolonged Fasting and PPARα Activation in Man. Cell Metab. 2008, 8, 169–174. [Google Scholar] [CrossRef] [Green Version]
- Adelman, R.; Saul, R.L.; Ames, B.N. Oxidative Damage to DNA: Relation to Species Metabolic Rate and Life Span. Proc. Natl. Acad. Sci. USA 1988, 85, 2706–2708. [Google Scholar] [CrossRef] [Green Version]
- Christodoulides, C.; Dyson, P.; Sprecher, D.; Tsintzas, K.; Karpe, F. Circulating Fibroblast Growth Factor 21 Is Induced by Peroxisome Proliferator-Activated Receptor Agonists but Not Ketosis in Man. J. Clin. Endocrinol. Metab. 2009, 94, 3594–3601. [Google Scholar] [CrossRef] [Green Version]
- Sánchez, J.; Palou, A.; Picó, C. Response to Carbohydrate and Fat Refeeding in the Expression of Genes Involved in Nutrient Partitioning and Metabolism: Striking Effects on Fibroblast Growth Factor-21 Induction. Endocrinology 2009, 150, 5341–5350. [Google Scholar] [CrossRef] [Green Version]
- Laeger, T.; Henagan, T.M.; Albarado, D.C.; Redman, L.M.; Bray, G.A.; Noland, R.C.; Münzberg, H.; Hutson, S.M.; Gettys, T.W.; Schwartz, M.W.; et al. FGF21 Is an Endocrine Signal of Protein Restriction. J. Clin. Investig. 2014, 124, 3913–3922. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stemmer, K.; Zani, F.; Habegger, K.M.; Neff, C.; Kotzbeck, P.; Bauer, M.; Yalamanchilli, S.; Azad, A.; Lehti, M.; Martins, P.J.F.; et al. FGF21 Is Not Required for Glucose Homeostasis, Ketosis or Tumour Suppression Associated with Ketogenic Diets in Mice. Diabetologia 2015, 58, 2414–2423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maida, A.; Zota, A.; Sjøberg, K.A.; Schumacher, J.; Sijmonsma, T.P.; Pfenninger, A.; Christensen, M.M.; Gantert, T.; Fuhrmeister, J.; Rothermel, U.; et al. A Liver Stress-Endocrine Nexus Promotes Metabolic Integrity during Dietary Protein Dilution. J. Clin. Investig. 2016, 126, 3263–3278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karusheva, Y.; Koessler, T.; Strassburger, K.; Markgraf, D.; Mastrototaro, L.; Jelenik, T.; Simon, M.-C.; Pesta, D.; Zaharia, O.-P.; Bódis, K.; et al. Short-Term Dietary Reduction of Branched-Chain Amino Acids Reduces Meal-Induced Insulin Secretion and Modifies Microbiome Composition in Type 2 Diabetes: A Randomized Controlled Crossover Trial. Am. J. Clin. Nutr. 2019, 110, 1098–1107. [Google Scholar] [CrossRef] [Green Version]
- Solon-Biet, S.M.; Cogger, V.C.; Pulpitel, T.; Heblinski, M.; Wahl, D.; McMahon, A.C.; Warren, A.; Durrant-Whyte, J.; Walters, K.A.; Krycer, J.R.; et al. Defining the Nutritional and Metabolic Context of FGF21 Using the Geometric Framework. Cell Metab. 2016, 24, 555–565. [Google Scholar] [CrossRef] [Green Version]
- Lees, E.K.; Król, E.; Grant, L.; Shearer, K.; Wyse, C.; Moncur, E.; Bykowska, A.S.; Mody, N.; Gettys, T.W.; Delibegovic, M. Methionine Restriction Restores a Younger Metabolic Phenotype in Adult Mice with Alterations in Fibroblast Growth Factor 21. Aging Cell 2014, 13, 817–827. [Google Scholar] [CrossRef] [Green Version]
- Wanders, D.; Forney, L.A.; Stone, K.P.; Burk, D.H.; Pierse, A.; Gettys, T.W. FGF21 Mediates the Thermogenic and Insulin-Sensitizing Effects of Dietary Methionine Restriction but Not Its Effects on Hepatic Lipid Metabolism. Diabetes 2017, 66, 858–867. [Google Scholar] [CrossRef] [Green Version]
- Yu, D.; Yang, S.E.; Miller, B.R.; Wisinski, J.A.; Sherman, D.S.; Brinkman, J.A.; Tomasiewicz, J.L.; Cummings, N.E.; Kimple, M.E.; Cryns, V.L.; et al. Short-term Methionine Deprivation Improves Metabolic Health via Sexually Dimorphic, MTORCI-independent Mechanisms. FASEB J. 2018, 32, 3471–3482. [Google Scholar] [CrossRef] [Green Version]
- Yap, Y.W.; Rusu, P.M.; Chan, A.Y.; Fam, B.C.; Jungmann, A.; Solon-Biet, S.M.; Barlow, C.K.; Creek, D.J.; Huang, C.; Schittenhelm, R.B.; et al. Restriction of Essential Amino Acids Dictates the Systemic Metabolic Response to Dietary Protein Dilution. Nat. Commun. 2020, 11, 2894. [Google Scholar] [CrossRef]
