
Mutational Landscapes of Normal Skin and Their Potential Implications in the Development of Skin Cancer: A Comprehensive Narrative Review
Abstract
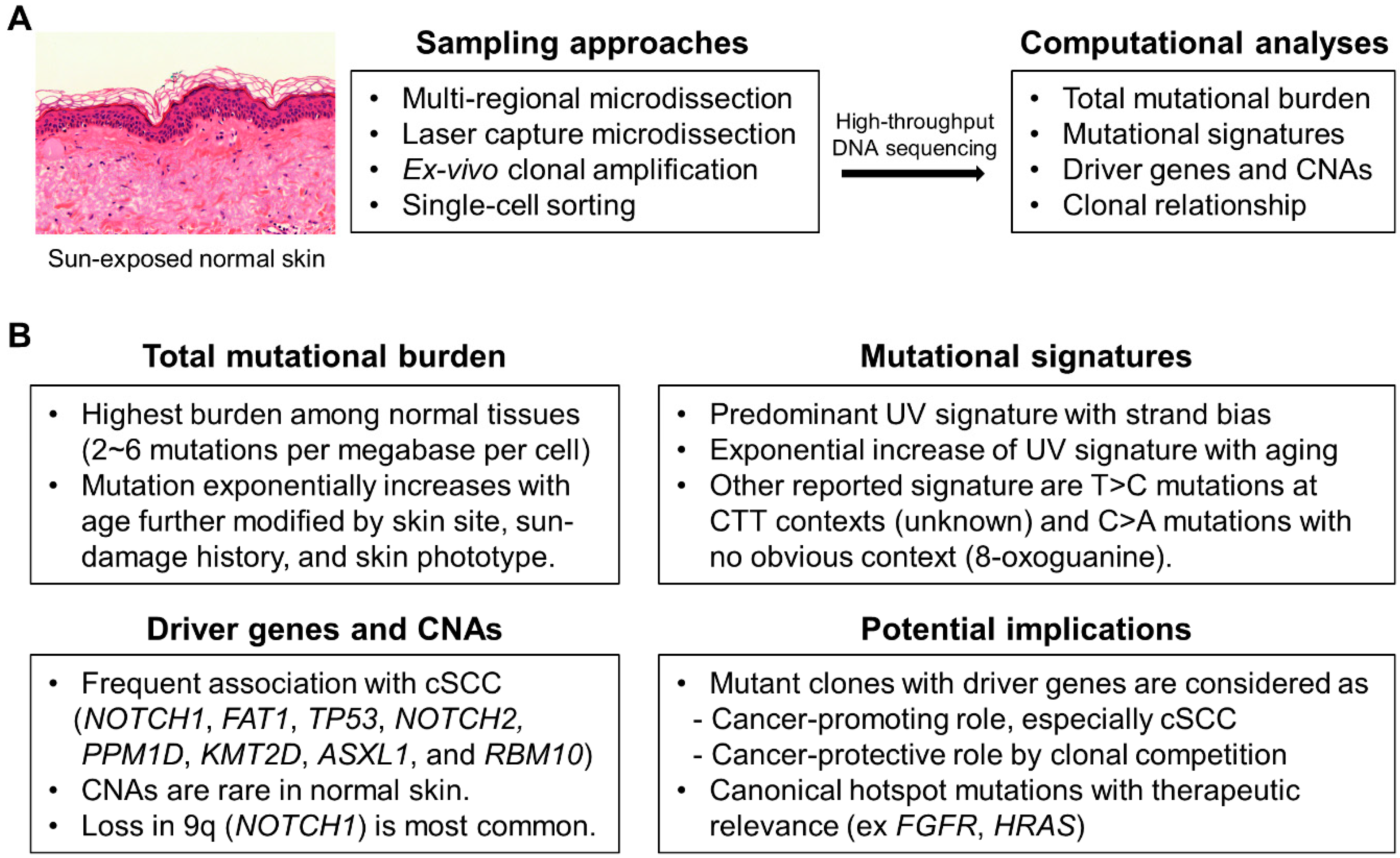
:1. Background
2. Mutational Landscapes of Normal Skin
3. Driver Genes under Positive Selection
4. Potential Implications of Driver Genes
5. Canonical Hotspot Mutations
6. Mutational Signatures
7. CNAs
8. Special Considerations for Genome Study of Normal Skin
9. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
References
- Kakiuchi, N.; Ogawa, S. Clonal expansion in non-cancer tissues. Nat. Rev. Cancer 2021, 21, 239–256. [Google Scholar] [CrossRef] [PubMed]
- Fiala, C.; Diamandis, E.P. Mutations in normal tissues—Some diagnostic and clinical implications. BMC Med. 2020, 18, 283. [Google Scholar] [CrossRef] [PubMed]
- Acha-Sagredo, A.; Ganguli, P.; Ciccarelli, F.D. Somatic variation in normal tissues: Friend or foe of cancer early detection? Ann. Oncol. 2022, 33, 1239–1249. [Google Scholar] [CrossRef]
- Martincorena, I. Somatic mutation and clonal expansions in human tissues. Genome. Med. 2019, 11, 35. [Google Scholar] [CrossRef] [PubMed]
- ICGC/TCGA Pan-Cancer Analysis of Whole Genomes Consortium. Pan-cancer analysis of whole genomes. Nature 2020, 578, 82–93. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Research, N.; Weinstein, J.N.; Collisson, E.A.; Mills, G.B.; Shaw, K.R.; Ozenberger, B.A.; Ellrott, K.; Shmulevich, I.; Sander, C.; Stuart, J.M. The Cancer Genome Atlas Pan-Cancer analysis project. Nat. Genet. 2013, 45, 1113–1120. [Google Scholar] [CrossRef]
- Lee-Six, H.; Olafsson, S.; Ellis, P.; Osborne, R.J.; Sanders, M.A.; Moore, L.; Georgakopoulos, N.; Torrente, F.; Noorani, A.; Goddard, M.; et al. The landscape of somatic mutation in normal colorectal epithelial cells. Nature 2019, 574, 532–537. [Google Scholar] [CrossRef]
- Moore, L.; Leongamornlert, D.; Coorens, T.H.H.; Sanders, M.A.; Ellis, P.; Dentro, S.C.; Dawson, K.J.; Butler, T.; Rahbari, R.; Mitchell, T.J.; et al. The mutational landscape of normal human endometrial epithelium. Nature 2020, 580, 640–646. [Google Scholar] [CrossRef]
- Yokoyama, A.; Kakiuchi, N.; Yoshizato, T.; Nannya, Y.; Suzuki, H.; Takeuchi, Y.; Shiozawa, Y.; Sato, Y.; Aoki, K.; Kim, S.K.; et al. Age-related remodelling of oesophageal epithelia by mutated cancer drivers. Nature 2019, 565, 312–317. [Google Scholar] [CrossRef]
- Brunner, S.F.; Roberts, N.D.; Wylie, L.A.; Moore, L.; Aitken, S.J.; Davies, S.E.; Sanders, M.A.; Ellis, P.; Alder, C.; Hooks, Y.; et al. Somatic mutations and clonal dynamics in healthy and cirrhotic human liver. Nature 2019, 574, 538–542. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, K.; Gowers, K.H.C.; Lee-Six, H.; Chandrasekharan, D.P.; Coorens, T.; Maughan, E.F.; Beal, K.; Menzies, A.; Millar, F.R.; Anderson, E.; et al. Tobacco smoking and somatic mutations in human bronchial epithelium. Nature 2020, 578, 266–272. [Google Scholar] [CrossRef] [PubMed]
