

Psychiatric Manifestations in Children and Adolescents with Inherited Metabolic Diseases

 , , and
, , and
Abstract
1. Introduction
2. Aim
3. Materials and Methods
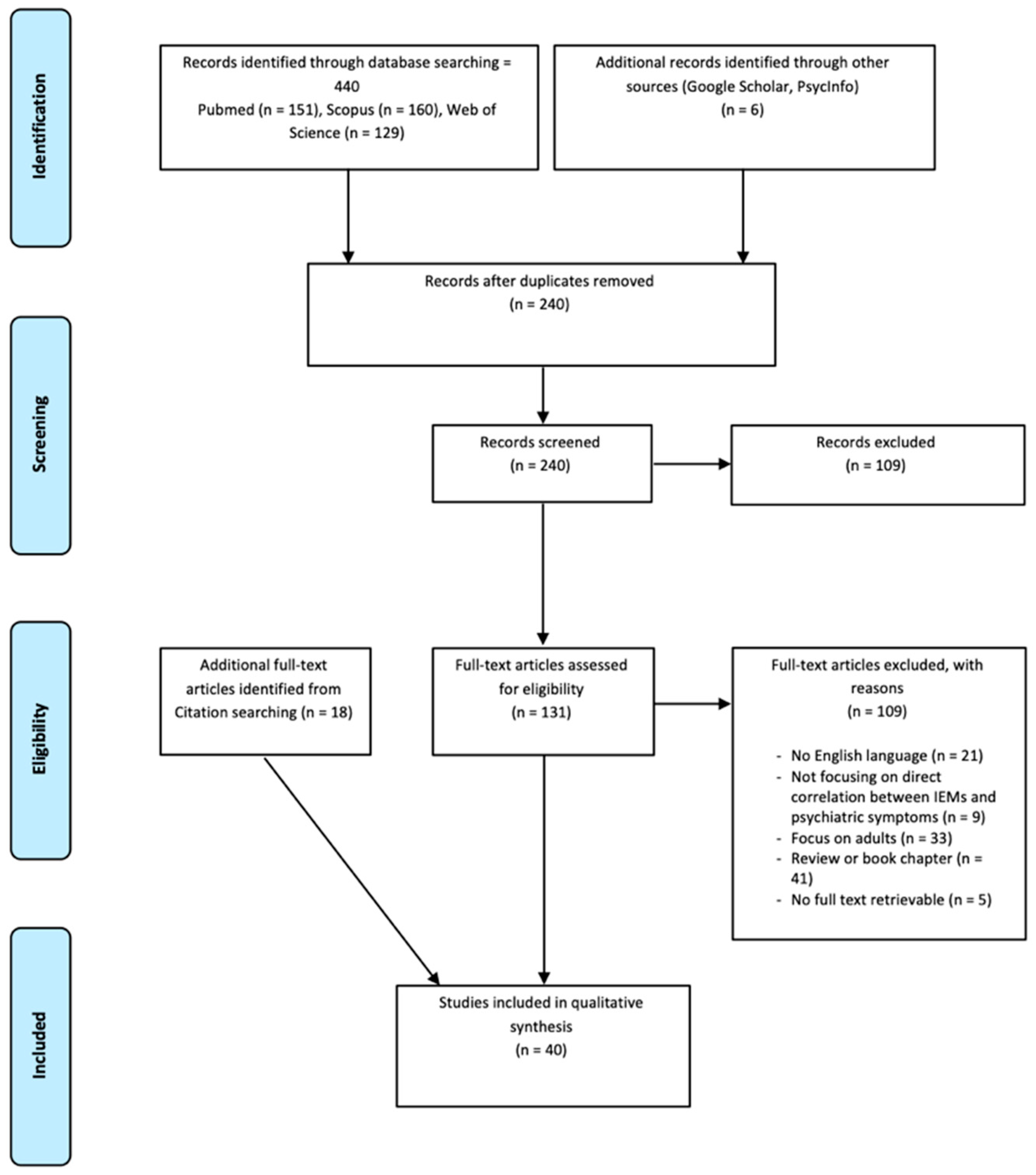
3.1. Database Search Strategy
3.2. Literature Search Strategy and Study Eligibility
3.3. Study Inclusion
4. Results
4.1. Epidemiology
4.2. Type of Onset
4.3. Psychiatric Phenomenology
4.3.1. Neurometabolic Diseases and Mood and Anxiety Disorders
- Phenylketonuria
- Tetrahydrobiopterin deficiencies
4.3.2. Neurometabolic Diseases and Schizophrenia-Spectrum Disorders
- Porphyrias
- Cerebrotendinous Xanthomatosis
- Niemann–Pick type C Disease
- Disorders of the Homocysteine Metabolism
- Urea
- Cycle Disorders
- Wilson’s Disease
4.3.3. Neurometabolic Diseases and Catatonia
4.3.4. Neurometabolic Diseases and Eating Disorders
- Methylmalonic Acidemia (MMA) and Propionic Acidemia (PA)
- Mitochondrial Neurogastrointestinal Encephalomyopathy
4.3.5. Neurometabolic Diseases and Self-Injurious Behaviors
- Lesch–Nyhan Syndrome
5. Discussion
5.1. Strengths and Limitations
5.2. Future Directions
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Simons, A.; Eyskens, F.; De Groof, A.; Van Diest, E.; Deboutte, D.; Vermeiren, R. Cognitive functioning and psychiatric disorders in children with a metabolic disease. Eur. Child Adolesc. Psychiatry 2006, 15, 207–213. [Google Scholar] [CrossRef] [PubMed]
- Simons, A.; Eyskens, F.; Glazemakers, I.; van West, D. Can psychiatric childhood disorders be due to inborn errors of metabolism? Psychiatry 2016, 26, 143–154. [Google Scholar] [CrossRef]
- Horvath, G.A.; Stowe, R.M.; Ferreira, C.R.; Blau, N. Clinical and biochemical footprints of inherited metabolic diseases. III. Psychiatric presentations. Mol. Genet. Metab. 2020, 130, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Walterfang, M.; Bonnot, O.; Mocellin, R.; Velakoulis, D. The neuropsychiatry of inborn errors of metabolism. J. Inherit. Metab. Dis. 2013, 36, 687–702. [Google Scholar] [CrossRef] [PubMed]
- Nia, S. Psychiatric signs and symptoms in treatable inborn errors of metabolism. J. Neurol. 2014, 261 (Suppl. 2), 559–568. [Google Scholar] [CrossRef] [PubMed]
- Sedel, F.; Baumann, N.; Turpin, J.; Lyon-Caen, O.; Saudubray, J.; Cohen, D. Psychiatric manifestations revealing inborn errors of metabolism in adolescents and adults. J. Inherit. Metab. Dis. 2007, 30, 631–641. [Google Scholar] [CrossRef]
- van de Burgt, N.; van Doesum, W.; Grevink, M.; van Niele, S.; de Koning, T.; Leibold, N.; Martinez-Martinez, P.; van Amelsvoort, T.; Cath, D. Psychiatric manifestations of inborn errors of metabolism: A systematic review. Neurosci. Biobehav. Rev. 2023, 144, 104970. [Google Scholar] [CrossRef]
- Ferreira, C.R.; Cassiman, D.; Blau, N. Clinical and biochemical footprints of inherited metabolic diseases. II. Metabolic liver diseases. Mol. Genet. Metab. 2019, 127, 117–121. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.; Zhou, Q.; Chao, Y.Q.; Zou, C.C. Clinical features and genetic analysis of childhood sitosterolemia: Two case reports and literature review. Medicine 2019, 98, e15013. [Google Scholar] [CrossRef]
- Saudubray, J.-M.; Garcia-Cazorla, A. An overview of inborn errors of metabolism affecting the brain: From neurodevelopment to neurodegenerative disorders. Dialogues Clin. Neurosci. 2018, 20, 301–325. [Google Scholar] [CrossRef]
- Ferreira, C.R.; Hoffmann, G.F.; Blau, N. Clinical and biochemical footprints of inherited metabolic diseases. I. Movement disorders. Mol. Genet. Metab. 2019, 127, 28–30. [Google Scholar] [CrossRef] [PubMed]
- Stenton, S.L.; Kremer, L.S.; Kopajtich, R.; Ludwig, C.; Prokisch, H. The diagnosis of inborn errors of metabolism by an integrative ‘multi-omics’ approach: A perspective encompassing genomics, transcriptomics, and proteomics. J. Inherit. Metab. Dis. 2020, 43, 25–35. [Google Scholar] [CrossRef] [PubMed]
- Marques, E.P.; Wyse, A.T. Creatine as a neuroprotector: An actor that can play many parts. Neurotox. Res. 2019, 36, 411–423. [Google Scholar] [CrossRef]
- Guerrero, R.B.; Kloke, K.M.; Salazar, D. Inborn errors of metabolism and the gastrointestinal tract. Gastroenterol. Clin. 2019, 48, 183–198. [Google Scholar] [CrossRef] [PubMed]
- Canton, M.; Le Gall, D.; Feillet, F.; Bonnemains, C.; Roy, A. Neuropsychological profile of children with early and continuously treated phenylketonuria: Systematic review and future approaches. J. Int. Neuropsychol. Soc. 2019, 25, 624–643. [Google Scholar] [CrossRef]
- Liberati, A.; Altman, D.G.; Tetzlaff, J.; Mulrow, C.; Gøtzsche, P.C.; Ioannidis, J.P.; Clarke, M.; Devereaux, P.J.; Kleijnen, J.; Moher, D. The PRISMA statement for reporting systematic reviews and meta-analyses of studies that evaluate health care interventions: Explanation and elaboration. Ann. Intern. Med. 2009, 151, W-65. [Google Scholar] [CrossRef] [PubMed]
- Abbott, M.H.; Folstein, S.E.; Abbey, H.; Pyeritz, R.E.; Opitz, J.M. Psychiatric manifestations of homocystinuria due to cystathionine β-synthase deficiency: Prevalence, natural history, and relationship to neurologic impairment and vitamin B6-responsiveness. Am. J. Med. Genet. 1987, 26, 959–969. [Google Scholar] [CrossRef] [PubMed]
- Al-Owain, M.; Colak, D.; Albakheet, A.; Al-Younes, B.; Al-Humaidi, Z.; Al-Sayed, M.; Al-Hindi, H.; Al-Sugair, A.; Al-Muhaideb, A.; Rahbeeni, Z.; et al. Clinical and biochemical features associated with BCS1L mutation. J. Inherit. Metab. Dis. 2013, 36, 813–820. [Google Scholar] [CrossRef] [PubMed]
- Alam, B.; Biswas, S.; Maria, M.; Rahman, A.; Rahman, H.; Das, P.P.; Rahman, S.; Kabir, K. Wilson’s Disease: An Uncommon Presentation. J. Med. 2012, 13, 103–105. [Google Scholar]
- Albokhari, D.; Ng, B.G.; Guberinic, A.; Daniel, E.J.P.; Engelhardt, N.M.; Barone, R.; Fiumara, A.; Garavelli, L.; Trimarchi, G.; Wolfe, L.; et al. ALG8-CDG: Molecular and phenotypic expansion suggests clinical management guidelines. J. Inherit. Metab. Dis. 2022, 45, 969–980. [Google Scholar] [CrossRef]
