Perspectives on Sotatercept in Pulmonary Arterial Hypertension

{kind=link}
Abstract
1. Introduction
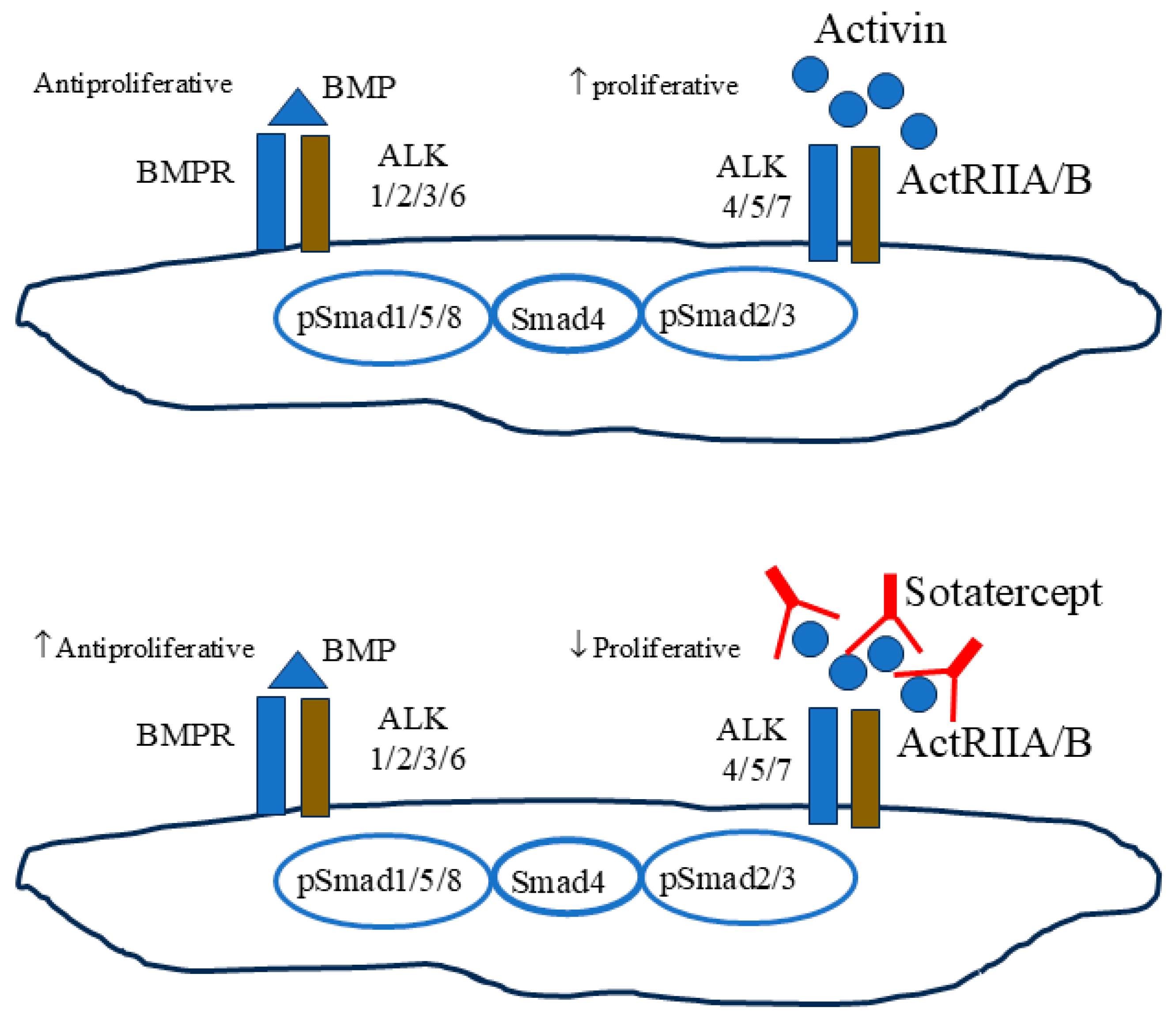
2. Targeting the Activin Pathway in PAH
3. Preclinical Evidence on Targeting the Activin Signaling Pathway in PAH Models
4. Interventional Studies and Ongoing Registered Trials on Activin Inhibitors in PAH Group 1
5. Gaps in Evidence and Issues with Existing RCTs
6. Could the Benefits of Sotatercept Therapy Apply to Non-Group 1 PH?
7. Conclusions and Future Perspectives
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Galiè, N.; Humbert, M.; Vachiery, J.L.; Gibbs, S.; Lang, I.; Torbicki, A.; Simonneau, G.; Peacock, A.; Vonk Noordegraaf, A.; Beghetti, M.; et al. 2015 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension: The Joint Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS): Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC), International Society for Heart and Lung Transplantation (ISHLT). Eur. Heart J. 2016, 37, 67–119. [Google Scholar] [CrossRef] [PubMed]
- Hoeper, M.M.; Badesch, D.B.; Ghofrani, H.A.; Gibbs, J.S.R.; Gomberg-Maitland, M.; McLaughlin, V.V.; Preston, I.R.; Souza, R.; Waxman, A.B.; Grünig, E.; et al. Phase 3 Trial of Sotatercept for Treatment of Pulmonary Arterial Hypertension. N. Engl. J. Med. 2023, 388, 1478–1490. [Google Scholar] [CrossRef] [PubMed]
- Jerkic, M.; Kabir, M.G.; Davies, A.; Yu, L.X.; McIntyre, B.A.; Husain, N.W.; Enomoto, M.; Sotov, V.; Husain, M.; Henkelman, M.; et al. Pulmonary hypertension in adult Alk1 heterozygous mice due to oxidative stress. Cardiovasc. Res. 2011, 92, 375–384. [Google Scholar] [CrossRef] [PubMed]
- Girerd, B.; Montani, D.; Coulet, F.; Sztrymf, B.; Yaici, A.; Jaïs, X.; Tregouet, D.; Reis, A.; Drouin-Garraud, V.; Fraisse, A.; et al. Clinical outcomes of pulmonary arterial hypertension in patients carrying an ACVRL1 (ALK1) mutation. Am. J. Respir. Crit. Care Med. 2010, 181, 851–861. [Google Scholar] [CrossRef] [PubMed]
- Bernard, D.J.; Chapman, S.C.; Woodruff, T.K. Mechanisms of inhibin signal transduction. Recent Prog. Horm. Res. 2001, 56, 417–450. [Google Scholar] [CrossRef]
- Vale, W.; Rivier, C.; Hsueh, A.; Campen, C.; Meunier, H.; Bicsak, T.; Vaughan, J.; Corrigan, A.; Bardin, W.; Sawchenko, P.; et al. Chemical and biological characterization of the inhibin family of protein hormones. Recent Prog. Horm. Res. 1988, 44, 1–34. [Google Scholar] [CrossRef]