- Zhang, Y.-K.J.; Yeager, R.L.; Tanaka, Y.; Klaassen, C.D. Enhanced Expression of Nrf2 in Mice Attenuates the Fatty Liver Produced by a Methionine- and Choline-Deficient Diet. Toxicol. Appl. Pharmacol. 2010, 245, 326–334. [Google Scholar] [CrossRef] [Green Version]
- Perrone, C.E.; Mattocks, D.A.L.; Plummer, J.D.; Chittur, S.V.; Mohney, R.; Vignola, K.; Orentreich, D.S.; Orentreich, N. Genomic and Metabolic Responses to Methionine-Restricted and Methionine-Restricted, Cysteine-Supplemented Diets in Fischer 344 Rat Inguinal Adipose Tissue, Liver and Quadriceps Muscle. J. Nutr. Nutr. 2012, 5, 132–157. [Google Scholar] [CrossRef] [PubMed]
- De Sousa-Coelho, A.L.; Marrero, P.F.; Haro, D. Activating Transcription Factor 4-Dependent Induction of FGF21 during Amino Acid Deprivation. Biochem. J. 2012, 443, 165–171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rudell, J.B.; Rechs, A.J.; Kelman, T.J.; Ross-Inta, C.M.; Hao, S.; Gietzen, D.W. The Anterior Piriform Cortex Is Sufficient for Detecting Depletion of an Indispensable Amino Acid, Showing Independent Cortical Sensory Function. J. Neurosci. 2011, 31, 1583–1590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salminen, A.; Kaarniranta, K.; Kauppinen, A. Integrated Stress Response Stimulates FGF21 Expression: Systemic Enhancer of Longevity. Cell. Signal. 2017, 40, 10–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laeger, T.; Albarado, D.C.; Burke, S.J.; Trosclair, L.; Hedgepeth, J.W.; Berthoud, H.-R.; Gettys, T.W.; Collier, J.J.; Münzberg, H.; Morrison, C.D. Metabolic Responses to Dietary Protein Restriction Require an Increase in FGF21 That Is Delayed by the Absence of GCN2. Cell Rep. 2016, 16, 707–716. [Google Scholar] [CrossRef] [Green Version]
- Sengupta, S.; Peterson, T.R.; Laplante, M.; Oh, S.; Sabatini, D.M. MTORC1 Controls Fasting-Induced Ketogenesis and Its Modulation by Ageing. Nature 2010, 468, 1100–1104. [Google Scholar] [CrossRef]
- Cornu, M.; Oppliger, W.; Albert, V.; Robitaille, A.M.; Trapani, F.; Quagliata, L.; Fuhrer, T.; Sauer, U.; Terracciano, L.; Hall, M.N. Hepatic MTORC1 Controls Locomotor Activity, Body Temperature, and Lipid Metabolism through FGF21. Proc. Natl. Acad. Sci. USA 2014, 111, 11592–11599. [Google Scholar] [CrossRef] [Green Version]
- Iizuka, K.; Takeda, J.; Horikawa, Y. Glucose Induces FGF21 MRNA Expression through ChREBP Activation in Rat Hepatocytes. FEBS Lett. 2009, 583, 2882–2886. [Google Scholar] [CrossRef] [Green Version]
- Yamashita, H.; Takenoshita, M.; Sakurai, M.; Bruick, R.K.; Henzel, W.J.; Shillinglaw, W.; Arnot, D.; Uyeda, K. A Glucose-Responsive Transcription Factor That Regulates Carbohydrate Metabolism in the Liver. Proc. Natl. Acad. Sci. USA 2001, 98, 9116–9121. [Google Scholar] [CrossRef] [Green Version]
- Kabashima, T.; Kawaguchi, T.; Wadzinski, B.E.; Uyeda, K. Xylulose 5-Phosphate Mediates Glucose-Induced Lipogenesis by Xylulose 5-Phosphate-Activated Protein Phosphatase in Rat Liver. Proc. Natl. Acad. Sci. USA 2003, 100, 5107–5112. [Google Scholar] [CrossRef] [Green Version]
- Li, M.V.; Chen, W.; Harmancey, R.N.; Nuotio-Antar, A.M.; Imamura, M.; Saha, P.; Taegtmeyer, H.; Chan, L. Glucose-6-Phosphate Mediates Activation of the Carbohydrate Responsive Binding Protein (ChREBP). Biochem. Biophys. Res. Commun. 2010, 395, 395–400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arden, C.; Tudhope, S.J.; Petrie, J.L.; Al-Oanzi, Z.H.; Cullen, K.S.; Lange, A.J.; Towle, H.C.; Agius, L. Fructose 2,6-Bisphosphate Is Essential for Glucose-Regulated Gene Transcription of Glucose-6-Phosphatase and Other ChREBP Target Genes in Hepatocytes. Biochem. J. 2012, 443, 111–123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdul-Wahed, A.; Guilmeau, S.; Postic, C. Sweet Sixteenth for ChREBP: Established Roles and Future Goals. Cell Metab. 2017, 26, 324–341. [Google Scholar] [CrossRef]
- Chalvon-Demersay, T.; Even, P.C.; Tomé, D.; Chaumontet, C.; Piedcoq, J.; Gaudichon, C.; Azzout-Marniche, D. Low-Protein Diet Induces, Whereas High-Protein Diet Reduces Hepatic FGF21 Production in Mice, but Glucose and Not Amino Acids up-Regulate FGF21 in Cultured Hepatocytes. J. Nutr. Biochem. 2016, 36, 60–67. [Google Scholar] [CrossRef] [PubMed]