- Lawson, A.R.J.; Abascal, F.; Coorens, T.H.H.; Hooks, Y.; O’Neill, L.; Latimer, C.; Raine, K.; Sanders, M.A.; Warren, A.Y.; Mahbubani, K.T.A.; et al. Extensive heterogeneity in somatic mutation and selection in the human bladder. Science 2020, 370, 75–82. [Google Scholar] [CrossRef] [PubMed]
- Colom, B.; Herms, A.; Hall, M.W.J.; Dentro, S.C.; King, C.; Sood, R.K.; Alcolea, M.P.; Piedrafita, G.; Fernandez-Antoran, D.; Ong, S.H.; et al. Mutant clones in normal epithelium outcompete and eliminate emerging tumours. Nature 2021, 598, 510–514. [Google Scholar] [CrossRef]
- Olafsson, S.; McIntyre, R.E.; Coorens, T.; Butler, T.; Jung, H.; Robinson, P.S.; Lee-Six, H.; Sanders, M.A.; Arestang, K.; Dawson, C.; et al. Somatic Evolution in Non-neoplastic IBD-Affected Colon. Cell 2020, 182, 672–684.e11. [Google Scholar] [CrossRef]
- Tall, A.R.; Fuster, J.J. Clonal hematopoiesis in cardiovascular disease and therapeutic implications. Nat. Cardiovasc. Res. 2022, 1, 116–124. [Google Scholar] [CrossRef]
- Vijg, J.; Dong, X. Pathogenic Mechanisms of Somatic Mutation and Genome Mosaicism in Aging. Cell 2020, 182, 12–23. [Google Scholar] [CrossRef]
- Martincorena, I.; Roshan, A.; Gerstung, M.; Ellis, P.; Van Loo, P.; McLaren, S.; Wedge, D.C.; Fullam, A.; Alexandrov, L.B.; Tubio, J.M.; et al. Tumor evolution. High burden and pervasive positive selection of somatic mutations in normal human skin. Science 2015, 348, 880–886. [Google Scholar] [CrossRef]
- Hernando, B.; Dietzen, M.; Parra, G.; Gil-Barrachina, M.; Pitarch, G.; Mahiques, L.; Valcuende-Cavero, F.; McGranahan, N.; Martinez-Cadenas, C. The effect of age on the acquisition and selection of cancer driver mutations in sun-exposed normal skin. Ann. Oncol. 2021, 32, 412–421. [Google Scholar] [CrossRef]
- Fowler, J.C.; King, C.; Bryant, C.; Hall, M.W.J.; Sood, R.; Ong, S.H.; Earp, E.; Fernandez-Antoran, D.; Koeppel, J.; Dentro, S.C.; et al. Selection of Oncogenic Mutant Clones in Normal Human Skin Varies with Body Site. Cancer Discov. 2021, 11, 340–361. [Google Scholar] [CrossRef] [PubMed]
- Wei, L.; Christensen, S.R.; Fitzgerald, M.E.; Graham, J.; Hutson, N.D.; Zhang, C.; Huang, Z.; Hu, Q.; Zhan, F.; Xie, J.; et al. Ultradeep sequencing differentiates patterns of skin clonal mutations associated with sun-exposure status and skin cancer burden. Sci. Adv. 2021, 7, eabd7703. [Google Scholar] [CrossRef]