- Anderson, L.T.; Ernst, M. Self-injury in Lesch-Nyhan disease. J. Autism Dev. Disord. 1994, 24, 67–81. [Google Scholar] [CrossRef] [PubMed]
- Bonnot, O.; Fraidakis, M.J.; Lucanto, R.; Chauvin, D.; Kelley, N.; Plaza, M.; Dubourg, O.; Lyon-Caen, O.; Sedel, F.; Cohen, D. Cerebrotendinous xanthomatosis presenting with severe externalized disorder: Improvement after one year of treatment with chenodeoxycholic acid. CNS Spectr. 2010, 15, 231–236. [Google Scholar] [CrossRef] [PubMed]
- Bonnot, O.; Gama, C.S.; Mengel, E.; Pineda, M.; Vanier, M.T.; Watson, L.; Watissée, M.; Schwierin, B.; Patterson, M.C. Psychiatric and neurological symptoms in patients with Niemann-Pick disease type C (NP-C): Findings from the International NPC Registry. World J. Biol. Psychiatry 2019, 20, 310–319. [Google Scholar] [CrossRef] [PubMed]
- Boon, F.F.; Ellis, C. Acute Intermittent Porphyria in a Children’s Psychiatric Hospital. J. Am. Acad. Child Adolesc. Psychiatry 1989, 28, 606–609. [Google Scholar] [CrossRef] [PubMed]
- Cappelletti, S.; Cotugno, G.; Goffredo, B.M.; Nicolò, R.; Bernabei, S.M.; Caviglia, S.; Di Ciommo, V. Cognitive findings and behavior in children and adolescents with phenylketonuria. J. Dev. Behav. Pediatr. 2013, 34, 392–398. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Dong, H.; Liu, Y.; He, R.; Song, J.; Jin, Y.; Li, M.; Liu, Y.; Liu, X.; Yan, H.; et al. Late-onset cblC deficiency around puberty: A retrospective study of the clinical characteristics, diagnosis, and treatment. Orphanet J. Rare Dis. 2022, 17, 330. [Google Scholar] [CrossRef]
- Dening, T.R.; Berrios, G.E. Wilson’s disease: Psychiatric symptoms in 195 cases. Arch. Gen. Psychiatry 1989, 46, 1126–1134. [Google Scholar] [CrossRef] [PubMed]
- Dening, T.R.; Berrios, G.E. Wilson’s disease: A longitudinal study of psychiatric symptoms. Biol. Psychiatry 1990, 28, 255–265. [Google Scholar] [CrossRef] [PubMed]
- Erlich, K.J. Case Report: Neuropsychiatric Symptoms in PKU Disease. J. Pediatr. Heal. Care 2019, 33, 718–721. [Google Scholar] [CrossRef]
- Fraidakis, M.J. Psychiatric manifestations in cerebrotendinous xanthomatosis. Transl. Psychiatry 2013, 3, e302. [Google Scholar] [CrossRef]
- Freeman, J.M.; Finkelstein, J.D.; Mudd, S.H. Folate-responsive homocystinuria and ‘schizophrenia’ A defect in methylation due to deficient 5, 10-methylenetetrahydrofolate reductase activity. N. Engl. J. Med. 1975, 292, 491–496. [Google Scholar] [CrossRef] [PubMed]
- Gardeitchik, T.; Humphrey, M.; Nation, J.; Boneh, A. Early clinical manifestations and eating patterns in patients with urea cycle disorders. J. Pediatr. 2012, 161, 328–332. [Google Scholar] [CrossRef] [PubMed]
- Giannitelli, M.; Consoli, A.; Raffin, M.; Jardri, R.; Levinson, D.F.; Cohen, D.; Laurent-Levinson, C. An overview of medical risk factors for childhood psychosis: Implications for research and treatment. Schizophr. Res. 2018, 192, 39–49. [Google Scholar] [CrossRef] [PubMed]
- Gökçen, C.; Işikay, S.; Yilmaz, K. L-2 Hydroxyglutaric aciduria presenting with anxiety symptoms. BMJ Case Rep. 2013, 2013. [Google Scholar] [CrossRef] [PubMed]
- Mazzei, D.H.; Rodriguez, S.M.; Moltó, H.P.; Izquierdo, J.R.; Baeza, I. A forgotten lethal psychosis: A case report. Eur. Child Adolesc. Psychiatry 2014, 23, 235–238. [Google Scholar] [CrossRef] [PubMed]
- Kevere, L.; Purvina, S.; Bauze, D.; Zeibarts, M.; Andrezina, R.; Piekuse, L.; Brekis, E.; Purvins, I. Homocysteine and MTHFR C677T polymorphism in children and adolescents with psychotic and mood disorders. Nord. J. Psychiatry 2014, 68, 129–136. [Google Scholar] [CrossRef] [PubMed]
- Kumar, B. Acute Intermittent Porphyria Presenting Solely with Psychosis: A Case Report and Discussion. Psychosomatics 2012, 53, 494–498. [Google Scholar] [CrossRef] [PubMed]
- Lahutte, B.; Cornic, F.; Bonnot, O.; Consoli, A.; An-Gourfinkel, I.; Amoura, Z.; Sedel, F.; Cohen, D. Multidisciplinary approach of organic catatonia in children and adolescents may improve treatment decision making. Prog. Neuro Psychopharmacol. Biol. Psychiatry 2008, 32, 1393–1398. [Google Scholar] [CrossRef] [PubMed]
- Libernini, L.; Lupis, C.; Mastrangelo, M.; Carrozzo, R.; Santorelli, F.M.; Inghilleri, M.; Leuzzi, V. Mitochondrial neurogastrointestinal encephalomyopathy: Novel pathogenic mutations in thymidine phosphorylase gene in two Italian brothers. Neuropediatrics 2012, 43, 201–208. [Google Scholar] [CrossRef]
- Mandoki, M.W.; Sumner, G.S. Psychiatric manifestations of hereditary coproporphyria in a child. J. Nerv. Ment. Dis. 1994, 182, 117–118. [Google Scholar] [CrossRef]
- Manti, F.; Nardecchia, F.; Banderali, G.; Burlina, A.; Carducci, C.; Carducci, C.; Donati, M.A.; Gueraldi, D.; Paci, S.; Pochiero, F.; et al. Long-term clinical outcome of 6-pyruvoyl-tetrahydropterin synthase-deficient patients. Mol. Genet. Metab. 2020, 131, 155–162. [Google Scholar] [CrossRef] [PubMed]
- Manti, F.; Nardecchia, F.; Chiarotti, F.; Carducci, C.; Carducci, C.; Leuzzi, V. Psychiatric disorders in adolescent and young adult patients with phenylketonuria. Mol. Genet. Metab. 2016, 117, 12–18. [Google Scholar] [CrossRef] [PubMed]
- Niwinski, P.; Remberk, B.; Rybakowski, F.; Rokicki, D. Psychiatric Symptoms as the First or Solitary Manifestation of Somatic Illnesses: Hyperammonaemia Type II. Neuropsychobiology 2021, 80, 271–275. [Google Scholar] [CrossRef] [PubMed]
- Pan, L.; Vockley, J. Neuropsychiatric Symptoms in Inborn Errors of Metabolism: Incorporation of Genomic and Metabolomic Analysis into Therapeutics and Prevention. Curr. Genet. Med. Rep. 2013, 1, 65–70. [Google Scholar] [CrossRef] [PubMed]
- Rahmandar, M.H.; Bawcom, A.; Romano, M.E.; Hamid, R. Cobalamin C deficiency in an adolescent with altered mental status and anorexia. Pediatrics 2014, 134, e1709–e1714. [Google Scholar] [CrossRef] [PubMed]