- Uebanso, T.; Taketani, Y.; Yamamoto, H.; Amo, K.; Ominami, H.; Arai, H.; Takei, Y.; Masuda, M.; Tanimura, A.; Harada, N.; et al. Paradoxical Regulation of Human FGF21 by Both Fasting and Feeding Signals: Is FGF21 a Nutritional Adaptation Factor? PLoS ONE 2011, 6, e22976. [Google Scholar] [CrossRef]
- von Holstein-Rathlou, S.; BonDurant, L.D.; Peltekian, L.; Naber, M.C.; Yin, T.C.; Claflin, K.E.; Urizar, A.I.; Madsen, A.N.; Ratner, C.; Holst, B.; et al. FGF21 Mediates Endocrine Control of Simple Sugar Intake and Sweet Taste Preference by the Liver. Cell Metab. 2016, 23, 335–343. [Google Scholar] [CrossRef] [Green Version]
- Fisher, F.M.; Kim, M.; Doridot, L.; Cunniff, J.C.; Parker, T.S.; Levine, D.M.; Hellerstein, M.K.; Hudgins, L.C.; Maratos-Flier, E.; Herman, M.A. A Critical Role for ChREBP-Mediated FGF21 Secretion in Hepatic Fructose Metabolism. Mol. Metab. 2017, 6, 14–21. [Google Scholar] [CrossRef]
- Hao, L.; Huang, K.-H.; Ito, K.; Sae-tan, S.; Lambert, J.D.; Ross, A.C. Fibroblast Growth Factor 21 (Fgf21) Gene Expression Is Elevated in the Liver of Mice Fed a High-Carbohydrate Liquid Diet and Attenuated by a Lipid Emulsion but Is Not Upregulated in the Liver of Mice Fed a High-Fat Obesogenic Diet. J. Nutr. 2016, 146, 184–190. [Google Scholar] [CrossRef] [Green Version]
- Dushay, J.R.; Toschi, E.; Mitten, E.K.; Fisher, F.M.; Herman, M.A.; Maratos-Flier, E. Fructose Ingestion Acutely Stimulates Circulating FGF21 Levels in Humans. Mol. Metab. 2015, 4, 51–57. [Google Scholar] [CrossRef]
- McGuinness, O.P.; Cherrington, A.D. Effects of Fructose on Hepatic Glucose Metabolism. Curr. Opin. Clin. Nutr. Metab. Care 2003, 6, 441–448. [Google Scholar] [CrossRef]
- Geidl-Flueck, B.; Gerber, P. Insights into the Hexose Liver Metabolism—Glucose versus Fructose. Nutrients 2017, 9, 1026. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.-S.; Krawczyk, S.A.; Doridot, L.; Fowler, A.J.; Wang, J.X.; Trauger, S.A.; Noh, H.-L.; Kang, H.J.; Meissen, J.K.; Blatnik, M.; et al. ChREBP Regulates Fructose-Induced Glucose Production Independently of Insulin Signaling. J. Clin. Investig. 2016, 126, 4372–4386. [Google Scholar] [CrossRef] [Green Version]
- Iroz, A.; Montagner, A.; Benhamed, F.; Levavasseur, F.; Polizzi, A.; Anthony, E.; Régnier, M.; Fouché, E.; Lukowicz, C.; Cauzac, M.; et al. A Specific ChREBP and PPARα Cross-Talk Is Required for the Glucose-Mediated FGF21 Response. Cell Rep. 2017, 21, 403–416. [Google Scholar] [CrossRef] [Green Version]
- Lin, Z.; Gong, Q.; Wu, C.; Yu, J.; Lu, T.; Pan, X.; Lin, S.; Li, X. Dynamic Change of Serum FGF21 Levels in Response to Glucose Challenge in Human. J. Clin. Endocrinol. Metab. 2012, 97, E1224–E1228. [Google Scholar] [CrossRef] [Green Version]
- Søberg, S.; Sandholt, C.H.; Jespersen, N.Z.; Toft, U.; Madsen, A.L.; von Holstein-Rathlou, S.; Grevengoed, T.J.; Christensen, K.B.; Bredie, W.L.P.; Potthoff, M.J.; et al. FGF21 Is a Sugar-Induced Hormone Associated with Sweet Intake and Preference in Humans. Cell Metab. 2017, 25, 1045–1053. [Google Scholar] [CrossRef] [Green Version]
- Zhao, C.; Liu, Y.; Xiao, J.; Liu, L.; Chen, S.; Mohammadi, M.; McClain, C.J.; Li, X.; Feng, W. FGF21 Mediates Alcohol-Induced Adipose Tissue Lipolysis by Activation of Systemic Release of Catecholamine in Mice. J. Lipid Res. 2015, 56, 1481–1491. [Google Scholar] [CrossRef] [Green Version]
- Desai, B.N.; Singhal, G.; Watanabe, M.; Stevanovic, D.; Lundasen, T.; Fisher, F.M.; Mather, M.L.; Vardeh, H.G.; Douris, N.; Adams, A.C.; et al. Fibroblast Growth Factor 21 (FGF21) Is Robustly Induced by Ethanol and Has a Protective Role in Ethanol Associated Liver Injury. Mol. Metab. 2017, 6, 1395–1406. [Google Scholar] [CrossRef] [PubMed]
- Song, P.; Zechner, C.; Hernandez, G.; Cánovas, J.; Xie, Y.; Sondhi, V.; Wagner, M.; Stadlbauer, V.; Horvath, A.; Leber, B.; et al. The Hormone FGF21 Stimulates Water Drinking in Response to Ketogenic Diet and Alcohol. Cell Metab. 2018, 27, 1338–1347. [Google Scholar] [CrossRef] [Green Version]