- Yizhak, K.; Aguet, F.; Kim, J.; Hess, J.M.; Kubler, K.; Grimsby, J.; Frazer, R.; Zhang, H.; Haradhvala, N.J.; Rosebrock, D.; et al. RNA sequence analysis reveals macroscopic somatic clonal expansion across normal tissues. Science 2019, 364, eaaw0726. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.; Fewings, E.; Chang, D.; Zeng, H.; Liu, S.; Jorapur, A.; Belote, R.L.; McNeal, A.S.; Tan, T.M.; Yeh, I.; et al. The genomic landscapes of individual melanocytes from human skin. Nature 2020, 586, 600–605. [Google Scholar] [CrossRef]
- Tokez, S.; Hollestein, L.; Louwman, M.; Nijsten, T.; Wakkee, M. Incidence of Multiple vs First Cutaneous Squamous Cell Carcinoma on a Nationwide Scale and Estimation of Future Incidences of Cutaneous Squamous Cell Carcinoma. JAMA Dermatol. 2020, 156, 1300–1306. [Google Scholar] [CrossRef] [PubMed]
- Ratushny, V.; Gober, M.D.; Hick, R.; Ridky, T.W.; Seykora, J.T. From keratinocyte to cancer: The pathogenesis and modeling of cutaneous squamous cell carcinoma. J. Clin. Invest 2012, 122, 464–472. [Google Scholar] [CrossRef] [PubMed]
- Ahmady, S.; Jansen, M.H.E.; Nelemans, P.J.; Kessels, J.; Arits, A.; de Rooij, M.J.M.; Essers, B.A.B.; Quaedvlieg, P.J.F.; Kelleners-Smeets, N.W.J.; Mosterd, K. Risk of Invasive Cutaneous Squamous Cell Carcinoma After Different Treatments for Actinic Keratosis: A Secondary Analysis of a Randomized Clinical Trial. JAMA Dermatol. 2022, 158, 634–640. [Google Scholar] [CrossRef] [PubMed]
- Saini, N.; Roberts, S.A.; Klimczak, L.J.; Chan, K.; Grimm, S.A.; Dai, S.; Fargo, D.C.; Boyer, J.C.; Kaufmann, W.K.; Taylor, J.A.; et al. The Impact of Environmental and Endogenous Damage on Somatic Mutation Load in Human Skin Fibroblasts. PLoS Genet. 2016, 12, e1006385. [Google Scholar] [CrossRef]
- Saini, N.; Giacobone, C.K.; Klimczak, L.J.; Papas, B.N.; Burkholder, A.B.; Li, J.L.; Fargo, D.C.; Bai, R.; Gerrish, K.; Innes, C.L.; et al. UV-exposure, endogenous DNA damage, and DNA replication errors shape the spectra of genome changes in human skin. PLoS Genet. 2021, 17, e1009302. [Google Scholar] [CrossRef] [PubMed]
- Ziegler, A.; Jonason, A.S.; Leffellt, D.J.; Simon, J.A.; Sharma, H.W.; Kimmelman, J.; Remington, L.; Jacks, T.; Brash, D.E. Sunburn and p53 in the onset of skin cancer. Nature 1994, 372, 773–776. [Google Scholar] [CrossRef]
- Jonason, A.S.; Kunala, S.; Price, G.J.; Restifo, R.J.; Spinelli, H.M.; Persing, J.A.; Leffell, D.J.; Tarone, R.E.; Brash, D.E. Frequent clones of p53-mutated keratinocytes in normal human skin. Proc. Natl. Acad. Sci. USA 1996, 93, 14025–14029. [Google Scholar] [CrossRef]
- Ståhl, P.L.; Stranneheim, H.; Asplund, A.; Berglund, L.; Pontén, F.; Lundeberg, J. Sun-Induced Nonsynonymous p53 Mutations Are Extensively Accumulated and Tolerated in Normal Appearing Human Skin. J. Investig. Dermatol. 2011, 131, 504–508. [Google Scholar] [CrossRef]