- Roze, E.; Gervais, D.; Demeret, S.; Ogier de Baulny, H.; Zittoun, J.; Benoist, J.F.; Said, G.; Pierrot-Deseilligny, C.; Bolgert, F. Neuropsychiatric disturbances in presumed late-onset cobalamin C disease. Arch. Neurol. 2003, 60, 1457–1462. [Google Scholar] [CrossRef] [PubMed]
- Sandu, S.; Jackowski-Dohrmann, S.; Ladner, A.; Haberhausen, M.; Bachmann, C. Niemann–Pick disease type C1 presenting with psychosis in an adolescent male. Eur. Child Adolesc. Psychiatry 2009, 18, 583–585. [Google Scholar] [CrossRef]
- Santosh, P.J.; Malhotra, S.; Raghunathan, M.; Mehra, Y.N. A study of P300 in melancholic depression—Correlation with psychotic features. Biol. Psychiatry 1994, 35, 474–479. [Google Scholar] [CrossRef]
- Serrano, M.; Martins, C.; Pérez-Dueñas, B.; Gómez-López, L.; Murgui, E.; Fons, C.; García-Cazorla, A.; Artuch, R.; Jara, F.; Arranz, J.A.; et al. Neuropsychiatric manifestations in late-onset urea cycle disorder patients. J. Child Neurol. 2009, 25, 352–358. [Google Scholar] [CrossRef]
- Smith, I.; Beasley, M.G.; Wolff, O.H.; Ades, A.E. Behavior disturbance in 8-year-old children with early treated phenylketonuria: Report from the MRC/DHSS Phenylketonuria Register. J. Pediatr. 1988, 112, 403–408. [Google Scholar] [CrossRef]
- Srinivas, K.; Sinha, S.; Taly, A.B.; Prashanth, L.K.; Arunodaya, G.R.; Janardhana Reddy, Y.C.; Khanna, S. Dominant psychiatric manifestations in Wilson’s disease: A diagnostic and therapeutic challenge! J. Neurol. Sci. 2008, 266, 104–108. [Google Scholar] [CrossRef] [PubMed]
- Tantawy, A.A.G.; Adly, A.A.M.; El-Din, N.Y.S.; Abdeen, M.S.E.D. Psychiatric manifestations in Egyptian Gaucher patients on enzyme replacement therapy. J. Psychosom. Res. 2019, 122, 75–81. [Google Scholar] [CrossRef]
- Valayannopoulos, V.; Boddaert, N.; Chabli, A.; Barbier, V.; Desguerre, I.; Philippe, A.; Afenjar, A.; Mazzuca, M.; Cheillan, D.; Munnich, A.; et al. Treatment by oral creatine, L-arginine and L-glycine in six severely affected patients with creatine transporter defect. J. Inherit. Metab. Dis. 2012, 35, 151–157. [Google Scholar] [CrossRef] [PubMed]
- Weglage, J.; Grenzebach, M.; Pietsch, M.; Feldmann, R.; Linnenbank, R.; Denecke, J.; Koch, H.G. Behavioural and emotional problems in early-treated adolescents with phenylketonuria in comparison with diabetic patients and healthy controls. J. Inherit. Metab. Dis. 2000, 23, 487–496. [Google Scholar] [CrossRef]
- Wouters, S.; De Meirleir, L.; Campforts, E.; Lampo, A. Psychosis in an adolescent girl: A common manifestation in Niemann-Pick Type C disease. Child Adolesc. Psychiatry Ment. Health 2014, 8, 20. [Google Scholar] [CrossRef]
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders, 5th ed.; Text Revision (DSM-5-TR); American Psychiatric Association: Washington, DC, USA, 2022. [Google Scholar]
- World Health Organization. Classificazione Statistica Internazionale delle Malattie e dei Problemi Sanitari Correlati (ICD-10); World Health Organization: Geneva, Switzerland, 2016; Volume 1. [Google Scholar]
- Lamon, J.M.; Frykholm, B.C.; Tschudy, D.P. Screening tests in acute porphyria. Arch. Neurol. 1977, 34, 709–712. [Google Scholar] [CrossRef]
- Nassogne, M.C.; Héron, B.; Touati, G.; Rabier, D.; Saudubray, J.M. Urea cycle defects: Management and outcome. J. Inherit. Metab. Dis. 2005, 28, 407–414. [Google Scholar] [CrossRef] [PubMed]
- Morris, M.S.; Fava, M.; Jacques, P.F.; Selhub, J.; Rosenberg, I.H. Depression and folate status in the US population. Psychother. Psychosom. 2003, 72, 80–87. [Google Scholar] [CrossRef]
- Manti, F.; Nardecchia, F.; Paci, S.; Chiarotti, F.; Carducci, C.; Carducci, C.; Dalmazzone, S.; Cefalo, G.; Salvatici, E.; Banderali, G.; et al. Predictability and inconsistencies in the cognitive outcome of early treated PKU patients. J. Inherit. Metab. Dis. 2017, 40, 793–799. [Google Scholar] [CrossRef]
- Gioia, G.A.; Isquith, P.K.; Guy, S.C.; Kenworthy, L. Test review behavior rating inventory of executive function. Child Neuropsychol. 2000, 6, 235–238. [Google Scholar] [CrossRef]
- Diamond, A. Executive functions. Annu. Rev. Psychol. 2013, 64, 135–168. [Google Scholar] [CrossRef]
- Lehto, J.E.; Juujärvi, P.; Kooistra, L.; Pulkkinen, L. Dimensions of executive functioning: Evidence from children. Br. J. Dev. Psychol. 2003, 21, 59–80. [Google Scholar] [CrossRef]
- Miyake, A.; Friedman, N.P.; Emerson, M.J.; Witzki, A.H.; Howerter, A.; Wager, T.D. The unity and diversity of executive functions and their contributions to complex ‘frontal lobe’ tasks: A latent variable analysis. Cogn. Psychol. 2000, 41, 49–100. [Google Scholar] [CrossRef] [PubMed]
- Ardila, A. Is ‘self-consciousness’ equivalent to ‘executive function’? Psychol. Neurosci. 2016, 9, 215. [Google Scholar] [CrossRef]
- Collins, A.; Koechlin, E. Reasoning, learning, and creativity: Frontal lobe function and human decision-making. PLoS Biol. 2012, 10, e1001293. [Google Scholar] [CrossRef] [PubMed]
- Lunt, L.; Bramham, J.; Morris, R.G.; Bullock, P.R.; Selway, R.P.; Xenitidis, K.; David, A.S. Prefrontal cortex dysfunction and ‘Jumping to Conclusions’: Bias or deficit? J. Neuropsychol. 2012, 6, 65–78. [Google Scholar] [CrossRef] [PubMed]
- Trimarco, B.; Manti, F.; Nardecchia, F.; Melogno, S.; Testa, M.; Meledandri, G.; Carducci, C.; Penge, R.; Leuzzi, V. Executive functioning, adaptive skills, emotional and behavioral profile: A comparison between autism spectrum disorder and phenylketonuria. Mol. Genet. Metab. Rep. 2020, 23, 100577. [Google Scholar] [CrossRef] [PubMed]
- Hyland, K. Abnormalities of biogenic amine metabolism. J. Inherit. Metab. Dis. 1993, 16, 676–690. [Google Scholar] [CrossRef] [PubMed]
- Blau, N.; Bonafé, L.; Thöny, B. Tetrahydrobiopterin deficiencies without hyperphenylalaninemia: Diagnosis and genetics of dopa-responsive dystonia and sepiapterin reductase deficiency. Mol. Genet. Metab. 2001, 74, 172–185. [Google Scholar] [CrossRef] [PubMed]