- Søberg, S.; Andersen, E.S.; Dalsgaard, N.B.; Jarlhelt, I.; Hansen, N.L.; Hoffmann, N.; Vilsbøll, T.; Chenchar, A.; Jensen, M.; Grevengoed, T.J.; et al. FGF21, a Liver Hormone That Inhibits Alcohol Intake in Mice, Increases in Human Circulation after Acute Alcohol Ingestion and Sustained Binge Drinking at Oktoberfest. Mol. Metab. 2018, 11, 96–103. [Google Scholar] [CrossRef]
- Kharitonenkov, A.; Shiyanova, T.L.; Koester, A.; Ford, A.M.; Micanovic, R.; Galbreath, E.J.; Sandusky, G.E.; Hammond, L.J.; Moyers, J.S.; Owens, R.A.; et al. FGF-21 as a Novel Metabolic Regulator. J. Clin. Investig. 2005, 115, 1627–1635. [Google Scholar] [CrossRef] [Green Version]
- Owen, B.M.; Ding, X.; Morgan, D.A.; Coate, K.C.; Bookout, A.L.; Rahmouni, K.; Kliewer, S.A.; Mangelsdorf, D.J. FGF21 Acts Centrally to Induce Sympathetic Nerve Activity, Energy Expenditure, and Weight Loss. Cell Metab. 2014, 20, 670–677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, X.; Boney-Montoya, J.; Owen, B.M.; Bookout, A.L.; Coate, K.C.; Mangelsdorf, D.J.; Kliewer, S.A. ΒKlotho Is Required for Fibroblast Growth Factor 21 Effects on Growth and Metabolism. Cell Metab. 2012, 16, 387–393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inagaki, T.; Lin, V.Y.; Goetz, R.; Mohammadi, M.; Mangelsdorf, D.J.; Kliewer, S.A. Inhibition of Growth Hormone Signaling by the Fasting-Induced Hormone FGF21. Cell Metab. 2008, 8, 77–83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarruf, D.A.; Thaler, J.P.; Morton, G.J.; German, J.; Fischer, J.D.; Ogimoto, K.; Schwartz, M.W. Fibroblast Growth Factor 21 Action in the Brain Increases Energy Expenditure and Insulin Sensitivity in Obese Rats. Diabetes 2010, 59, 1817–1824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coskun, T.; Bina, H.A.; Schneider, M.A.; Dunbar, J.D.; Hu, C.C.; Chen, Y.; Moller, D.E.; Kharitonenkov, A. Fibroblast Growth Factor 21 Corrects Obesity in Mice. Endocrinology 2008, 149, 6018–6027. [Google Scholar] [CrossRef] [PubMed]
- Larson, K.R.; Chaffin, A.T.-B.; Goodson, M.L.; Fang, Y.; Ryan, K.K. Fibroblast Growth Factor-21 Controls Dietary Protein Intake in Male Mice. Endocrinology 2019, 160, 1069–1080. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Lloyd, D.J.; Hale, C.; Stanislaus, S.; Chen, M.; Sivits, G.; Vonderfecht, S.; Hecht, R.; Li, Y.-S.; Lindberg, R.A.; et al. Fibroblast Growth Factor 21 Reverses Hepatic Steatosis, Increases Energy Expenditure, and Improves Insulin Sensitivity in Diet-Induced Obese Mice. Diabetes 2009, 58, 250–259. [Google Scholar] [CrossRef] [Green Version]
- Samms, R.J.; Smith, D.P.; Cheng, C.C.; Antonellis, P.P.; Perfield, J.W.; Kharitonenkov, A.; Gimeno, R.E.; Adams, A.C. Discrete Aspects of FGF21 In Vivo Pharmacology Do Not Require UCP1. Cell Rep. 2015, 11, 991–999. [Google Scholar] [CrossRef] [Green Version]
- Cummings, N.E.; Williams, E.M.; Kasza, I.; Konon, E.N.; Schaid, M.D.; Schmidt, B.A.; Poudel, C.; Sherman, D.S.; Yu, D.; Arriola Apelo, S.I.; et al. Restoration of Metabolic Health by Decreased Consumption of Branched-Chain Amino Acids: A Low BCAA Diet Restores Metabolic Health. J. Physiol. 2018, 596, 623–645. [Google Scholar] [CrossRef] [Green Version]
- Hill, C.M.; Laeger, T.; Albarado, D.C.; McDougal, D.H.; Berthoud, H.-R.; Münzberg, H.; Morrison, C.D. Low Protein-Induced Increases in FGF21 Drive UCP1-Dependent Metabolic but Not Thermoregulatory Endpoints. Sci. Rep. 2017, 7, 8209. [Google Scholar] [CrossRef]
