
Clinical and Genetic Correlation in Neurocristopathies: Bridging a Precision Medicine Gap

 ,
,  ,
,  ,
,  ,
, 

Abstract
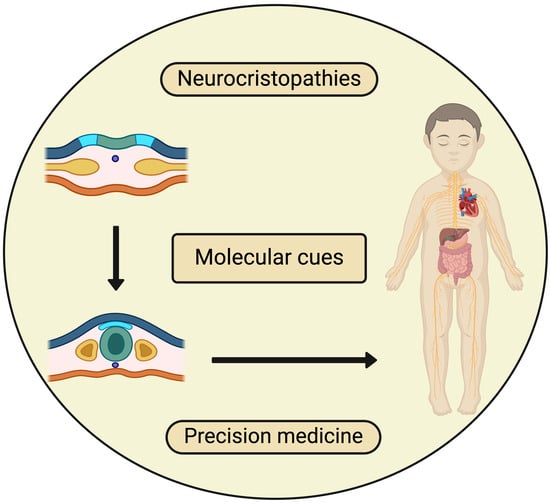
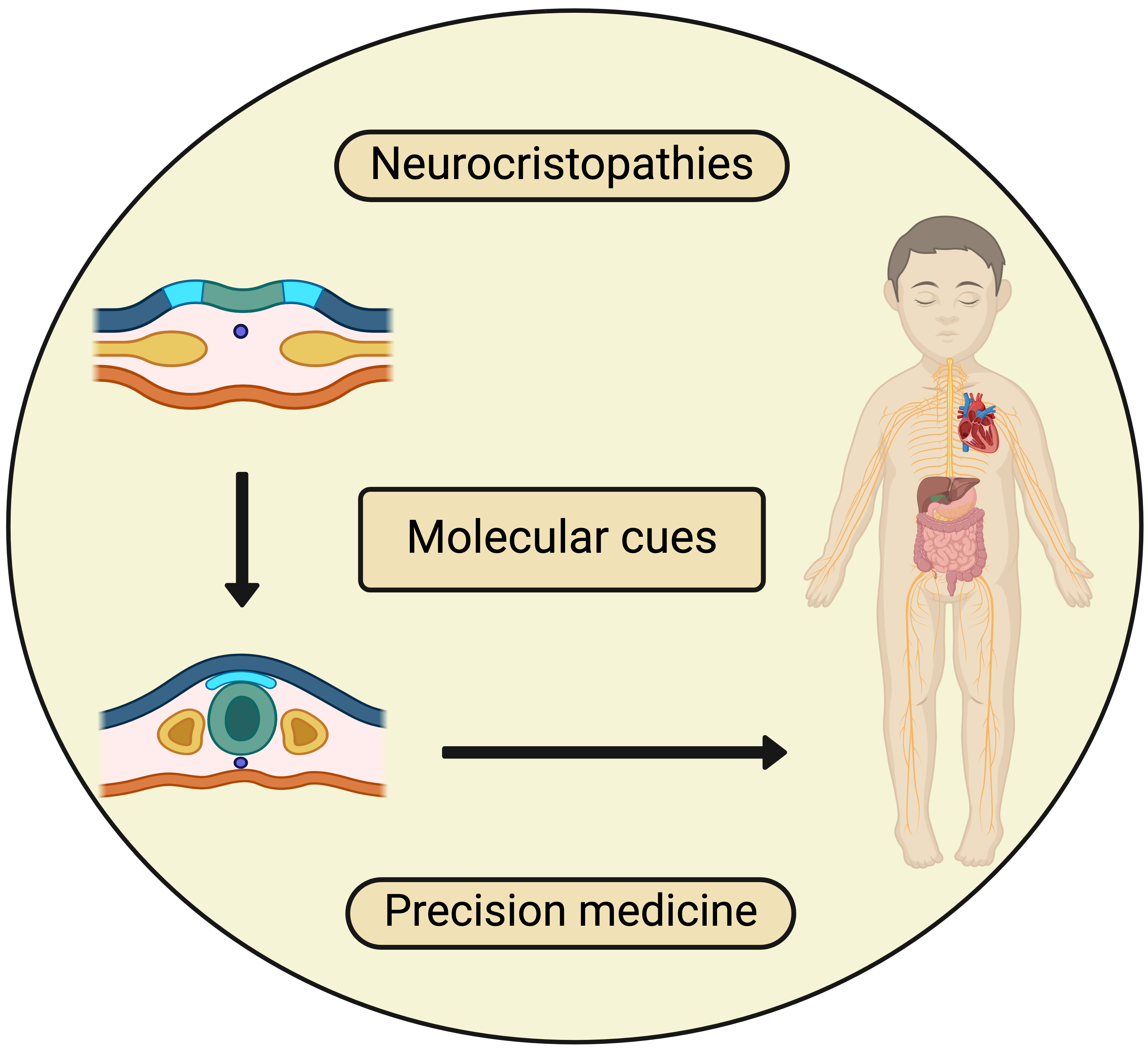
:
1. Introduction
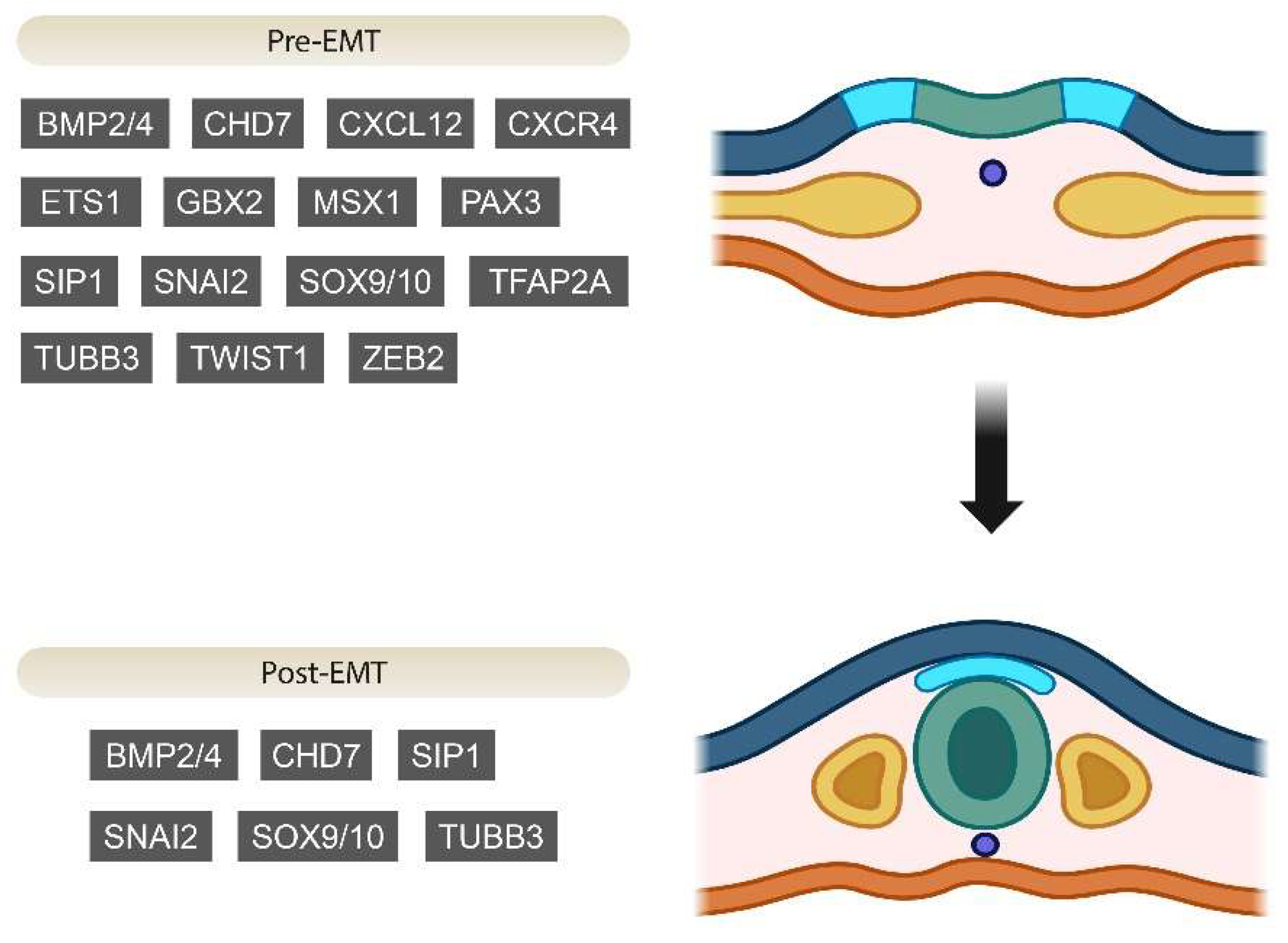
2. Genes Involved in EMT during Neural Crest Development
2.1. Pre-EMT Genes
2.2. Post-EMT Genes and Regulation
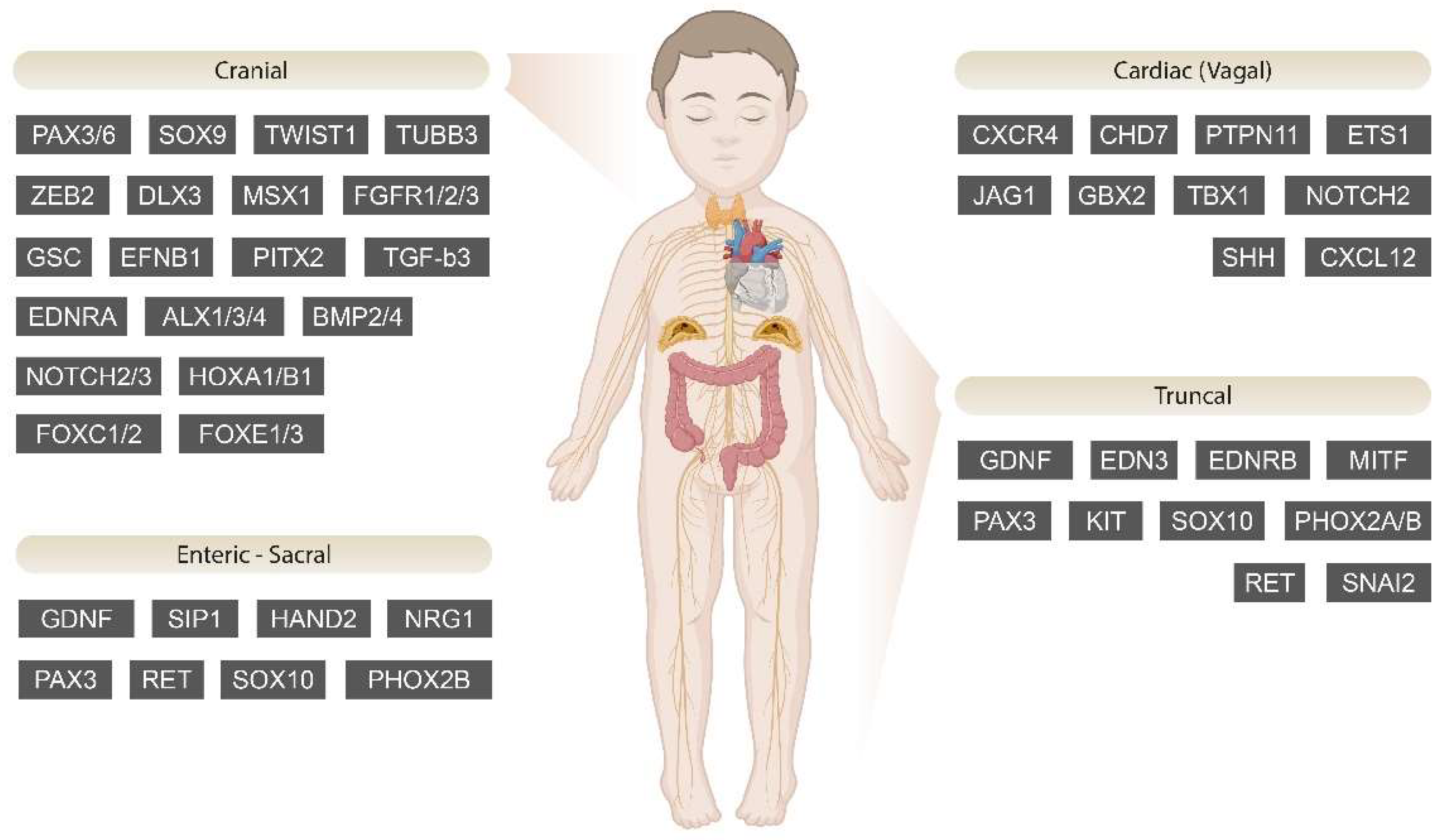
3. NCPs
3.1. Cranial NCPs
3.1.1. Goldenhar Syndrome
3.1.2. Axenfeld–Rieger Syndrome (ARS)
3.1.3. Craniosynostosis
3.1.4. Craniofacial–Deafness–Hand Syndrome (CFDS)
3.1.5. Tricho-Dento-Osseous Syndrome (TDO)
3.1.6. Peter’s Anomaly
3.1.7. Bamforth–Lazarus Syndrome
3.1.8. Branchio-Oculo-Facial Syndrome (BOFS)
3.1.9. Cerebral Autosomal Dominant Arteriopathy with Subcortical Infarcts and Leukoencephalopathy (CADASIL)
3.1.10. Congenital Aniridia
3.1.11. Frontonasal Dysplasia (FND)
3.1.12. Hajdu–Cheney Syndrome
3.1.13. Moebius Syndrome (MBS)
3.1.14. Pierre Robin Sequence (PRS)
3.1.15. Mowat–Wilson Syndrome
3.1.16. SAMS Disorder