- Opladen, T.; Hoffmann, G.F.; Blau, N. An international survey of patients with tetrahydrobiopterin deficiencies presenting with hyperphenylalaninaemia. J. Inherit. Metab. Dis. 2012, 35, 963–973. [Google Scholar] [CrossRef]
- Horvath, G.A.; Stockler-Ipsiroglu, S.G.; Salvarinova-Zivkovic, R.; Lillquist, Y.P.; Connolly, M.; Hyland, K.; Blau, N.; Rupar, T.; Waters, P.J. Autosomal recessive GTP cyclohydrolase I deficiency without hyperphenylalaninemia: Evidence of a phenotypic continuum between dominant and recessive forms. Mol. Genet. Metab. 2008, 94, 127–131. [Google Scholar] [CrossRef]
- van Os, J.; Kapur, S. Schizophrenia. Lancet 2009, 374, 635–645. [Google Scholar] [CrossRef] [PubMed]
- Johnstone, E.C.; MacMillan, J.F.; Crow, T.J. The occurrence of organic disease of possible or probable aetiological significance in a population of 268 cases of first episode schizophrenia. Psychol. Med. 1987, 17, 371–379. [Google Scholar] [CrossRef] [PubMed]
- Bonnot, O.; Herrera, P.M.; Tordjman, S.; Walterfang, M. Secondary psychosis induced by metabolic disorders. Front. Cell. Neurosci. 2015, 9, 177. [Google Scholar] [CrossRef] [PubMed]
- Merritt, J.; Tanguturi, Y.; Fuchs, C.; Cundiff, A.W. Medical etiologies of secondary psychosis in children and adolescents. Child Adolesc. Psychiatr. Clin. 2020, 29, 29–42. [Google Scholar] [CrossRef] [PubMed]
- Karim, Z.; Lyoumi, S.; Nicolas, G.; Deybach, J.-C.; Gouya, L.; Puy, H. Porphyrias: A 2015 update. Clin. Res. Hepatol. Gastroenterol. 2015, 39, 412–425. [Google Scholar] [CrossRef]
- Vakili, R.; Armanpoor, P. Acute intermittent porphyria: A diagnostic challenge. Iran. J. Pediatr. 2016, 26, e5238. [Google Scholar] [CrossRef] [PubMed]
- Duque-Serrano, L.; Patarroyo-Rodriguez, L.; Gotlib, D.; Molano-Eslava, J.C. Psychiatric aspects of acute porphyria: A comprehensive review. Curr. Psychiatry Rep. 2018, 20, 5. [Google Scholar] [CrossRef] [PubMed]
- Moghadasian, M.H.; Salen, G.; Frohlich, J.J.; Scudamore, C.H. Cerebrotendinous xanthomatosis: A rare disease with diverse manifestations. Arch. Neurol. 2002, 59, 527–529. [Google Scholar] [CrossRef]
- Patterson, M.C.; Hendriksz, C.J.; Walterfang, M.; Sedel, F.; Vanier, M.T.; Wijburg, F. NP-C Guidelines Working Group. Recommendations for the diagnosis and management of Niemann–Pick disease type C: An update. Mol. Genet. Metab. 2012, 106, 330–344. [Google Scholar] [CrossRef]
- Patterson, M.C.; Vecchio, D.; Prady, H.; Abel, L.; Wraith, E.J. Miglustat for treatment of Niemann-Pick C disease: A randomised controlled study. Lancet Neurol. 2007, 6, 765–772. [Google Scholar] [CrossRef]
- Morris, A.A.M.; Kožich, V.; Santra, S.; Andria, G.; Ben-Omran, T.I.; Chakrapani, A.B.; Crushell, E.; Henderson, M.J.; Hochuli, M.; Huemer, M.; et al. Guidelines for the diagnosis and management of cystathionine beta-synthase deficiency. J. Inherit. Metab. Dis. 2017, 40, 49–74. [Google Scholar] [CrossRef] [PubMed]
- Carrillo-Carrasco, N.; Chandler, R.J.; Venditti, C.P. Combined methylmalonic acidemia and homocystinuria, cblC type. I. Clinical presentations, diagnosis and management. J. Inherit. Metab. Dis. 2012, 35, 91–102. [Google Scholar] [CrossRef] [PubMed]
- Saudubray, J.-M.; Baumgartner, M.R.; Walter, J.H. Inborn Metabolic Diseases: Diagnosis and Treatment; Springer: Berlin/Heidelberg, Germany, 2016. [Google Scholar]
- Mew, N.A.; Simpson, K.L.; Gropman, A.L.; Lanpher, B.C.; Chapman, K.A.; Summar, M.L. Urea Cycle Disorders Overview. In GeneReviews® [Internet]; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Cox, D.W. Disorders of copper transport. Br. Med. Bull. 1999, 55, 544–555. [Google Scholar] [CrossRef] [PubMed]
- Litwin, T.; Dusek, P.; Szafrański, T.; Dzieżyc, K.; Członkowska, A.; Rybakowski, J.K. Psychiatric manifestations in Wilson’s disease: Possibilities and difficulties for treatment. Ther. Adv. Psychopharmacol. 2018, 8, 199–211. [Google Scholar] [CrossRef]
- Mulligan, C.; Bronstein, J.M. Wilson disease: An overview and approach to management. Neurol. Clin. 2020, 38, 417–432. [Google Scholar] [CrossRef]
- Shah, N.; Kumar, D. Wilson’s disease, psychosis, and ECT. J. ECT 1997, 13, 278–279. [Google Scholar]
- Cornic, F.; Consoli, A.; Tanguy, M.L.; Bonnot, O.; Périsse, D.; Tordjman, S.; Laurent, C.; Cohen, D. Association of adolescent catatonia with increased mortality and morbidity: Evidence from a prospective follow-up study. Schizophr. Res. 2009, 113, 233–240. [Google Scholar] [CrossRef]
- Cohen, D.; Nicolas, J.D.; Flament, M.F.; Périsse, D.; Dubos, P.F.; Bonnot, O.; Speranza, M.; Graindorge, C.; Tordjman, S.; Mazet, P. Clinical relevance of chronic catatonic schizophrenia in children and adolescents: Evidence from a prospective naturalistic study. Schizophr. Res. 2005, 76, 301–308. [Google Scholar] [CrossRef]
- Cohen, D.; Flament, M.; Dubos, P.-F.; Basquin, M. Case series: Catatonic syndrome in young people. J. Am. Acad. Child Adolesc. Psychiatry 1999, 38, 1040–1046. [Google Scholar] [CrossRef]
- de Clérambault, G.G. Psychoses a Base D’automatisme et Syndrome D’automatisme; Masson et Cie: Paris, France, 1927. [Google Scholar]
- Zhou, X.; Cui, Y.; Han, J. Methylmalonic acidemia: Current status and research priorities. Intractable Rare Dis. Res. 2018, 7, 73–78. [Google Scholar] [CrossRef]
- Shchelochkov, O.A.; Carrillo, N.; Venditti, C. Propionic Acidemia. In GeneReviews® [Internet]; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Pillai, N.R.; Stroup, B.M.; Poliner, A.; Rossetti, L.; Rawls, B.; Shayota, B.J.; Soler-Alfonso, C.; Tunuguntala, H.P.; Goss, J.; Craigen, W.; et al. Liver transplantation in propionic and methylmalonic acidemia: A single center study with literature review. Mol. Genet. Metab. 2019, 128, 431–443. [Google Scholar] [CrossRef] [PubMed]
- Touati, G.; Valayannopoulos, V.; Mention, K.; de Lonlay, P.; Jouvet, P.; Depondt, E.; Assoun, M.; Souberbielle, J.C.; Rabier, D.; Ogier de Baulny, H.; et al. Methylmalonic and propionic acidurias: Management without or with a few supplements of specific amino acid mixture. J. Inherit. Metab. Dis. 2006, 29, 288–298. [Google Scholar] [CrossRef] [PubMed]