- Hill, C.M.; Laeger, T.; Dehner, M.; Albarado, D.C.; Clarke, B.; Wanders, D.; Burke, S.J.; Collier, J.J.; Qualls-Creekmore, E.; Solon-Biet, S.M.; et al. FGF21 Signals Protein Status to the Brain and Adaptively Regulates Food Choice and Metabolism. Cell Rep. 2019, 27, 2934–2947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adams, A.C.; Halstead, C.A.; Hansen, B.C.; Irizarry, A.R.; Martin, J.A.; Myers, S.R.; Reynolds, V.L.; Smith, H.W.; Wroblewski, V.J.; Kharitonenkov, A. LY2405319, an Engineered FGF21 Variant, Improves the Metabolic Status of Diabetic Monkeys. PLoS ONE 2013, 8, e65763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Talukdar, S.; Zhou, Y.; Li, D.; Rossulek, M.; Dong, J.; Somayaji, V.; Weng, Y.; Clark, R.; Lanba, A.; Owen, B.M.; et al. A Long-Acting FGF21 Molecule, PF-05231023, Decreases Body Weight and Improves Lipid Profile in Non-Human Primates and Type 2 Diabetic Subjects. Cell Metab. 2016, 23, 427–440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stanislaus, S.; Hecht, R.; Yie, J.; Hager, T.; Hall, M.; Spahr, C.; Wang, W.; Weiszmann, J.; Li, Y.; Deng, L.; et al. A Novel Fc-FGF21 With Improved Resistance to Proteolysis, Increased Affinity Toward β-Klotho, and Enhanced Efficacy in Mice and Cynomolgus Monkeys. Endocrinology 2017, 158, 1314–1327. [Google Scholar] [CrossRef] [PubMed]
- Larson, K.R.; Russo, K.A.; Fang, Y.; Mohajerani, N.; Goodson, M.L.; Ryan, K.K. Sex Differences in the Hormonal and Metabolic Response to Dietary Protein Dilution. Endocrinology 2017, 158, 3477–3487. [Google Scholar] [CrossRef] [PubMed]
- Simpson, S.J.; Raubenheimer, D. Obesity: The Protein Leverage Hypothesis. Obes. Rev. 2005, 6, 133–142. [Google Scholar] [CrossRef]
- Sørensen, A.; Mayntz, D.; Raubenheimer, D.; Simpson, S.J. Protein-Leverage in Mice: The Geometry of Macronutrient Balancing and Consequences for Fat Deposition. Obesity 2008, 16, 566–571. [Google Scholar] [CrossRef]
- Chu, A.Y.; Workalemahu, T.; Paynter, N.P.; Rose, L.M.; Giulianini, F.; Tanaka, T.; Ngwa, J.S.; Qi, Q.; Curhan, G.C.; Rimm, E.B.; et al. Novel Locus Including FGF21 Is Associated with Dietary Macronutrient Intake. Hum. Mol. Genet. 2013, 22, 1895–1902. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, T.; Ngwa, J.S.; van Rooij, F.J.; Zillikens, M.C.; Wojczynski, M.K.; Frazier-Wood, A.C.; Houston, D.K.; Kanoni, S.; Lemaitre, R.N.; Luan, J.; et al. Genome-Wide Meta-Analysis of Observational Studies Shows Common Genetic Variants Associated with Macronutrient Intake. Am. J. Clin. Nutr. 2013, 97, 1395–1402. [Google Scholar] [CrossRef] [Green Version]
- Clarke, T.-K.; Adams, M.J.; Davies, G.; Howard, D.M.; Hall, L.S.; Padmanabhan, S.; Murray, A.D.; Smith, B.H.; Campbell, A.; Hayward, C.; et al. Genome-Wide Association Study of Alcohol Consumption and Genetic Overlap with Other Health-Related Traits in UK Biobank (N = 112 117). Mol. Psychiatry 2017, 22, 1376–1384. [Google Scholar] [CrossRef] [Green Version]
- Schumann, G.; Liu, C.; O’Reilly, P.; Gao, H.; Song, P.; Xu, B.; Ruggeri, B.; Amin, N.; Jia, T.; Preis, S.; et al. KLB Is Associated with Alcohol Drinking, and Its Gene Product β-Klotho Is Necessary for FGF21 Regulation of Alcohol Preference. Proc. Natl. Acad. Sci. USA 2016, 113, 14372–14377. [Google Scholar] [CrossRef] [Green Version]
- Talukdar, S.; Owen, B.M.; Song, P.; Hernandez, G.; Zhang, Y.; Zhou, Y.; Scott, W.T.; Paratala, B.; Turner, T.; Smith, A.; et al. FGF21 Regulates Sweet and Alcohol Preference. Cell Metab. 2016, 23, 344–349. [Google Scholar] [CrossRef] [Green Version]
- Holstein-Rathlou, S.; Gillum, M.P. Fibroblast Growth Factor 21: An Endocrine Inhibitor of Sugar and Alcohol Appetite. J. Physiol. 2019, 597, 3539–3548. [Google Scholar] [CrossRef] [Green Version]
- Simpson, S.J.; Raubenheimer, D. The Nature of Nutrition: A Unifying Framework from Animal Adaptation to Human Obesity; Princeton University Press: Princeton, NJ, USA, 2012; ISBN 978-1-4008-4280-3. [Google Scholar]
- Hsuchou, H.; Pan, W.; Kastin, A.J. The Fasting Polypeptide FGF21 Can Enter Brain from Blood. Peptides 2007, 28, 2382–2386. [Google Scholar] [CrossRef] [Green Version]
- Jensen-Cody, S.O.; Flippo, K.H.; Claflin, K.E.; Yavuz, Y.; Sapouckey, S.A.; Walters, G.C.; Usachev, Y.M.; Atasoy, D.; Gillum, M.P.; Potthoff, M.J. FGF21 Signals to Glutamatergic Neurons in the Ventromedial Hypothalamus to Suppress Carbohydrate Intake. Cell Metab. 2020, 32, 273–286. [Google Scholar] [CrossRef] [PubMed]