- Meunier, H.; Cajander, S.B.; Roberts, V.J.; Rivier, C.; Sawchenko, P.E.; Hsueh, A.J.; Vale, W. Rapid changes in the expression of inhibin alpha-, beta A-, and beta B-subunits in ovarian cell types during the rat estrous cycle. Mol. Endocrinol. 1988, 2, 1352–1363. [Google Scholar] [CrossRef]
- Santibanez, J.F.; Blanco, F.J.; Garrido-Martin, E.M.; Sanz-Rodriguez, F.; del Pozo, M.A.; Bernabeu, C. Caveolin-1 interacts and cooperates with the transforming growth factor-beta type I receptor ALK1 in endothelial caveolae. Cardiovasc. Res. 2008, 77, 791–799. [Google Scholar] [CrossRef]
- Yong, H.E.; Murthi, P.; Wong, M.H.; Kalionis, B.; Cartwright, J.E.; Brennecke, S.P.; Keogh, R.J. Effects of normal and high circulating concentrations of activin A on vascular endothelial cell functions and vasoactive factor production. Pregnancy Hypertens. 2015, 5, 346–353. [Google Scholar] [CrossRef]
- Sugimoto, K.; Yokokawa, T.; Misaka, T.; Kaneshiro, T.; Yamada, S.; Yoshihisa, A.; Nakazato, K.; Takeishi, Y. Endothelin-1 Upregulates Activin Receptor-Like Kinase-1 Expression via Gi/RhoA/Sp-1/Rho Kinase Pathways in Human Pulmonary Arterial Endothelial Cells. Front. Cardiovasc. Med. 2021, 8, 648981. [Google Scholar] [CrossRef]
- Yung, L.M.; Yang, P.; Joshi, S.; Augur, Z.M.; Kim, S.S.J.; Bocobo, G.A.; Dinter, T.; Troncone, L.; Chen, P.S.; McNeil, M.E.; et al. ACTRIIA-Fc rebalances activin/GDF versus BMP signaling in pulmonary hypertension. Sci. Transl. Med. 2020, 12, eaaz5660. [Google Scholar] [CrossRef] [PubMed]
- Joshi, S.R.; Liu, J.; Bloom, T.; Karaca Atabay, E.; Kuo, T.H.; Lee, M.; Belcheva, E.; Spaits, M.; Grenha, R.; Maguire, M.C.; et al. Sotatercept analog suppresses inflammation to reverse experimental pulmonary arterial hypertension. Sci. Rep. 2022, 12, 7803. [Google Scholar] [CrossRef]
- Yung, L.M.; Nikolic, I.; Paskin-Flerlage, S.D.; Pearsall, R.S.; Kumar, R.; Yu, P.B. A Selective Transforming Growth Factor-β Ligand Trap Attenuates Pulmonary Hypertension. Am. J. Respir. Crit. Care Med. 2016, 194, 1140–1151. [Google Scholar] [CrossRef] [PubMed]
- Thomas, M.; Docx, C.; Holmes, A.M.; Beach, S.; Duggan, N.; England, K.; Leblanc, C.; Lebret, C.; Schindler, F.; Raza, F.; et al. Activin-like kinase 5 (ALK5) mediates abnormal proliferation of vascular smooth muscle cells from patients with familial pulmonary arterial hypertension and is involved in the progression of experimental pulmonary arterial hypertension induced by monocrotaline. Am. J. Pathol. 2009, 174, 380–389. [Google Scholar] [CrossRef] [PubMed]
