
General Treatment and Ophthalmic Management of Peters’ Anomaly

Abstract
1. Introduction
2. Etiology
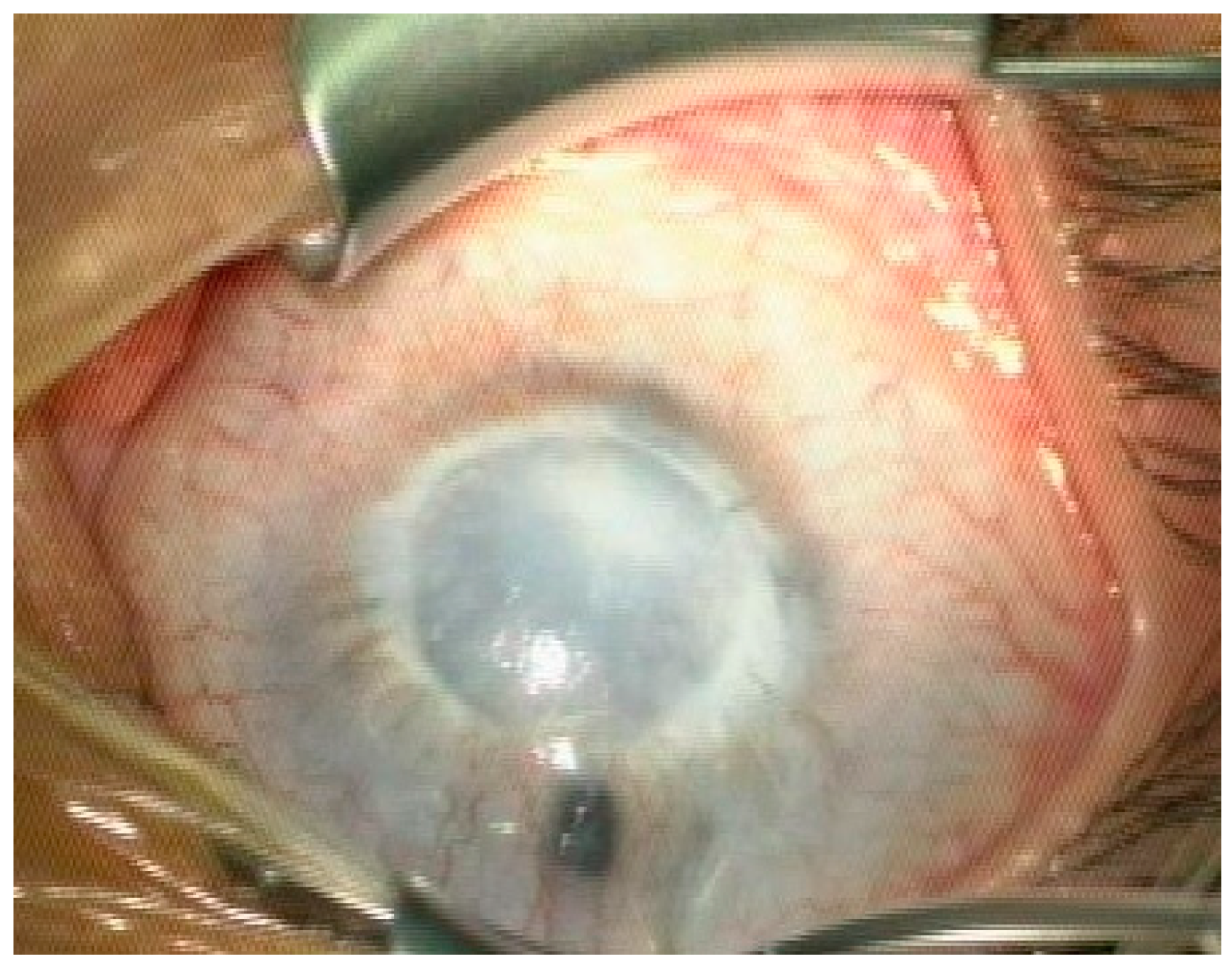
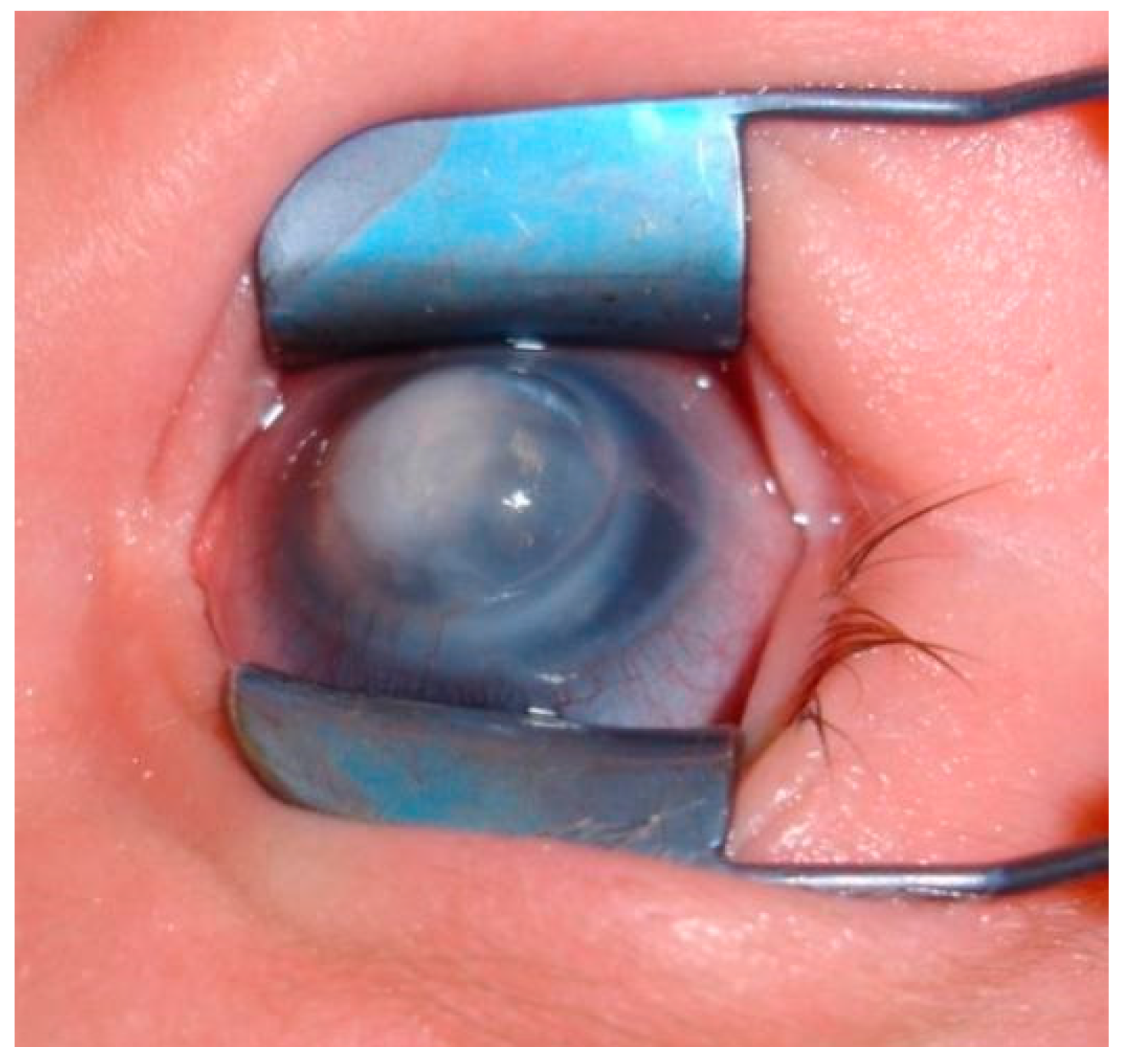
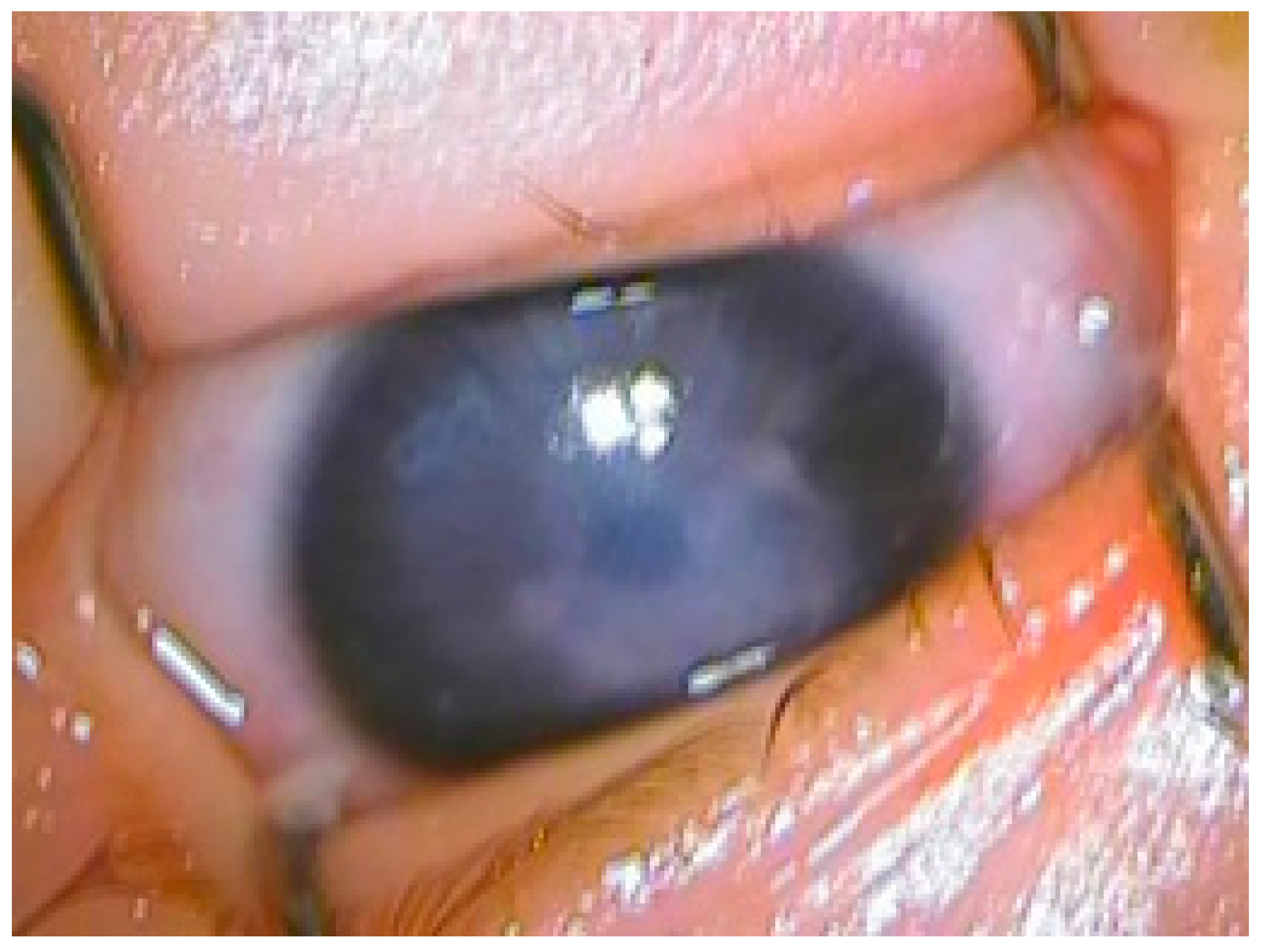
3. Clinical Findings
4. Examination and Diagnosis
5. Treatment
5.1. Penetrating Keratoplasty (PK)
5.2. Rehabilitation
5.3. Timing of Surgery
6. Results
7. Reasons of Graft Failure
8. Alternatives to Penetrating Keratoplasty
Partial Corneal Opacity or Disqualification from PK
9. Pediatric Keratoprosthesis
10. Summary
11. Methods of Literature Research
Author Contributions
Funding
Conflicts of Interest
References
- Kurilec, J.M.; Zaidman, G.W. Incidence of Peters Anomaly and Congenital Corneal Opacities Interfering with Vision in the United States. Cornea 2014, 33, 848–850. [Google Scholar] [CrossRef] [PubMed]
- Spierer, O.; Cavuoto, K.M.; Suwannaraj, S.; McKeown, C.A.; Chang, T.C. Outcome of Optical Iridectomy in Peters Anomaly. Graefes Arch. Clin. Exp. Ophthalmol. 2018, 256, 1679–1683. [Google Scholar] [CrossRef] [PubMed]
- Bhandari, R.; Ferri, S.; Whittaker, B.; Liu, M.; Lazzaro, D.R. Peters Anomaly: Review of the Literature. Cornea 2011, 30, 939–944. [Google Scholar] [CrossRef] [PubMed]
- Shigeyasu, C.; Yamada, M.; Mizuno, Y.; Yokoi, T.; Nishina, S.; Azuma, N. Clinical Features of Anterior Segment Dysgenesis Associated with Congenital Corneal Opacities. Cornea 2012, 31, 293–298. [Google Scholar] [CrossRef] [PubMed]
- Cruysberg, J.R.M. Misspelling of Peters Anomaly. Am. J. Ophthalmol. 2003, 135, 260. [Google Scholar] [CrossRef]
- Peters, A. Ueber Angeborene Defektbildung der Descemetschen Membran. Klin. Mbl. Augenheilkd. 1906, 44, 27–40. [Google Scholar]