- Kurosu, H.; Choi, M.; Ogawa, Y.; Dickson, A.S.; Goetz, R.; Eliseenkova, A.V.; Mohammadi, M.; Rosenblatt, K.P.; Kliewer, S.A.; Kuro-o, M. Tissue-Specific Expression of ΒKlotho and Fibroblast Growth Factor (FGF) Receptor Isoforms Determines Metabolic Activity of FGF19 and FGF21. J. Biol. Chem. 2007, 282, 26687–26695. [Google Scholar] [CrossRef] [Green Version]
- Ogawa, Y.; Kurosu, H.; Yamamoto, M.; Nandi, A.; Rosenblatt, K.P.; Goetz, R.; Eliseenkova, A.V.; Mohammadi, M.; Kuro-o, M. ΒKlotho Is Required for Metabolic Activity of Fibroblast Growth Factor 21. Cell Biol. 2007, 104, 7432–7437. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, M.; Uehara, Y.; Motomura-Matsuzaka, K.; Oki, J.; Koyama, Y.; Kimura, M.; Asada, M.; Komi-Kuramochi, A.; Oka, S.; Toru, T. ΒKlotho Is Required for Fibroblast Growth Factor (FGF) 21 Signaling through FGF Receptor (FGFR) 1c and FGFR3c. Mol. Endocrinol. 2008, 22, 1006–1014. [Google Scholar] [CrossRef] [Green Version]
- Tacer, K.F.; Bookout, A.L.; Ding, X.; Kurosu, H.; John, G.B.; Wang, L.; Goetz, R.; Mohammadi, M.; Kuro-o, M.; Mangelsdorf, D.J.; et al. Research Resource: Comprehensive Expression Atlas of the Fibroblast Growth Factor System in Adult Mouse. Mol. Endocrinol. 2010, 24, 2050–2064. [Google Scholar] [CrossRef] [Green Version]
- Owen, B.M.; Mangelsdorf, D.J.; Kliewer, S.A. Tissue-Specific Actions of the Metabolic Hormones FGF15/19 and FGF21. Trends Endocrinol. Metab. 2015, 26, 22–29. [Google Scholar] [CrossRef] [Green Version]
- Bookout, A.L.; de Groot, M.H.M.; Owen, B.M.; Lee, S.; Gautron, L.; Lawrence, H.L.; Ding, X.; Elmquist, J.K.; Takahashi, J.S.; Mangelsdorf, D.J.; et al. FGF21 Regulates Metabolism and Circadian Behavior by Acting on the Nervous System. Nat. Med. 2013, 19, 1147–1152. [Google Scholar] [CrossRef]
- Liang, Q.; Zhong, L.; Zhang, J.; Wang, Y.; Bornstein, S.R.; Triggle, C.R.; Ding, H.; Lam, K.S.L.; Xu, A. FGF21 Maintains Glucose Homeostasis by Mediating the Cross Talk Between Liver and Brain During Prolonged Fasting. Diabetes 2014, 63, 4064–4075. [Google Scholar] [CrossRef] [Green Version]
- Hultman, K.; Scarlett, J.M.; Baquero, A.F.; Cornea, A.; Zhang, Y.; Salinas, C.B.G.; Brown, J.; Morton, G.J.; Whalen, E.J.; Grove, K.L.; et al. The Central Fibroblast Growth Factor Receptor/Beta Klotho System: Comprehensive Mapping in Mus Musculus and Comparisons to Nonhuman Primate and Human Samples Using an Automated in Situ Hybridization Platform. J. Comp. Neurol. 2019, 527, 2069–2085. [Google Scholar] [CrossRef]
- Flippo, K.H.; Jensen-Cody, S.O.; Claflin, K.E.; Potthoff, M.J. FGF21 Signaling in Glutamatergic Neurons Is Required for Weight Loss Associated with Dietary Protein Dilution. Sci. Rep. 2020, 10, 19521. [Google Scholar] [CrossRef]
- Recinella, L.; Leone, S.; Ferrante, C.; Chiavaroli, A.; Di Nisio, C.; Martinotti, S.; Vacca, M.; Brunetti, L.; Orlando, G. Effects of Central Fibroblast Growth Factor 21 (FGF21) in Energy Balance. J. Biol. Regul. Homeost. Agents 2017, 31, 603–613. [Google Scholar]
- Špolcová, A.; Holubová, M.; Mikulášková, B.; Nagelová, V.; Štofková, A.; Lacinová, Z.; Jurčovičová, J.; Haluzík, M.; Maletínská, L.; Železná, B. Changes in FGF21 Serum Concentrations and Liver MRNA Expression in an Experimental Model of Complete Lipodystrophy and Insulin-Resistant Diabetes. Physiol. Res. 2014, 63, 483–490. [Google Scholar] [CrossRef]
- Leng, G.; Sabatier, N. Oxytocin—The Sweet Hormone? Trends Endocrinol. Metab. 2017, 28, 365–376. [Google Scholar] [CrossRef] [Green Version]
- Sinclair, M.S.; Perea-Martinez, I.; Abouyared, M.; St. John, S.J.; Chaudhari, N. Oxytocin Decreases Sweet Taste Sensitivity in Mice. Physiol. Behav. 2015, 141, 103–110. [Google Scholar] [CrossRef] [Green Version]
- Palmer, A.A.; Sharpe, A.L.; Burkhart-Kasch, S.; McKinnon, C.S.; Coste, S.C.; Stenzel-Poore, M.P.; Phillips, T.J. Corticotropin-Releasing Factor Overexpression Decreases Ethanol Drinking and Increases Sensitivity to the Sedative Effects of Ethanol. Psychopharmacology 2004, 176, 386–397. [Google Scholar] [CrossRef]
- Yuan, Y.; Wu, W.; Chen, M.; Cai, F.; Fan, C.; Shen, W.; Sun, W.; Hu, J. Reward Inhibits Paraventricular CRH Neurons to Relieve Stress. Curr. Biol. 2019, 29, 1243–1251. [Google Scholar] [CrossRef] [Green Version]