3.2. Cardiac (Vagal) NCPs
3.2.1. DiGeorge Syndrome
3.2.2. CHARGE Syndrome
3.2.3. Velocardiofacial Syndrome
3.2.4. Fetal Alcohol Spectrum Disorders
3.2.5. Alagille Syndrome
3.2.6. Noonan–LEOPARD Syndrome
3.2.7. Jacobsen Syndrome
3.3. Truncal NCPs
3.3.1. Waardenburg Syndrome (WS)
3.3.2. Piebaldism
3.3.3. Oculocutaneous Albinism (OCA)
3.3.4. Congenital Central Hypoventilation Syndrome (CCHS)
3.4. Enteric–Sacral NCPs
Hirschsprung Disease (HSCR)
4. Cancers
4.1. Melanocytic Cancers
Malignant Melanoma
4.2. Schwann Cell Cancers
Schwannoma
4.3. Sympathetic Cell Cancers
4.3.1. Neuroblastoma
4.3.2. Pheochromocytomas–Paragangliomas (PPGLs)
4.4. Cancers from Multiple Lineages
Familial Medullary Thyroid Carcinomas
5. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
Abbreviations
| ALK | ALK receptor tyrosine kinase |
| ALX | ALX homeobox |
| AMELX | Amelogenin X-linked |
| ARS | Axenfeld–Rieger Syndrome |
| ARTN | Artemin |
| ATRX | ATRX chromatin remodeler |
| B3GLCT | Beta 3-glucosyltransferase |
| BCOR | BCL6 corepressor |