- Martincorena, I.; Raine, K.M.; Gerstung, M.; Dawson, K.J.; Haase, K.; Van Loo, P.; Davies, H.; Stratton, M.R.; Campbell, P.J. Universal Patterns of Selection in Cancer and Somatic Tissues. Cell 2017, 171, 1029–1041.e21. [Google Scholar] [CrossRef]
- Wijewardhane, N.; Dressler, L.; Ciccarelli, F.D. Normal Somatic Mutations in Cancer Transformation. Cancer Cell 2021, 39, 125–129. [Google Scholar] [CrossRef]
- Kim, Y.S.; Bang, C.H.; Chung, Y.J. Mutational Landscape of Normal Human Skin: Clues to Understanding Early-Stage Carcinogenesis in Keratinocyte Neoplasia. J. Invest Dermatol. 2023, 143, 1187–1196.e9. [Google Scholar] [CrossRef] [PubMed]
- Colom, B.; Alcolea, M.P.; Piedrafita, G.; Hall, M.W.J.; Wabik, A.; Dentro, S.C.; Fowler, J.C.; Herms, A.; King, C.; Ong, S.H.; et al. Spatial competition shapes the dynamic mutational landscape of normal esophageal epithelium. Nat. Genet. 2020, 52, 604–614. [Google Scholar] [CrossRef]
- Hall, M.W.J.; Jones, P.H.; Hall, B.A. Relating evolutionary selection and mutant clonal dynamics in normal epithelia. J. R. Soc. Interface 2019, 16, 20190230. [Google Scholar] [CrossRef]
- Simons, B.D. Deep sequencing as a probe of normal stem cell fate and preneoplasia in human epidermis. Proc. Natl. Acad. Sci. USA 2016, 113, 128–133. [Google Scholar] [CrossRef] [PubMed]
- Murai, K.; Skrupskelyte, G.; Piedrafita, G.; Hall, M.; Kostiou, V.; Ong, S.H.; Nagy, T.; Cagan, A.; Goulding, D.; Klein, A.M.; et al. Epidermal Tissue Adapts to Restrain Progenitors Carrying Clonal p53 Mutations. Cell Stem. Cell 2018, 23, 687–699.e8. [Google Scholar] [CrossRef] [PubMed]
- Murai, K.; Dentro, S.; Ong, S.H.; Sood, R.; Fernandez-Antoran, D.; Herms, A.; Kostiou, V.; Abnizova, I.; Hall, B.A.; Gerstung, M.; et al. p53 mutation in normal esophagus promotes multiple stages of carcinogenesis but is constrained by clonal competition. Nat. Commun. 2022, 13, 6206. [Google Scholar] [CrossRef]
- Weiss, J.M.; Hunter, M.V.; Cruz, N.M.; Baggiolini, A.; Tagore, M.; Ma, Y.; Misale, S.; Marasco, M.; Simon-Vermot, T.; Campbell, N.R.; et al. Anatomic position determines oncogenic specificity in melanoma. Nature 2022, 604, 354–361. [Google Scholar] [CrossRef]
- Venkatachalam, A.; Pikarsky, E.; Ben-Neriah, Y. Putative homeostatic role of cancer driver mutations. Trends Cell Biol. 2022, 32, 8–17. [Google Scholar] [CrossRef]
- Abby, E.; Dentro, S.C.; Hall, M.W.J.; Fowler, J.C.; Ong, S.H.; Sood, R.; Herms, A.; Piedrafita, G.; Abnizova, I.; Siebel, C.W.; et al. Notch1 mutations drive clonal expansion in normal esophageal epithelium but impair tumor growth. Nat. Genet. 2023, 55, 232–245. [Google Scholar] [CrossRef]