- Matsui, S.; Sasaki, T.; Kohno, D.; Yaku, K.; Inutsuka, A.; Yokota-Hashimoto, H.; Kikuchi, O.; Suga, T.; Kobayashi, M.; Yamanaka, A.; et al. Neuronal SIRT1 Regulates Macronutrient-Based Diet Selection through FGF21 and Oxytocin Signalling in Mice. Nat. Commun. 2018, 9, 4604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wise, R.A. Role of Brain Dopamine in Food Reward and Reinforcement. Philos. Trans. R. Soc. B Biol. Sci. 2006, 361, 1149–1158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, O.; Iwai, Y.; Narukawa, M.; Ishikawa, A.W.; Ishii, K.K.; Murata, K.; Yoshimura, Y.; Touhara, K.; Misaka, T.; Minokoshi, Y.; et al. Hypothalamic Neuronal Circuits Regulating Hunger-Induced Taste Modification. Nat. Commun. 2019, 10, 4560. [Google Scholar] [CrossRef] [PubMed]
- Jastrzębska, K.; Walczak, M.; Cieślak, P.E.; Szumiec, Ł.; Turbasa, M.; Engblom, D.; Błasiak, T.; Parkitna, J.R. Loss of NMDA Receptors in Dopamine Neurons Leads to the Development of Affective Disorder-like Symptoms in Mice. Sci. Rep. 2016, 6, 37171. [Google Scholar] [CrossRef] [PubMed]
- Eisenhardt, M.; Leixner, S.; Luján, R.; Spanagel, R.; Bilbao, A. Glutamate Receptors within the Mesolimbic Dopamine System Mediate Alcohol Relapse Behavior. J. Neurosci. 2015, 35, 15523–15538. [Google Scholar] [CrossRef] [Green Version]
- Gremel, C.M.; Cunningham, C.L. Involvement of Amygdala Dopamine and Nucleus Accumbens NMDA Receptors in Ethanol-Seeking Behavior in Mice. Neuropsychopharmacology 2009, 34, 1443–1453. [Google Scholar] [CrossRef]
- Chaumontet, C.; Recio, I.; Fromentin, G.; Benoit, S.; Piedcoq, J.; Darcel, N.; Tomé, D. The Protein Status of Rats Affects the Rewarding Value of Meals Due to Their Protein Content. J. Nutr. 2018, 148, 989–998. [Google Scholar] [CrossRef]
- Fulton, S. Appetite and Reward. Front. Neuroendocrinol. 2010, 31, 85–103. [Google Scholar] [CrossRef]
- Liu, Q.; Tabuchi, M.; Liu, S.; Kodama, L.; Horiuchi, W.; Daniels, J.; Chiu, L.; Baldoni, D.; Wu, M.N. Branch-Specific Plasticity of a Bifunctional Dopamine Circuit Encodes Protein Hunger. Science 2017, 356, 534–539. [Google Scholar] [CrossRef] [Green Version]
- Vucetic, Z.; Totoki, K.; Schoch, H.; Whitaker, K.W.; Hill-Smith, T.; Lucki, I.; Reyes, T.M. Early Life Protein Restriction Alters Dopamine Circuitry. Neuroscience 2010, 168, 359–370. [Google Scholar] [CrossRef] [Green Version]
- Papageorgiou, G.K.; Baudonnat, M.; Cucca, F.; Walton, M.E. Mesolimbic Dopamine Encodes Prediction Errors in a State-Dependent Manner. Cell Rep. 2016, 15, 221–228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, A.H.; Aston-Jones, G. Circuit Projection from Suprachiasmatic Nucleus to Ventral Tegmental Area: A Novel Circadian Output Pathway. Eur. J. Neurosci. 2009, 29, 748–760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ulrich-Lai, Y.M.; Ryan, K.K. Neuroendocrine Circuits Governing Energy Balance and Stress Regulation: Functional Overlap and Therapeutic Implications. Cell Metab. 2014, 19, 910–925. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Könner, A.C.; Janoschek, R.; Plum, L.; Jordan, S.D.; Rother, E.; Ma, X.; Xu, C.; Enriori, P.; Hampel, B.; Barsh, G.S.; et al. Insulin Action in AgRP-Expressing Neurons Is Required for Suppression of Hepatic Glucose Production. Cell Metab. 2007, 5, 438–449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coppari, R.; Ichinose, M.; Lee, C.E.; Pullen, A.E.; Kenny, C.D.; McGovern, R.A.; Tang, V.; Liu, S.M.; Ludwig, T.; Chua, S.C.; et al. The Hypothalamic Arcuate Nucleus: A Key Site for Mediating Leptin’s Effects on Glucose Homeostasis and Locomotor Activity. Cell Metab. 2005, 1, 63–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asrih, M.; Veyrat-Durebex, C.; Poher, A.-L.; Lyautey, J.; Rohner-Jeanrenaud, F.; Jornayvaz, F.R. Leptin as a Potential Regulator of FGF21. Cell. Physiol. Biochem. 2016, 38, 1218–1225. [Google Scholar] [CrossRef]