- Lara, M.C.; Valentino, M.L.; Torres-Torronteras, J.; Hirano, M.; Martí, R. Mitochondrial neurogastrointestinal encephalomyopathy (MNGIE): Biochemical features and therapeutic approaches. Biosci. Rep. 2007, 27, 151–163. [Google Scholar] [CrossRef] [PubMed]
- Hirano, M.; Lagier-Tourenne, C.; Valentino, M.L.; Martí, R.; Nishigaki, Y. Thymidine phosphorylase mutations cause instability of mitochondrial DNA. Gene 2005, 354, 152–156. [Google Scholar] [CrossRef] [PubMed]
- Garone, C.; Tadesse, S.; Hirano, M. Clinical and genetic spectrum of mitochondrial neurogastrointestinal encephalomyopathy. Brain 2011, 134, 3326–3332. [Google Scholar] [CrossRef] [PubMed]
- Bardosi, A.; Creutzfeldt, W.; DiMauro, S.; Felgenhauer, K.; Friede, R.L.; Goebel, H.H.; Kohlschütter, A.; Mayer, G.; Rahlf, G.; Servidei, S. Myo-, neuro-, gastrointestinal encephalopathy (MNGIE syndrome) due to partial deficiency of cytochrome-c-oxidase: A new mitochondrial multisystem disorder. Acta Neuropathol. 1987, 74, 248–258. [Google Scholar] [CrossRef] [PubMed]
- Hirano, M. Mitochondrial Neurogastrointestinal Encephalopathy Disease. In GeneReviews® [Internet]; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Lara, M.C.; Weiss, B.; Illa, I.; Madoz, P.; Massuet, L.; Andreu, A.L.; Valentino, M.L.; Anikster, Y.; Hirano, M.; Martí, R. Infusion of platelets transiently reduces nucleoside overload in MNGIE. Neurology 2006, 67, 1461–1463. [Google Scholar] [CrossRef] [PubMed]
- Halter, J.; Schüpbach, W.; Casali, C.; Elhasid, R.; Fay, K.; Hammans, S.; Illa, I.; Kappeler, L.; Krähenbühl, S.; Lehmann, T.; et al. Allogeneic hematopoietic SCT as treatment option for patients with mitochondrial neurogastrointestinal encephalomyopathy (MNGIE): A consensus conference proposal for a standardized approach. Bone Marrow Transplant. 2011, 46, 330–337. [Google Scholar] [CrossRef] [PubMed]
- Huisman, S.; Mulder, P.; Kuijk, J.; Kerstholt, M.; van Eeghen, A.; Leenders, A.; van Balkom, I.; Oliver, C.; Piening, S.; Hennekam, R. Self-injurious behavior. Neurosci. Biobehav. Rev. 2018, 84, 483–491. [Google Scholar] [CrossRef]
- Gahr, M.; Plener, P.L.; Kölle, M.A.; Freudenmann, R.W.; Schönfeldt-Lecuona, C. Self-mutilation induced by psychotropic substances: A systematic review. Psychiatry Res. 2012, 200, 977–983. [Google Scholar] [CrossRef]
- Honings, S.; Drukker, M.; Groen, R.; van Os, J. Psychotic experiences and risk of self-injurious behaviour in the general population: A systematic review and meta-analysis. Psychol. Med. 2016, 46, 237–251. [Google Scholar] [CrossRef] [PubMed]
- Fischer, J.-F.; Mainka, T.; Worbe, Y.; Pringsheim, T.; Bhatia, K.; Ganos, C. Self-injurious behaviour in movement disorders: Systematic review. J. Neurol. Neurosurg. Psychiatry 2020, 91, 712–719. [Google Scholar] [CrossRef]
- Lewers, J.; Ceballos-Picot, I.; Shirley, T.; Mockel, L.; Egami, K.; Jinnah, H. Consequences of impaired purine recycling in dopaminergic neurons. Neuroscience 2008, 152, 761–772. [Google Scholar] [CrossRef]
- Visser, J.E.; Bär, P.R.; Jinnah, H.A. Lesch–Nyhan disease and the basal ganglia. Brain Res. Rev. 2000, 32, 449–475. [Google Scholar] [CrossRef]
- Doucet, B.P.; Jegatheesan, D.; Burke, J. Late diagnosis of Lesch-Nyhan disease variant. Case Rep. 2013, 2013, bcr2013201997. [Google Scholar] [CrossRef] [PubMed]
- Fu, R.; Sutcliffe, D.; Zhao, H.; Huang, X.; Schretlen, D.J.; Benkovic, S.; Jinnah, H.A. Clinical severity in Lesch–Nyhan disease: The role of residual enzyme and compensatory pathways. Mol. Genet. Metab. 2015, 114, 55–61. [Google Scholar] [CrossRef] [PubMed]
- Jinnah, H.A.; Ceballos-Picot, I.; Torres, R.J.; Visser, J.E.; Schretlen, D.J.; Verdu, A.; Laróvere, L.E.; Chen, C.J.; Cossu, A.; Wu, C.H.; et al. Attenuated variants of Lesch-Nyhan disease. Brain 2010, 133, 671–689. [Google Scholar] [CrossRef]
- Fu, R.; Ceballos-Picot, I.; Torres, R.J.; Larovere, L.E.; Yamada, Y.; Nguyen, K.V.; Hegde, M.; Visser, J.E.; Schretlen, D.J.; Nyhan, W.L.; et al. Genotype–phenotype correlations in neurogenetics: Lesch-Nyhan disease as a model disorder. Brain 2014, 137, 1282–1303. [Google Scholar] [CrossRef]
- Fu, R.; Chen, C.-J.; Jinnah, H. Genotypic and phenotypic spectrum in attenuated variants of Lesch–Nyhan disease. Mol. Genet. Metab. 2014, 112, 280–285. [Google Scholar] [CrossRef]
- Page, T.; Nyhan, W.L. The spectrum of HPRT deficiency: An update. In Purine and Pyrimidine Metabolism in Man VI. Advances in Experimental Medicine and Biology; Springer: Boston, MA, USA, 1989; pp. 129–133. [Google Scholar]
- Jinnah, H.A. Lesch-Nyhan disease: From mechanism to model and back again. Dis. Model. Mech. 2009, 2, 116–121. [Google Scholar] [CrossRef]
- Wong, D.F.; Harris, J.C.; Naidu, S.; Yokoi, F.; Marenco, S.; Dannals, R.F.; Ravert, H.T.; Yaster, M.; Evans, A.; Rousset, O.; et al. Dopamine transporters are markedly reduced in Lesch-Nyhan disease in vivo. Proc. Natl. Acad. Sci. USA 1996, 93, 5539–5543. [Google Scholar] [CrossRef] [PubMed]
- Torres, R.J.; Puig, J.G. Hypoxanthine-guanine phosophoribosyltransferase (HPRT) deficiency: Lesch-Nyhan syndrome. Orphanet J. Rare Dis. 2007, 2, 48. [Google Scholar]
- Taira, T.; Kobayashi, T.; Hori, T. Disappearance of self-mutilation in a patient with Lesch—Nyhan syndrome after bilateral chronic stimulation of the globus pallidus internus: Case report. J. Neurosurg. 2003, 98, 414–416. [Google Scholar] [CrossRef] [PubMed]
- van de Burgt, N.; van Koningsbruggen, S.; Behrens, L.; Leibold, N.; Martinez-Martinez, P.; Mannens, M.; van Amelsvoort, T. Screening for inborn errors of metabolism in psychotic patients using Next Generation Sequencing. J. Psychiatr. Res. 2021, 138, 125–129. [Google Scholar] [CrossRef]


{kind=link}
| Study | N. of Patients | Age | IEM | Psychiatric Manifestations | Red Flags/Other Manifestations | Type of Onset of PMs | Time of Onset of PMs |