- Chang, M.T.; Bhattarai, T.S.; Schram, A.M.; Bielski, C.M.; Donoghue, M.T.A.; Jonsson, P.; Chakravarty, D.; Phillips, S.; Kandoth, C.; Penson, A.; et al. Accelerating Discovery of Functional Mutant Alleles in Cancer. Cancer Discov. 2018, 8, 174–183. [Google Scholar] [CrossRef] [PubMed]
- Gorbunova, V.; Seluanov, A.; Mao, Z.; Hine, C. Changes in DNA repair during aging. Nucleic Acids Res. 2007, 35, 7466–7474. [Google Scholar] [CrossRef] [PubMed]
- Brash, D.E. UV signature mutations. Photochem. Photobiol. 2015, 91, 15–26. [Google Scholar] [CrossRef] [PubMed]
- Besaratinia, A.; Kim, S.I.; Bates, S.E.; Pfeifer, G.P. Riboflavin activated by ultraviolet A1 irradiation induces oxidative DNA damage-mediated mutations inhibited by vitamin C. Proc. Natl. Acad. Sci. USA 2007, 104, 5953–5958. [Google Scholar] [CrossRef]
- Alexandrov, L.B.; Kim, J.; Haradhvala, N.J.; Huang, M.N.; Tian Ng, A.W.; Wu, Y.; Boot, A.; Covington, K.R.; Gordenin, D.A.; Bergstrom, E.N.; et al. The repertoire of mutational signatures in human cancer. Nature 2020, 578, 94–101. [Google Scholar] [CrossRef] [PubMed]
- Speer, R.M.; Nandi, S.P.; Cooper, K.L.; Zhou, X.; Yu, H.; Guo, Y.; Hudson, L.G.; Alexandrov, L.B.; Liu, K.J. Arsenic is a potent co-mutagen of ultraviolet light. Commun Biol. 2023, 6, 1273. [Google Scholar] [CrossRef] [PubMed]
- Inman, G.J.; Wang, J.; Nagano, A.; Alexandrov, L.B.; Purdie, K.J.; Taylor, R.G.; Sherwood, V.; Thomson, J.; Hogan, S.; Spender, L.C.; et al. The genomic landscape of cutaneous SCC reveals drivers and a novel azathioprine associated mutational signature. Nat. Commun. 2018, 9, 3667. [Google Scholar] [CrossRef]
- Gumaste, P.V.; Penn, L.A.; Cymerman, R.M.; Kirchhoff, T.; Polsky, D.; McLellan, B. Skin cancer risk in BRCA1/2 mutation carriers. Br. J. Dermatol. 2015, 172, 1498–1506. [Google Scholar] [CrossRef]
- Buhigas, C.; Warren, A.Y.; Leung, W.K.; Whitaker, H.C.; Luxton, H.J.; Hawkins, S.; Kay, J.; Butler, A.; Xu, Y.; Woodcock, D.J.; et al. The architecture of clonal expansions in morphologically normal tissue from cancerous and non-cancerous prostates. Mol. Cancer 2022, 21, 183. [Google Scholar] [CrossRef]
- Schneider, M.P.; Cullen, A.E.; Pangonyte, J.; Skelton, J.; Major, H.; Van Oudenhove, E.; Garcia, M.J.; Chaves Urbano, B.; Piskorz, A.M.; Brenton, J.D.; et al. scAbsolute: Measuring single-cell ploidy and replication status. Genome Biol. 2024, 25, 62. [Google Scholar] [CrossRef]
- Qin, F.; Cai, G.; Amos, C.I.; Xiao, F. A statistical learning method for simultaneous copy number estimation and subclone clustering with single-cell sequencing data. Genome Res. 2024, 34, 85–93. [Google Scholar] [CrossRef] [PubMed]
- Knouse, K.A.; Wu, J.; Amon, A. Assessment of megabase-scale somatic copy number variation using single-cell sequencing. Genome Res. 2016, 26, 376–384. [Google Scholar] [CrossRef]