- Zaiman, A.L.; Podowski, M.; Medicherla, S.; Gordy, K.; Xu, F.; Zhen, L.; Shimoda, L.A.; Neptune, E.; Higgins, L.; Murphy, A.; et al. Role of the TGF-beta/Alk5 signaling pathway in monocrotaline- induced pulmonary hypertension. Am. J. Respir. Crit. Care Med. 2008, 177, 896–905. [Google Scholar] [CrossRef] [PubMed]
- Tarras, S.; Gadia, C.; Meister, L.; Roldan, E.; Gregorios, J.B. Homozygous protein C deficiency in a newborn. Clinicopathologic correlation. Arch. Neurol. 1988, 45, 214–216. [Google Scholar] [CrossRef]
- Guignabert, C.; Savale, L.; Boucly, A.; Thuillet, R.; Tu, L.; Ottaviani, M.; Rhodes, C.J.; De Groote, P.; Prévot, G.; Bergot, E.; et al. Serum and Pulmonary Expression Profiles of the Activin Signaling System in Pulmonary Arterial Hypertension. Circulation 2023, 147, 1809–1822. [Google Scholar] [CrossRef]
- Humbert, M.; McLaughlin, V.; Gibbs, J.S.R.; Gomberg-Maitland, M.; Hoeper, M.M.; Preston, I.R.; Souza, R.; Waxman, A.; Escribano Subias, P.; Feldman, J.; et al. Sotatercept for the Treatment of Pulmonary Arterial Hypertension. N. Engl. J. Med. 2021, 384, 1204–1215. [Google Scholar] [CrossRef]
- Liao, K.; Mackenzie, H.; Ait-Oudhia, S.; Manimaran, S.; Zeng, Y.; Akers, T.; Yun, T.; de Oliveira Pena, J. The Impact of Immunogenicity on the Pharmacokinetics, Efficacy, and Safety of Sotatercept in a Phase III Study of Pulmonary Arterial Hypertension. Clin. Pharmacol. Ther. 2024, 115, 478–487. [Google Scholar] [CrossRef]
- Waxman, A.B.; Systrom, D.M.; Manimaran, S.; de Oliveira Pena, J.; Lu, J.; Rischard, F.P. SPECTRA Phase 2b Study: Impact of Sotatercept on Exercise Tolerance and Right Ventricular Function in Pulmonary Arterial Hypertension. Circ. Heart Fail. 2024, 17, e011227. [Google Scholar] [CrossRef]
- Galiè, N.; Barberà, J.A.; Frost, A.E.; Ghofrani, H.A.; Hoeper, M.M.; McLaughlin, V.V.; Peacock, A.J.; Simonneau, G.; Vachiery, J.L.; Grünig, E.; et al. Initial Use of Ambrisentan plus Tadalafil in Pulmonary Arterial Hypertension. N. Engl. J. Med. 2015, 373, 834–844. [Google Scholar] [CrossRef] [PubMed]
- Souza, R.; Badesch, D.B.; Ghofrani, H.A.; Gibbs, J.S.R.; Gomberg-Maitland, M.; McLaughlin, V.V.; Preston, I.R.; Waxman, A.B.; Grünig, E.; Kopeć, G.; et al. Effects of sotatercept on haemodynamics and right heart function: Analysis of the STELLAR trial. Eur. Respir. J. 2023, 62, 2301107. [Google Scholar] [CrossRef] [PubMed]
- Rubin, L.J.; Naeije, R. Sotatercept for pulmonary arterial hypertension: Something old and something new. Eur. Respir. J. 2023, 61, 2201972. [Google Scholar] [CrossRef] [PubMed]