|---|---|---|---|---|---|---|---|
| Abbott et al., 1987 [17] | 63 | 19 years (range 8–30 years, SD 11 years) | Homocystinuria due to CBSd | Behavior and personality disorders, depression, obsessive compulsive disorder | / | Chronic | 20 years (range 10–31 years, SD 11 years) |
| Al-Owain et al., 2013 [18] | 9 | 14.9 years (range 7½-28 years) | MDs due to BCS1L gene mutation | Hypomania, anxiety, intermittent psychosis, attention deficit hyperactivity disorder | Lactic acidosis, cortical visual dysfunction | Chronic | / |
| Alam et al., 2012 [19] | 1 | 15 years | WD | Behavioral disorders, irrelevant speech, attention deficit | Corneal Kayser–Fleischer ring, dystonia, tremor, dysphagia | Chronic | 15 years |
| Albokhari et al., 2022 [20] | 7 | 5.14 years (range 3–9 years) | CDG | Aggressive behavior, oppositional defiant disorder, obsessive compulsive disorder, attention deficit hyperactivity disorder | Hypotonia, protein-losing enteropathy, hepatic involvement, developmental delay, skeletal abnormalities | Chronic | / |
| Anderson and Ernst, 1994 [21] | 40 | 13.9 years (range 2–32 years) | LNS | SIBs | Developmental delay, teething and renal problems | Chronic | 3 years (range 1–10 years, SD 1.96 years) |
| Bonnot et al., 2010 [22] | 2 | / | CTX | Aggressive behavior, oppositional defiant disorder learning disabilities, attention deficit hyperactivity disorder | Diarrhea, cataract, pes cavus and hammer toes, progressive neurological dysfunction, cognitive impairment | Chronic | 11 years (range 9–13 years) |
| Bonnot et al., 2019 [23] | 44 | 29.2 years (range 1.9–69.8 years) | NPC | Psychotic breaks, mood and anxiety disorders, impaired impulse control | Ataxia, dysarthria, supranuclear gaze palsy, epilepsy, dysphagia, cataplexy, cognitive impairment, visceral symptoms | Acute and Chronic | 17.9 years (range 2.5–67.9 years) |
| Boon and Ellis, 1989 [24] | 2 | 8.5 years (range 6–11 years) | AIP | Psychotic breaks (auditory and visual hallucinations, bizarre and disorganized thought and behavior) | Abdominal pain, diarrhea, nausea, and emesis | Acute | 8.5 years (range 6–11 years) |
| Cappelletti et al., 2013 [25] | 35 | 11.5 years (range 4–24 years, SD 6.2 years) | PKU | Mood and anxiety disorders | Executive functions deficit | Chronic | / |
| Chen et al., 2022 [26] | 56 | 12 years (range 10–20 years) | Late-onset cblC deficiency | Psychotic symptoms (hallucinations, bizarre and disorganized thought) and behavioral disorders | Movement disorders, mental regression, cardiovascular diseases, pulmonary hypertension, visual impairments | Acute and Chronic | / |
| Dening and Berrios 1989 [27] | 195 | 19.7 years (SD 8.7 years) | WD | Irritability, personality changes, aggressive behavior, depression, catatonia | Movement disorders, corneal Kayser–Fleischer ring, hepatic disorders, cognitive impairment | Chronic | / |
| Dening and Berrios 1990 [28] | 129 | 18.8 years (SD 7.9 years) | WD | Depression, irritability, personality changes, aggressive behavior | Movement disorders, corneal Kayser–Fleischer ring, hepatic disorders, cognitive impairment | Chronic | / |
| Erlich, 2019 [29] | 1 | 15 years | PKU | Generalized anxiety disorder, depression, history of suicidal ideation | Mild conductive hearing loss, obesity, blonde hair, blue eyes, fair complexion | Chronic | / |
| Fraidakis, 2013 [30] | 6 | 35 years (range 17–58 years) | CTX | Oppositional defiant behaviors and deficits in attention, impulsivity, aggressiveness, anxiety, depression | Diarrhea, pes cavus and hammer toes, progressive neurological dysfunction, cognitive impairment | Chronic | 31.8 years (range 6–50 years) |
| Freeman et al., 1975 [31] | 1 | 15 years | MTHFR deficiency | Psychotic symptoms (hallucinations, delusions, flat affect), catatonic posturing, withdrawal, anorexia | Tremor, cognitive impairment | Chronic | 15 years |
| Gardeitchik et al., 2012 [32] | 90 | 1.62 years (range 0.11–43.14 years) | UCDs | Confusion, behavioral problems, irritability, protein aversion, food refusal, adverse reactions to high-protein foods | Emesis, lethargy, altered consciousness, developmental delay, failure to thrive, hypotonia | Acute | / |
| Giannitelli et al. 2018 [33] | 160 | Childhood and adolescence | NPC, Hunter syndrome | Psychotic symptoms | / | Acute and chronic | Childhood and adolescence |
| Gökçen et al., 2013 [34] | 1 | 13 years | L-2 HGA | Anxiety | Learning disorders, intentional tremors, truncal ataxia, titubation, dysmetria, dysdiadochokinesia, dysarthria | Acute | 13 years |
| Hidalgo Mazzei et al., 2014 [35] | 1 | 17 years | Homocystinuria due to CBSd | Psychotic symptoms (hallucinations, delusional thoughts), mutism, anxiety, psychomotor agitation | Ectopia lentis, thrombopulmonary embolism | Acute | 17 years |
| Kevere et al., 2014 [36] | 116 | 15.56 years | MTHFR deficiency, hyperomocisteinemia | Schizophrenia-spectrum and mood disorders | Cognitive disturbances | Acute and chronic | / |
| Kumar, 2012 [37] | 1 | 15 years | AIP | Psychotic symptoms (bizarre and disorganized thought and behavior) | None | Acute | 14 years |
| Lahutte et al., 2008 [38] | 3 | 15.7 years (range 14–17 years) | Storage disease | Catatonia, psychotic symptoms | / | Acute | / |
| Libernini et al., 2012 [39] | 1 | 15 years | MNGIE | Mood disorder, anorexia nervosa symptoms | Hypotonia, diverticulosis of duodenum and ileum, muscular hypotrophy, reduced deep tendon reflexes, claw feet, hyporexia, weight loss, emesis | Chronic | 15 years |
| Mandoki and Sumner, 1994 [40] | 1 | 9 years | HC | Aggressive behavior, personality disorder (explosive type), hallucinations | Stomachaches, headaches, sleep disturbances | Acute | 9 years |
| Manti et al., 2020 [41] | 28 | 19.9 years (range 5–44 years, SD 10.9 years) | PTPsd | Generalized anxiety disorder; depressive disorder; obsessive compulsive disorder, attention deficit hyperactivity disorder | Movement disorders, intellectual disability, autonomic dysfunction, sleep disorders, epilepsy, scoliosis | Chronic | / |