- Bermejo, E.; Martínez-Frías, M.L. Congenital Eye Malformations: Clinical-Epidemiological Analysis of 1,124,654 Consecutive Births in Spain. Am. J. Med. Genet. 1998, 75, 497–504. [Google Scholar] [CrossRef]
- Huang, C.; O’Hara, M.; Mannis, M.J. Primary Pediatric Keratoplasty: Indications and Outcomes. Cornea 2009, 28, 1003–1008. [Google Scholar] [CrossRef]
- Rezende, R.A.; Uchoa, U.B.C.; Uchoa, R.; Rapuano, C.J.; Laibson, P.R.; Cohen, E.J. Congenital Corneal Opacities in a Cornea Referral Practice. Cornea 2004, 23, 565–570. [Google Scholar] [CrossRef]
- Vanathi, M.; Panda, A.; Vengayil, S.; Chaudhuri, Z.; Dada, T. Pediatric Keratoplasty. Surv. Ophthalmol. 2009, 54, 245–271. [Google Scholar] [CrossRef]
- Lowe, M.T.; Keane, M.C.; Coster, D.J.; Williams, K.A. The Outcome of Corneal Transplantation in Infants, Children, and Adolescents. Ophthalmology 2011, 118, 492–497. [Google Scholar] [CrossRef] [PubMed]
- Nischal, K.K. A New Approach to the Classification of Neonatal Corneal Opacities. Curr. Opin. Ophthalmol. 2012, 23, 344–354. [Google Scholar] [CrossRef] [PubMed]
- Mataftsi, A.; Islam, L.; Kelberman, D.; Sowden, J.C.; Nischal, K.K. Chromosome Abnormalities and the Genetics of Congenital Corneal Opacification. Mol. Vis. 2011, 17, 1624–1640. [Google Scholar] [PubMed]
- Weh, E.; Reis, L.M.; Happ, H.C.; Levin, A.V.; Wheeler, P.G.; David, K.L.; Carney, E.; Angle, B.; Hauser, N.; Semina, E.V. Whole Exome Sequence Analysis of Peters Anomaly. Hum. Genet. 2014, 133, 1497–1511. [Google Scholar] [CrossRef] [PubMed]
- Reis, L.M.; Houssin, N.S.; Zamora, C.; Abdul-Rahman, O.; Kalish, J.M.; Zackai, E.H.; Plageman, T.F.; Semina, E.V. Novel Variants in CDH2 Are Associated with a New Syndrome Including Peters Anomaly. Clin. Genet. 2020, 97, 502–508. [Google Scholar] [CrossRef]