- Sun, C.; Kathuria, K.; Emery, S.B.; Kim, B.; Burbulis, I.E.; Shin, J.H.; Weinberger, D.R.; Moran, J.V.; Kidd, J.M.; Mills, R.E.; et al. Mapping recurrent mosaic copy number variation in human neurons. Nat. Commun. 2024, 15, 4220. [Google Scholar] [CrossRef] [PubMed]
- Abyzov, A.; Mariani, J.; Palejev, D.; Zhang, Y.; Haney, M.S.; Tomasini, L.; Ferrandino, A.F.; Rosenberg Belmaker, L.A.; Szekely, A.; Wilson, M.; et al. Somatic copy number mosaicism in human skin revealed by induced pluripotent stem cells. Nature 2012, 492, 438–442. [Google Scholar] [CrossRef]
- Knouse, K.A.; Wu, J.; Whittaker, C.A.; Amon, A. Single cell sequencing reveals low levels of aneuploidy across mammalian tissues. Proc. Natl. Acad. Sci. USA 2014, 111, 13409–13414. [Google Scholar] [CrossRef] [PubMed]
- Pei, X.M.; Yeung, M.H.Y.; Wong, A.N.N.; Tsang, H.F.; Yu, A.C.S.; Yim, A.K.Y.; Wong, S.C.C. Targeted Sequencing Approach and Its Clinical Applications for the Molecular Diagnosis of Human Diseases. Cells 2023, 12, 493. [Google Scholar] [CrossRef] [PubMed]
- Koboldt, D.C. Best practices for variant calling in clinical sequencing. Genome Med. 2020, 12, 91. [Google Scholar] [CrossRef]
- Li, R.; Di, L.; Li, J.; Fan, W.; Liu, Y.; Guo, W.; Liu, W.; Liu, L.; Li, Q.; Chen, L.; et al. A body map of somatic mutagenesis in morphologically normal human tissues. Nature 2021, 597, 398–403. [Google Scholar] [CrossRef] [PubMed]
- Seferbekova, Z.; Lomakin, A.; Yates, L.R.; Gerstung, M. Spatial biology of cancer evolution. Nat. Rev. Genet. 2022, 24, 295–313. [Google Scholar] [CrossRef]

{kind=link}
| Ref (Year) | Patient (n) | Study Methods (Sample (n)) | Main Findings |
|---|---|---|---|
| [17] (2015) | 4 | TDS of small biopsies of eyelid skin (0.8 to 4.7 mm2) (n = 234) | Average mutation burden of 2-6/Mb per cell and the major mutational signature of UV Significantly mutated genes of NOTCH1-3, FAT1, TP53, and RBM10. Positively selected mutations in 18–32% of normal skin cells: ~140 cancer drivers per cm2 NOTCH1 is the most frequent gene with CNAs (27/234) |
| [22] (2020) | 6 | TDS of flow-sorted melanocytes after tissue culture of skin biopsy (n = 159) | Mutation burden is higher in intermittently sun-exposed skin than in chronically sun-exposed skin. Mutation burden is higher in melanocytes adjacent to a skin cancer than in cancer-free donors. Melanocytes from normal skin commonly contained pathogenic (weakly oncogenic) mutations. Phylogenetic analyses identified groups of clonally related melanocytes in the same skin regions. |
| [19] (2021) | 35 | TDS of small biopsies of normal skin (2 mm2) (n = 1261) | Most mutations were caused by UV, but there are differences in DNA-repair processes between sites. TP53 preferentially positively selected in the head and FAT1 in the leg. Fine-scale mapping revealed 10% of clones had CNAs. Mutations in the upper hair follicle resembled adjacent skin, but the lower follicle did not. |