- Humbert, M.; McLaughlin, V.; Gibbs, J.S.R.; Gomberg-Maitland, M.; Hoeper, M.M.; Preston, I.R.; Souza, R.; Waxman, A.B.; Ghofrani, H.A.; Escribano Subias, P.; et al. Sotatercept for the treatment of pulmonary arterial hypertension: PULSAR open-label extension. Eur. Respir. J. 2023, 61, 2201347. [Google Scholar] [CrossRef]
- Komrokji, R.; Garcia-Manero, G.; Ades, L.; Prebet, T.; Steensma, D.P.; Jurcic, J.G.; Sekeres, M.A.; Berdeja, J.; Savona, M.R.; Beyne-Rauzy, O.; et al. Sotatercept with long-term extension for the treatment of anaemia in patients with lower-risk myelodysplastic syndromes: A phase 2, dose-ranging trial. Lancet Haematol. 2018, 5, e63–e72. [Google Scholar] [CrossRef]
- Bose, P.; Masarova, L.; Pemmaraju, N.; Bledsoe, S.D.; Daver, N.G.; Jabbour, E.J.; Kadia, T.M.; Estrov, Z.; Kornblau, S.M.; Andreeff, M.; et al. Sotatercept for anemia of myelofibrosis: A phase II investigator-initiated study. Haematologica 2024, 109, 2660–2664. [Google Scholar] [CrossRef]
- Quatredeniers, M.; Mendes-Ferreira, P.; Santos-Ribeiro, D.; Nakhleh, M.K.; Ghigna, M.R.; Cohen-Kaminsky, S.; Perros, F. Iron Deficiency in Pulmonary Arterial Hypertension: A Deep Dive into the Mechanisms. Cells 2021, 10, 477. [Google Scholar] [CrossRef]
- Krasuski, R.A.; Hart, S.A.; Smith, B.; Wang, A.; Harrison, J.K.; Bashore, T.M. Association of anemia and long-term survival in patients with pulmonary hypertension. Int. J. Cardiol. 2011, 150, 291–295. [Google Scholar] [CrossRef]
- Horwich, T.B.; Fonarow, G.C.; Hamilton, M.A.; MacLellan, W.R.; Borenstein, J. Anemia is associated with worse symptoms, greater impairment in functional capacity and a significant increase in mortality in patients with advanced heart failure. J. Am. Coll. Cardiol. 2002, 39, 1780–1786. [Google Scholar] [CrossRef]
- Swedberg, K.; Young, J.B.; Anand, I.S.; Cheng, S.; Desai, A.S.; Diaz, R.; Maggioni, A.P.; McMurray, J.J.; O’Connor, C.; Pfeffer, M.A.; et al. Treatment of anemia with darbepoetin alfa in systolic heart failure. N. Engl. J. Med. 2013, 368, 1210–1219. [Google Scholar] [CrossRef]
- Karamanian, V.A.; Harhay, M.; Grant, G.R.; Palevsky, H.I.; Grizzle, W.E.; Zamanian, R.T.; Ihida-Stansbury, K.; Taichman, D.B.; Kawut, S.M.; Jones, P.L. Erythropoietin upregulation in pulmonary arterial hypertension. Pulm. Circ. 2014, 4, 269–279. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Z.; Li, K.; Shen, C.; Ma, Y.; Guo, L. Erythropoietin improves pulmonary hypertension by promoting the homing and differentiation of bone marrow mesenchymal stem cells in lung tissue. Hum. Cell. 2024, 37, 214–228. [Google Scholar] [CrossRef] [PubMed]