| BLS | Bamborth–Lazarus Syndrome |
| BMP | Bone morphogenetic protein |
| BOFS | Branchio-oculo-facial syndrome |
| BRAF | B-Raf proto-oncogene |
| CADASIL | Cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy |
| CCHS | Congenital central hypoventilation syndrome |
| CDH | Cadherin |
| CDK13 | Cyclin-dependent kinase 13 |
| CDX2 | Caudal type homeobox 2 |
| CFDS | Craniofacial–deafness–hand syndrome |
| CFNS | Craniofrontonasal syndrome |
| CHD7 | Chromodomain helicase DNA-binding protein 7 |
| COL | Collagen |
| CRBP1 | Retinol-binding protein 1 |
| CTF | C terminal fragment |
| CTF | C-Terminal Fragment |
| Cxcl12 | C-X-C motif chemokine ligand 12 |
| Cxcr4 | C-X-C motif chemokine receptor 4 |
| CYP1B1 | Cytochrome P450 family 1 subfamily B member 1 |
| DBH | Dopamine beta-hydroxylase |
| DCDC1 | Doublecortin domain-containing 1 |
| Dkk | Dickkopf WNT signaling pathway inhibitor |
| DLX | Distal-less homeobox |
| DOPA | Dihydroxyphenylalanine |
| ECE1 | Endothelin-converting enzyme 1 |
| ECG | Electrocardiography |