| Manti et al., 2016 [42] | 46 | 22.6 years (range 12–44 years, SD 7.35) | Early-treated PKU | Generalized anxiety disorder, depressive disorder, personality disorder, specific phobia, selective mutism | None | Chronic | 14.5 years (range 6–32 years) |
| Niwinski et al., 2021 [43] | 1 | 15 years | OCT deficiency | Social anxiety disorder, pervasive developmental disorder, selective mutism, atypical anorexia nervosa | Protein aversion | Chronic | 13 years |
| Pan et al., 2013 [44] | 2 | 19 years-youth | GTPCH deficiency, MTHFR deficiency | Major depressive disorder, suicidal ideation, non-suicidal self-injury | / | Acute and Chronic | 14 years |
| Rahmandar et al., 2014 [45] | 1 | 13 years | CblC deficiency | Psychotic symptoms (hallucinations, altered mental status), anorexia symptoms | Seizures, ataxia, urinary infections | Acute | 13 years |
| Roze et al., 2003 [46] | 2 | 20 years (range 16–24 years) | CblC deficiency | Psychotic symptoms (delusions, dissociative symptoms, auditory and visual hallucinations) | Progressive neuropathy, urinary incontinence, unsteady gait, areflexic paraparesis | Chronic | 16 years |
| Sandu et al., 2009 [47] | 1 | 17 years | NPC | Psychotic symptoms (auditory and visual hallucinations, paranoid delusions), obsessive compulsive symptoms, aggressive behavior | Neonatal jaundice, developmental delay, absence epilepsy, dysdiadochokinesia, ataxia, dysarthria | Acute | 16 years |
| Santosh et al., 1994 [48] | 1 | 14 years | AIP | Psychosis, catatonia | Headache, intellectual disability, muteness, cramp-like epigastric pain, insomnia, echolalia | Acute | 14 years |
| Serrano et al., 2009 [49] | 9 | 10.6 years (range 4–27 years) | UCDs | Eating disorders, PICA, anxiety, confusion, disorientation, delirium-like state, behavioral and personality disorders, tic disorder | Gastrointestinal symptoms, protein aversion, cerebellar syndrome, loss of consciousness, coma, development delay, strabismus, hepatomegaly, autism traits | Acute and chronic | 5,8 years (range 2–15 years) |
| Simons et al., 2006 [1] | 53 | Range 0–18 years | PKU, MDs, MMA, storage diseases, HGA, Homocystinuria, OCT deficiency | Behavior, mood and anxiety disorders, attention deficit hyperactivity disorder, oppositional defiant disorder | Cognitive impairment | Chronic | / |
| Smith et al., 1988 [50] | 544 | 8 years | Early-treated PKU | Defiant behavior, hyperactivity, anxiety | / | Chronic | / |
| Srinivas et al., 2008 [51] | 15 | 19.8 years (SD 5.8 years) | WD | Mood (both depressive and manic manifestations) and anxiety disorders, schizophreniform-like symptoms | Neuropathy, Parkinson’s disease, and cognitive decline | Acute | / |
| Tantawy et al., 2019 [52] | 22 | 14.6 years (range 6–29 years, SD 6.2 years) | GD | Depression, generalized anxiety disorder, attention deficit hyperactivity disorder, schizophrenia-like disorder, suicidality, tic disorder | / | Chronic | 2 years (range 1–10 years) |
| Valayannopoulos et al., 2012 [53] | 6 | 7.1 years (range 2–16 years) | CTP deficiency due to SLC6A8 gene mutation | Psychotic symptoms (hallucinations), behavior disturbance, anxiety, hyperactivity | Myoclonic and absence epilepsy, intellectual disability, muscular hypotonia, autistic traits | Chronic | / |
| Weglage et al., 2000 [54] | 42 | 14.7 years (range 10–18 years, SD 2.9) | PKU | Behavioral, mood and anxiety disorders | / | Chronic | / |
| Wouters et al., 2014 [55] | 1 | 16 years | NPC | Psychotic symptoms (auditory and visual hallucinations, paranoid delusions), agitation, anxiety, hyperactivity | Cognitive regression, ataxia, swallowing problems, vertical supranuclear gaze palsy, splenomegaly | Acute | 16 years |
| TIME OF ONSET | metabolic condition | psychiatric signs |
|---|---|---|
| Infancy–Early childhood: 1 month–5 years-old | PKU and biopterin metabolism errors Urea cycle disorders Methylmalonic and propionic acidemia Congenital disorders of glycosylation Lesch–Nyhan syndrome Gaucher disease | Aggressive behavior, oppositional defiant disorder, obsessive compulsive disorder, attention deficit hyperactivity disorder, mood and anxiety disorders, eating disorders, SIBs, tic disorder |
| Childhood–Adolescence: 5–15 years-old | PKU and biopterin metabolism errors Urea cycle disorders Porphyrias Mitochondrial disorders Cerebrotendinous xanthomatosis Homocystinuria Creatine transporter deficiency | Psychosis, mood and anxiety disorders, catatonia, eating disorders, aggressive behavior, oppositional defiant disorder, obsessive compulsive disorder, attention deficit hyperactivity disorder |
| Late Adolescence: 15–18 years-old | PKU and biopterin metabolism errors Cerebrotendinous xanthomatosis Homocystinuria Niemann–Pick type C Wilson disease MNGIE | Psychosis, mood and anxiety disorders, catatonia, eating disorders, aggressive behavior |
| MOOD AND ANXIETY DISORDERS | SCHIZOPHRENIA-SPECTRUM DISORDERS | CATATONIA | EATING DISORDERS | SELF-INJURIOUS BEHAVIORS | |
|---|---|---|---|---|---|
| Inborn errors of the metabolism (IEMs) | PKU BH4 deficiencies L-2 HGA MTHFR deficiency CBSd MDs CTX GD WD CDG Acute porphirias NPC | Acute porphirias CTX NPC CBSd MTHFR deficiency CblC WD UCDs MDs | Acute porphirias CTX Lysosomal storage diseases WD UCDs Homocysteine metabolism disorders | UCDs MMA PA MNGIE | LNS PKU PTPsd Hepatolenticular degeneration |
| DISEASES | PSYCHIATRIC SIGNS | NEUROLOGICAL SIGNS | SYSTEMIC SIGNS |
|---|---|---|---|
| ACUTE PORPHYRIAS | Psychosis, mood disorders (namely depressive type), confusion, catatonia. | Acute peripheral neuropathy, dysautonomia, epilepsy. | Severe abdominal pain, nausea, emesis, and constipation. Cutaneous signs. |
| CEREBROTENDINOUS XANTHOMATOSIS | Schizophreniform attacks. Attention deficit hyperactivity disorder. | Progressive spastic paraparesis, cerebellar ataxia, polyneuropathy. Epilepsy, cognitive impairment, dementia. | Xanthomas, juvenile cataract, chronic diarrhea. |