- Zaidman, G.W.; Flanagan, J.K.; Furey, C.C. Long-Term Visual Prognosis in Children after Corneal Transplant Surgery for Peters Anomaly Type I. Am. J. Ophthalmol. 2007, 144, 104–108. [Google Scholar] [CrossRef]
- Traboulsi, E.I.; Maumenee, I.H. Peters’ Anomaly and Associated Congenital Malformations. Arch. Ophthalmol. 1992, 110, 1739–1742. [Google Scholar] [CrossRef]
- Ozeki, H.; Shirai, S.; Nozaki, M.; Sakurai, E.; Mizuno, S.; Ashikari, M.; Matsunaga, N.; Ogura, Y. Ocular and Systemic Features of Peters’ Anomaly. Graefes Arch. Clin. Exp. Ophthalmol. 2000, 238, 833–839. [Google Scholar] [CrossRef]
- Chang, J.W.; Kim, J.H.; Kim, S.-J.; Yu, Y.S. Long-Term Clinical Course and Visual Outcome Associated with Peters’ Anomaly. Eye 2012, 26, 1237–1242. [Google Scholar] [CrossRef]
- Rao, K.V.; Fernandes, M.; Gangopadhyay, N.; Vemuganti, G.K.; Krishnaiah, S.; Sangwan, V.S. Outcome of Penetrating Keratoplasty for Peters Anomaly. Cornea 2008, 27, 749–753. [Google Scholar] [CrossRef]
- Neilan, E.; Pikman, Y.; Kimonis, V.E. Peters Anomaly in Association with Multiple Midline Anomalies and a Familial Chromosome 4 Inversion. Ophthalmic Genet. 2006, 27, 63–65. [Google Scholar] [CrossRef]
- Cook, C.S.; Sulik, K.K. Keratolenticular Dysgenesis (Peters’ Anomaly) as a Result of Acute Embryonic Insult during Gastrulation. J. Pediatr. Ophthalmol. Strabismus 1988, 25, 60–66. [Google Scholar] [CrossRef] [PubMed]
- Cook, C.S.; Nowotny, A.Z.; Sulik, K.K. Fetal Alcohol Syndrome. Eye Malformations in a Mouse Model. Arch. Ophthalmol. 1987, 105, 1576–1581. [Google Scholar] [CrossRef] [PubMed]
- Majander, A.S.; Lindahl, P.M.; Vasara, L.K.; Krootila, K. Anterior Segment Optical Coherence Tomography in Congenital Corneal Opacities. Ophthalmology 2012, 119, 2450–2457. [Google Scholar] [CrossRef] [PubMed]