- Hoeper, M.M.; Humbert, M.; Souza, R.; Idrees, M.; Kawut, S.M.; Sliwa-Hahnle, K.; Jing, Z.C.; Gibbs, J.S. A global view of pulmonary hypertension. Lancet Respir. Med. 2016, 4, 306–322. [Google Scholar] [CrossRef] [PubMed]
- Madonna, R.; Biondi, F.; Alberti, M.; Ghelardoni, S.; Mattii, L.; D’Alleva, A. Cardiovascular outcomes and molecular targets for the cardiac effects of Sodium-Glucose Cotransporter 2 Inhibitors: A systematic review. Biomed. Pharmacother. 2024, 175, 116650. [Google Scholar] [CrossRef]
- Madonna, R.; Biondi, F.; Ghelardoni, S.; D’Alleva, A.; Quarta, S.; Massaro, M. Pulmonary hypertension associated to left heart disease: Phenotypes and treatment. Eur. J. Intern. Med. 2024, in press. [Google Scholar] [CrossRef]
- Roh, J.D.; Hobson, R.; Chaudhari, V.; Quintero, P.; Yeri, A.; Benson, M.; Xiao, C.; Zlotoff, D.; Bezzerides, V.; Houstis, N.; et al. Activin type II receptor signaling in cardiac aging and heart failure. Sci. Transl. Med. 2019, 11, eaau8680. [Google Scholar] [CrossRef]
- Liu, M.; Mao, C.; Li, J.; Han, F.; Yang, P. Effects of the Activin A-Follistatin System on Myocardial Cell Apoptosis through the Endoplasmic Reticulum Stress Pathway in Heart Failure. Int. J. Mol. Sci. 2017, 18, 374. [Google Scholar] [CrossRef]
- Yndestad, A.; Ueland, T.; Øie, E.; Florholmen, G.; Halvorsen, B.; Attramadal, H.; Simonsen, S.; Frøland, S.S.; Gullestad, L.; Christensen, G.; et al. Elevated levels of activin A in heart failure: Potential role in myocardial remodeling. Circulation 2004, 109, 1379–1385. [Google Scholar] [CrossRef] [PubMed]
- Tan, S.M.; Zhang, Y.; Connelly, K.A.; Gilbert, R.E.; Kelly, D.J. Targeted inhibition of activin receptor-like kinase 5 signaling attenuates cardiac dysfunction following myocardial infarction. Am. J. Physiol. Heart Circ. Physiol. 2010, 298, H1415–H1425. [Google Scholar] [CrossRef]
- Swinnen, K.; Quarck, R.; Godinas, L.; Belge, C.; Delcroix, M. Learning from registries in pulmonary arterial hypertension: Pitfalls and recommendations. Eur. Respir. Rev. 2019, 28, 190050. [Google Scholar] [CrossRef]
- Yazici, H. Beware of registries for their biases. Bull. NYU Hosp. Jt. Dis. 2012, 70, 95–98. [Google Scholar] [PubMed]
- Krumholz, H.M. Registries and selection bias: The need for accountability. Circ. Cardiovasc. Qual. Outcomes 2009, 2, 517–518. [Google Scholar] [CrossRef] [PubMed]