- Burdakov, D.; Luckman, S.M.; Verkhratsky, A. Glucose-Sensing Neurons of the Hypothalamus. Philos. Trans. R. Soc. B Biol. Sci. 2005, 360, 2227–2235. [Google Scholar] [CrossRef] [Green Version]
- Koekkoek, L.L.; Mul, J.D.; la Fleur, S.E. Glucose-Sensing in the Reward System. Front. Neurosci. 2017, 11, 716. [Google Scholar] [CrossRef] [Green Version]
- Blouet, C.; Jo, Y.-H.; Li, X.; Schwartz, G.J. Mediobasal Hypothalamic Leucine Sensing Regulates Food Intake through Activation of a Hypothalamus-Brainstem Circuit. J. Neurosci. 2009, 29, 8302–8311. [Google Scholar] [CrossRef]
- Blouet, C.; Schwartz, G.J. Brainstem Nutrient Sensing in the Nucleus of the Solitary Tract Inhibits Feeding. Cell Metab. 2012, 16, 579–587. [Google Scholar] [CrossRef] [Green Version]
- Maurin, A.-C.; Benani, A.; Lorsignol, A.; Brenachot, X.; Parry, L.; Carraro, V.; Guissard, C.; Averous, J.; Jousse, C.; Bruhat, A.; et al. Hypothalamic EIF2α Signaling Regulates Food Intake. Cell Rep. 2014, 6, 438–444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tong, X.; Muchnik, M.; Chen, Z.; Patel, M.; Wu, N.; Joshi, S.; Rui, L.; Lazar, M.A.; Yin, L. Transcriptional Repressor E4-Binding Protein 4 (E4BP4) Regulates Metabolic Hormone Fibroblast Growth Factor 21 (FGF21) during Circadian Cycles and Feeding. J. Biol. Chem. 2010, 285, 36401–36409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, H.; Xia, F.; Lam, K.S.; Wang, Y.; Bao, Y.; Zhang, J.; Gu, Y.; Zhou, P.; Lu, J.; Jia, W.; et al. Circadian Rhythm of Circulating Fibroblast Growth Factor 21 Is Related to Diurnal Changes in Fatty Acids in Humans. Clin. Chem. 2011, 57, 691–700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leibowitz, S.F.; Alexander, J.T. Hypothalamic Serotonin in Control of Eating Behavior, Meal Size, and Body Weight. Biol. Psychiatry 1998, 44, 851–864. [Google Scholar] [CrossRef]
- Challet, E. The Circadian Regulation of Food Intake. Nat. Rev. Endocrinol. 2019, 15, 393–405. [Google Scholar] [CrossRef]
- Kliewer, S.A.; Mangelsdorf, D.J. A Dozen Years of Discovery: Insights into the Physiology and Pharmacology of FGF21. Cell Metab. 2019, 29, 246–253. [Google Scholar] [CrossRef] [Green Version]
- Shao, W.; Jin, T. Hepatic Hormone FGF21 and Its Analogues in Clinical Trials. Chronic Dis. Transl. Med. 2021, in press. [Google Scholar] [CrossRef]
- Talukdar, S.; Kharitonenkov, A. FGF19 and FGF21: In NASH We Trust. Mol. Metab. 2021, 46, 101152. [Google Scholar] [CrossRef]
- Zarei, M.; Pizarro-Delgado, J.; Barroso, E.; Palomer, X.; Vázquez-Carrera, M. Targeting FGF21 for the Treatment of Nonalcoholic Steatohepatitis. Trends Pharmacol. Sci. 2020, 41, 199–208. [Google Scholar] [CrossRef]
- Ferrer-Curriu, G.; Guitart-Mampel, M.; Rupérez, C.; Zamora, M.; Crispi, F.; Villarroya, F.; Fernández-Solà, J.; Garrabou, G.; Planavila, A. The Protective Effect of Fibroblast Growth Factor-21 in Alcoholic Cardiomyopathy: A Role in Protecting Cardiac Mitochondrial Function. J. Pathol. 2021, 253, 198–208. [Google Scholar] [CrossRef]
- Jung, H.-W.; Park, J.H.; Kim, D.A.; Jang, I.-Y.; Park, S.J.; Lee, J.Y.; Lee, S.; Kim, J.H.; Yi, H.-S.; Lee, E.; et al. Association between Serum FGF21 Level and Sarcopenia in Older Adults. Bone 2021, 145, 115877. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, C.-T.; Chaffin, A.T.; Ryan, K.K. Fibroblast Growth Factor 21 Facilitates the Homeostatic Control of Feeding Behavior. J. Clin. Med. 2022, 11, 580. https://doi.org/10.3390/jcm11030580
Wu C-T, Chaffin AT, Ryan KK. Fibroblast Growth Factor 21 Facilitates the Homeostatic Control of Feeding Behavior. Journal of Clinical Medicine. 2022; 11(3):580. https://doi.org/10.3390/jcm11030580
Chicago/Turabian StyleWu, Chih-Ting, Aki T. Chaffin, and Karen K. Ryan. 2022. "Fibroblast Growth Factor 21 Facilitates the Homeostatic Control of Feeding Behavior" Journal of Clinical Medicine 11, no. 3: 580. https://doi.org/10.3390/jcm11030580
APA StyleWu, C. -T., Chaffin, A. T., & Ryan, K. K. (2022). Fibroblast Growth Factor 21 Facilitates the Homeostatic Control of Feeding Behavior. Journal of Clinical Medicine, 11(3), 580. https://doi.org/10.3390/jcm11030580