- Nischal, K.K.; Naor, J.; Jay, V.; MacKeen, L.D.; Rootman, D.S. Clinicopathological Correlation of Congenital Corneal Opacification Using Ultrasound Biomicroscopy. Br. J. Ophthalmol. 2002, 86, 62–69. [Google Scholar] [CrossRef] [PubMed]
- Hong, J.; Yang, Y.; Cursiefen, C.; Mashaghi, A.; Wu, D.; Liu, Z.; Sun, X.; Dana, R.; Xu, J. Optimising Keratoplasty for Peters’ Anomaly in Infants Using Spectral-Domain Optical Coherence Tomography. Br. J. Ophthalmol. 2017, 101, 820–827. [Google Scholar] [CrossRef] [PubMed]
- Najjar, D.M.; Christiansen, S.P.; Bothun, E.D.; Summers, C.G. Strabismus and Amblyopia in Bilateral Peters Anomaly. J. AAPOS 2006, 10, 193–197. [Google Scholar] [CrossRef]
- Salik, I.; Gupta, A.; Tara, A.; Zaidman, G.; Barst, S. Peters Anomaly: A 5-Year Experience. Paediatr. Anaesth. 2020, 30, 577–583. [Google Scholar] [CrossRef]
- Yang, L.L.H.; Lambert, S.R.; Drews-Botsch, C.; Stulting, R.D. Long-Term Visual Outcome of Penetrating Keratoplasty in Infants and Children with Peters Anomaly. J. AAPOS 2009, 13, 175–180. [Google Scholar] [CrossRef]
- Basdekidou, C.; Dureau, P.; Edelson, C.; De Laage De Meux, P.; Caputo, G. Should Unilateral Congenital Corneal Opacities in Peters’ Anomaly Be Grafted? Eur. J. Ophthalmol. 2011, 21, 695–699. [Google Scholar] [CrossRef]
- Reidy, J.J. Penetrating Keratoplasty in Infancy and Early Childhood. Curr. Opin. Ophthalmol. 2001, 12, 258–261. [Google Scholar] [CrossRef] [PubMed]
- Elbaz, U.; Strungaru, H.; Mireskandari, K.; Stephens, D.; Ali, A. Long-Term Visual Outcomes and Clinical Course of Patients With Peters Anomaly. Cornea 2021, 40, 822–830. [Google Scholar] [CrossRef] [PubMed]