- Davis, K.K.; Lilienfeld, D.E.; Doyle, R.L. Increased mortality in African Americans with idiopathic pulmonary arterial hypertension. J. Natl. Med. Assoc. 2008, 100, 69–72. [Google Scholar] [CrossRef]
- Gabler, N.B.; French, B.; Strom, B.L.; Liu, Z.; Palevsky, H.I.; Taichman, D.B.; Kawut, S.M.; Halpern, S.D. Race and sex differences in response to endothelin receptor antagonists for pulmonary arterial hypertension. Chest 2012, 141, 20–26. [Google Scholar] [CrossRef]
- Boucly, A.; Weatherald, J.; Savale, L.; de Groote, P.; Cottin, V.; Prévot, G.; Chaouat, A.; Picard, F.; Horeau-Langlard, D.; Bourdin, A.; et al. External validation of a refined four-stratum risk assessment score from the French pulmonary hypertension registry. Eur. Respir. J. 2022, 59, 2102419. [Google Scholar] [CrossRef]
- Rossi, R.; Talarico, M.; Schepis, F.; Coppi, F.; Sgura, F.A.; Monopoli, D.E.; Minici, R.; Boriani, G. Effects of sildenafil on right ventricle remodelling in Portopulmonary hypertension. Pulm. Pharmacol. Ther. 2021, 70, 102071. [Google Scholar] [CrossRef]
- Lippi, G.; Franchini, M.; Favaloro, E.J. Thrombotic complications of erythropoiesis-stimulating agents. Semin. Thromb. Hemost. 2010, 36, 537–549. [Google Scholar] [CrossRef] [PubMed]
- Tapson, V.F.; Jing, Z.C.; Xu, K.F.; Pan, L.; Feldman, J.; Kiely, D.G.; Kotlyar, E.; McSwain, C.S.; Laliberte, K.; Arneson, C.; et al. Oral treprostinil for the treatment of pulmonary arterial hypertension in patients receiving background endothelin receptor antagonist and phosphodiesterase type 5 inhibitor therapy (the FREEDOM-C2 study): A randomized controlled trial. Chest 2013, 144, 952–958. [Google Scholar] [CrossRef]
- Pulido, T.; Adzerikho, I.; Channick, R.N.; Delcroix, M.; Galiè, N.; Ghofrani, H.A.; Jansa, P.; Jing, Z.C.; Le Brun, F.O.; Mehta, S.; et al. Macitentan and morbidity and mortality in pulmonary arterial hypertension. N. Engl. J. Med. 2013, 369, 809–818. [Google Scholar] [CrossRef]
- Studer, S.M.; Gilkin, R.J., Jr. Clinical trial designs in PAH: Shifting from functional measurements to long-term clinical outcomes. Am. J. Manag. Care. 2014, 20 (Suppl. S6), S115–S122. [Google Scholar] [PubMed]
- Tremblay, É.; Gosselin, C.; Mai, V.; Lajoie, A.C.; Kilo, R.; Weatherald, J.; Lacasse, Y.; Bonnet, S.; Lega, J.C.; Provencher, S. Assessment of Clinical Worsening End Points as a Surrogate for Mortality in Pulmonary Arterial Hypertension: A Systematic Review and Meta-Analysis of Randomized Controlled Trials. Circulation 2022, 146, 597–612. [Google Scholar] [CrossRef] [PubMed]