| NIEMANN–PICK TYPE C DISEASE | Psychosis, behavioral disorders, depression, autism-spectrum-like disorders. | Supranuclear gaze palsy. Progressive ataxia, epilepsy, spasticity, dysarthria, dysphagia, deafness, cataplexy. | Hepatosplenomegaly. Cherry-red spot on the macula or gray pigmentation around the fovea in the fundus oculi. |
| HOMOCYSTEINE METABOLISM DISORDERS | Personality and behavioral (aggressiveness) disorders, depression, obsessive compulsive disorder, schizophrenic-like disorders. | Focal neurological signs, epilepsy, movement disorders, peripheral neuropathy, pyramidal signs, strokes. | Thromboembolic events. Lenticular ectopia, severe myopia, optic atrophy. Atherothrombotic disease. |
| UREA CYCLE DISORDERS | Mood disorders or psychosis, eating disorders. | Stroke-like episodes (diplopia, hemiparesis) coma, epilepsy, pyramidal signs. | Nausea, vomiting, and headache. |
| WILSON’S DISEASE | Mood disorders (depressive and manic manifestations), personality changes, aggressive behavior, schizophrenia-like presentation, catatonia. | Dysarthria, ataxia, hypomimia and motor clumsiness. Spasticity, seizures, gait, and movement disorders. Dysphagia and autonomic dysfunction. | Corneal Kayser–Fleischer ring. Chronic liver disease. |
| 1 MONTH–5 YEAR-OLD | 5–15-YEAR-OLD | 15–18-YEAR-OLD | ||||
|---|---|---|---|---|---|---|
| IEMs | Red Flags/Other Manifestations | IEMs | Red Flags/Other Manifestations | IEMs | Red Flags/Other Manifestations | |
| MOOD AND ANXIETY DISORDERS | PKU and biopterin metabolism errors; congenital disorders of glycosylation; Gaucher disease | Developmental delay; blonde hair; blue eyes; fair complexion; hypotonia; protein-losing enteropathy; hepatic involvement; skeletal abnormalities | PKU and biopterin metabolism errors; porphyrias; mitochondrial disorders; cerebrotendinous xanthomatosis; homocystinuria; creatine transporter deficiency; hydroxyglutaric aciduria | Executive functions’ deficit; intellectual disability; learning disorders; cerebellar signs; movement disorders; autonomic dysfunction; abdominal pain; nausea; emesis; cutaneous signs; epilepsy; neuropathy; xanthomas; juvenile cataract; thromboembolic events; autism traits | PKU and biopterin metabolism errors; cerebrotendinous xanthomatosis; homocystinuria; Niemann–Pick type C; Wilson disease | Movement disorder; intellectual disability; autonomic dysfunction; epilepsy; executive functions deficit; supranuclear gaze palsy; hepatosplenomegaly; xanthomas; juvenile cataract; thromboembolic events; corneal Kayser–Fleischer ring; chronic liver disease |
| SCHIZOPHRENIA-SPECTRUM DISORDERS | Urea cycle disorders; porphyrias; mitochondrial disorders; cerebrotendinous xanthomatosis; homocystinuria; creatine transporter deficiency | Stroke-like episodes; epilepsy, pyramidal signs; nausea; emesis; autonomic dysfunction; abdominal pain; cutaneous signs; xanthomas; juvenile cataract; thromboembolic events; autism traits | Cerebrotendinous xanthomatosis; homocystinuria; Niemann–Pick type C; Wilson disease | Supranuclear gaze palsy; hepatosplenomegaly; xanthomas; juvenile cataract; thromboembolic events; corneal Kayser–Fleischer ring; chronic liver disease; cerebellar signs; movement disorders; autonomic dysfunction | ||
| CATATONIA | Urea cycle disorders; porphyrias; cerebrotendinous xanthomatosis; homocystinuria | Stroke-like episodes; epilepsy; pyramidal signs; nausea; emesis; autonomic dysfunction; abdominal pain; cutaneous signs; xanthomas; juvenile cataract; thromboembolic events | Cerebrotendinous xanthomatosis; homocystinuria; Wilson disease | Xanthomas; juvenile cataract; thromboembolic events; corneal Kayser–Fleischer ring; chronic liver disease; cerebellar signs; movement disorders; autonomic dysfunction | ||
| EATING DISORDERS | Urea cycle disorders; methylmalonic and propionic acidemia | Protein aversion; emesis; altered consciousness; developmental delay; hypotonia; gastrointestinal symptoms; cerebellar syndrome; strabismus; hepatomegaly; autism traits | Urea cycle disorders | Protein aversion; emesis; altered consciousness; hypotonia; gastrointestinal symptoms; cerebellar syndrome; strabismus; hepatomegaly; autism traits | MNGIE | Hypotonia; duodenum and ileum diverticulosis; muscular hypotrophy; reduced deep-tendon reflexes; claw feet; hyporexia; weight loss; emesis |
| SIBs | PKU and biopterin metabolism errors; Lesch–Nyhan syndrome | Developmental delay; teething and renal problems | ||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Baglioni, V.; Bozza, F.; Lentini, G.; Beatrice, A.; Cameli, N.; Colacino Cinnante, E.M.; Terrinoni, A.; Nardecchia, F.; Pisani, F. Psychiatric Manifestations in Children and Adolescents with Inherited Metabolic Diseases. J. Clin. Med. 2024, 13, 2190. https://doi.org/10.3390/jcm13082190
Baglioni V, Bozza F, Lentini G, Beatrice A, Cameli N, Colacino Cinnante EM, Terrinoni A, Nardecchia F, Pisani F. Psychiatric Manifestations in Children and Adolescents with Inherited Metabolic Diseases. Journal of Clinical Medicine. 2024; 13(8):2190. https://doi.org/10.3390/jcm13082190
Chicago/Turabian StyleBaglioni, Valentina, Fabiola Bozza, Giuliana Lentini, Annachiara Beatrice, Noemi Cameli, Elisa Maria Colacino Cinnante, Arianna Terrinoni, Francesca Nardecchia, and Francesco Pisani. 2024. "Psychiatric Manifestations in Children and Adolescents with Inherited Metabolic Diseases" Journal of Clinical Medicine 13, no. 8: 2190. https://doi.org/10.3390/jcm13082190
APA StyleBaglioni, V., Bozza, F., Lentini, G., Beatrice, A., Cameli, N., Colacino Cinnante, E. M., Terrinoni, A., Nardecchia, F., & Pisani, F. (2024). Psychiatric Manifestations in Children and Adolescents with Inherited Metabolic Diseases. Journal of Clinical Medicine, 13(8), 2190. https://doi.org/10.3390/jcm13082190