- Wagoner, M.D.; Al-Ghamdi, A.H.; Al-Rajhi, A.A. Bacterial Keratitis after Primary Pediatric Penetrating Keratoplasty. Am. J. Ophthalmol. 2007, 143, 1045–1047. [Google Scholar] [CrossRef] [PubMed]
- Hashemi, H.; Ghaffari, R.; Mohebi, M. Posterior Lamellar Keratoplasty (DSAEK) in Peters Anomaly. Cornea 2012, 31, 1201–1205. [Google Scholar] [CrossRef] [PubMed]
- Jünemann, A.; Gusek, G.C.; Naumann, G.O. Optical sector iridectomy: An alternative to perforating keratoplasty in Peters’ anomaly. Klin. Mbl. Augenheilkd. 1996, 209, 117–124. [Google Scholar] [CrossRef]
- Sundaresh, K.; Jethani, J.; Vijayalakshmi, P. Optical Iridectomy in Children with Corneal Opacities. J. AAPOS 2008, 12, 163–165. [Google Scholar] [CrossRef]
- Zaidman, G.W.; Rabinowitz, Y.; Forstot, S.L. Optical Iridectomy for Corneal Opacities in Peter’s Anomaly. J. Cataract. Refract. Surg. 1998, 24, 719–722. [Google Scholar] [CrossRef]
- Alkatan, H.M.; Al Dhaheri, H.; Al Harby, M. Terminology of Peters’ Anomaly Variants: Summary of Histopathological Findings in 6 Corneas and Detailed Clinicopathological Correlation in 2 Cases. Saudi J. Ophthalmol. 2019, 33, 277–282. [Google Scholar] [CrossRef]
- Agarwal, T.; Sharma, N.; Jhanji, V.; Vajpayee, R.B. Computer Simulation-Assisted Rotational Autokeratoplasty with Pupillary Enlargement for Management of Cases with Partial Corneal Opacification. Br. J. Ophthalmol. 2010, 94, 24–25. [Google Scholar] [CrossRef]
- Aldave, A.J.; Sangwan, V.S.; Basu, S.; Basak, S.K.; Hovakimyan, A.; Gevorgyan, O.; Kharashi, S.A.; Jindan, M.A.; Tandon, R.; Mascarenhas, J.; et al. International Results with the Boston Type I Keratoprosthesis. Ophthalmology 2012, 119, 1530–1538. [Google Scholar] [CrossRef]
- Aquavella, J.V.; Gearinger, M.D.; Akpek, E.K.; McCormick, G.J. Pediatric Keratoprosthesis. Ophthalmology 2007, 114, 989–994. [Google Scholar] [CrossRef] [PubMed]