- Verhamme, F.M.; Bracke, K.R.; Amatngalim, G.D.; Verleden, G.M.; Van Pottelberge, G.R.; Hiemstra, P.S.; Joos, G.F.; Brusselle, G.G. Role of activin-A in cigarette smoke-induced inflammation and COPD. Eur. Respir. J. 2014, 43, 1028–1041. [Google Scholar] [CrossRef] [PubMed]
- Zhou, G.; Gui, X.; Chen, R.; Fu, X.; Ji, X.; Ding, H. Elevated serum Activin A in chronic obstructive pulmonary disease with skeletal muscle wasting. Clinics (Sao Paulo) 2019, 74, e981. [Google Scholar] [CrossRef] [PubMed]
- Scaroni, C.; Orlandini, E.; Venturi Pasini, C.; Gangemi, M.; Mantero, F. HLA and hormonal studies in 5 patients with late-onset 21-hydroxylase deficiency syndrome (21OHDS). J. Endocrinol. Investig. 1986, 9, 65–70. [Google Scholar] [CrossRef] [PubMed]
- Batt, J.; Ahmed, S.S.; Correa, J.; Bain, A.; Granton, J. Skeletal muscle dysfunction in idiopathic pulmonary arterial hypertension. Am. J. Respir. Cell Mol. Biol. 2014, 50, 74–86. [Google Scholar] [CrossRef]
- McGettrick, M.; Peacock, A. Group 3 pulmonary hypertension: Challenges and opportunities. Glob. Cardiol. Sci. Pract. 2020, 2020, e202006. [Google Scholar] [CrossRef]
- Waxman, A.; Restrepo-Jaramillo, R.; Thenappan, T.; Ravichandran, A.; Engel, P.; Bajwa, A.; Allen, R.; Feldman, J.; Argula, R.; Smith, P.; et al. Inhaled Treprostinil in Pulmonary Hypertension Due to Interstitial Lung Disease. N. Engl. J. Med. 2021, 384, 325–334. [Google Scholar] [CrossRef]
- Humbert, M.; Kovacs, G.; Hoeper, M.M.; Badagliacca, R.; Berger, R.M.F.; Brida, M.; Carlsen, J.; Coats, A.J.S.; Escribano-Subias, P.; Ferrari, P.; et al. 2022 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension. Eur Heart J. 2022, 43, 3618–3731. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Madonna, R.; Biondi, F. Perspectives on Sotatercept in Pulmonary Arterial Hypertension. J. Clin. Med. 2024, 13, 6463. https://doi.org/10.3390/jcm13216463
Madonna R, Biondi F. Perspectives on Sotatercept in Pulmonary Arterial Hypertension. Journal of Clinical Medicine. 2024; 13(21):6463. https://doi.org/10.3390/jcm13216463
Chicago/Turabian StyleMadonna, Rosalinda, and Filippo Biondi. 2024. "Perspectives on Sotatercept in Pulmonary Arterial Hypertension" Journal of Clinical Medicine 13, no. 21: 6463. https://doi.org/10.3390/jcm13216463
APA StyleMadonna, R., & Biondi, F. (2024). Perspectives on Sotatercept in Pulmonary Arterial Hypertension. Journal of Clinical Medicine, 13(21), 6463. https://doi.org/10.3390/jcm13216463