| [18] (2021) | 123 | TDS of small biopsies of normal skin (n = 123) | Exponential accumulation of UV-related somatic mutations with age, matching skin cancer incidence. The increase of mutational burden is in turn modified by an individual’s skin phototype. Somatic mutations preferentially accumulated in cSCC cancer genes and clonally expanded with age. Mutational signature analyses suggest a loss of fidelity in transcription-coupled repair later in life. |
| [20] (2021) | 13 | TDS of small biopsies of normal skin from postmortem donors (n = 450) | Mutation burden and driver genes were significantly different between SE and NE areas. Hotspot mutations in TP53, NOTCH1, and GRM3 significantly associated with UV exposure. Mutational counts in cSCC were associated with the UV-induced mutation counts in skin with the difference mostly conferred by the low-frequency mutations. |
| [27] (2021) | WGS of single-cell derived clonal lineages of fibroblasts and melanocytes (n = 21) | Each skin cell carries from 402 to 14,029 base substitutions, 7 to 71 indels and 1 to 14 structural variants per cell including known cancer drivers. UV-induced mutations were prominent in many samples even in sun-shielded skin, and were age-independent. Spontaneous deamination of methylated cytosines and indels characteristic of DNA replication errors were also found with a linear increase with age. | |
| [31] (2023) | 39 | WES of small biopsies of normal skin (9 mm2) (n = 39) | Median mutation burden was higher in exposed skin (10.4/Mb) than non-exposed skin (0.25/Mb). Significantly mutated genes of NOTCH1, FAT1, TP53, PPM1D, KMT2D, and ASXL1. Single mutational signature in normal skin with components of UV and aging. Rare instances of copy-neutral loss of heterozygosity in 9q (n = 2) and 6q (n = 1). |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Riew, T.-R.; Kim, Y.-S. Mutational Landscapes of Normal Skin and Their Potential Implications in the Development of Skin Cancer: A Comprehensive Narrative Review. J. Clin. Med. 2024, 13, 4815. https://doi.org/10.3390/jcm13164815
Riew T-R, Kim Y-S. Mutational Landscapes of Normal Skin and Their Potential Implications in the Development of Skin Cancer: A Comprehensive Narrative Review. Journal of Clinical Medicine. 2024; 13(16):4815. https://doi.org/10.3390/jcm13164815
Chicago/Turabian StyleRiew, Tae-Ryong, and Yoon-Seob Kim. 2024. "Mutational Landscapes of Normal Skin and Their Potential Implications in the Development of Skin Cancer: A Comprehensive Narrative Review" Journal of Clinical Medicine 13, no. 16: 4815. https://doi.org/10.3390/jcm13164815