- Nallasamy, S.; Colby, K. Keratoprosthesis: Procedure of Choice for Corneal Opacities in Children? Semin. Ophthalmol. 2010, 25, 244–248. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| OPHTHALMIC | microphthalmia, iridocorneal adhesion, corectopia, keratolenticular touch, anterior polar cataract, aniridia, aphakia, iris hypoplasia, iris coloboma, chorioretinal coloboma, staphyloma, retinal dysplasia, ptosis, persistent hyperplasic primary vitreous, optic nerve hypoplasia, foveal hypoplasia, macular pigment epitheliopathy, sclerocornea |
| SYSTEMIC | developmental delay, central nervous system defects, craniofacial abnormalities, microcephaly, hypopituitarism, dwarfism, cardiac malformations, skeletal deformities, genitourinary malformations, ear defects, cleft lip and palate, fetal alcohol syndrome, autism |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wowra, B.; Dobrowolski, D.; Parekh, M.; Wylęgała, E. General Treatment and Ophthalmic Management of Peters’ Anomaly. J. Clin. Med. 2024, 13, 532. https://doi.org/10.3390/jcm13020532
Wowra B, Dobrowolski D, Parekh M, Wylęgała E. General Treatment and Ophthalmic Management of Peters’ Anomaly. Journal of Clinical Medicine. 2024; 13(2):532. https://doi.org/10.3390/jcm13020532
Chicago/Turabian StyleWowra, Bogumil, Dariusz Dobrowolski, Mohit Parekh, and Edward Wylęgała. 2024. "General Treatment and Ophthalmic Management of Peters’ Anomaly" Journal of Clinical Medicine 13, no. 2: 532. https://doi.org/10.3390/jcm13020532
APA StyleWowra, B., Dobrowolski, D., Parekh, M., & Wylęgała, E. (2024). General Treatment and Ophthalmic Management of Peters’ Anomaly. Journal of Clinical Medicine, 13(2), 532. https://doi.org/10.3390/jcm13020532