| EDN3 | Endothelin 3 |
| EDNRB | Endothelin receptor type B |
| EFNB1 | Ephrin-B1 |
| EGF | Epidermal growth factor |
| EGR2 | Early growth response 2 |
| ELP4 | Elongator acetyltransferase complex subunit 4 |
| EMT | Epithelial–mesenchymal transition |
| ETS1 | ETS proto-oncogene 1, transcription factor |
| FGF | Fibroblast growth factor |
| FGFR | Fibroblast growth factor receptor |
| FND | Frontonasal dysplasia |
| FOXC1 | Forkhead box C1 |
| FOXD3 | Forkhead box D3 |
| FOXF | Forkhead box F |
| FOXI1/2 | Forkhead box I1/2 |
| GATA | GATA binding domain |
| GBX2 | Gastrulation brain homeobox 2 |
| GFRA1 | Glial cell-derived neurotrophic factor family receptor alpha 1 |
| GLI1 | GLI family zinc finger 1 |
| GSC | Goosecoid |
| GSX1 | GS homeobox 1 |
| HCCS | Holocytochrome c synthase |
| HCS | Hajdu–Cheney syndrome |
| HOX | Homeobox |
| HSCR | Hirschsprung disease |
| iPSCs | Induced pluripotent stem cells |
| IVF | In vitro fertilization |
| JAG1 | Jagged canonical Notch ligand 1 |
| KIT | KIT proto-oncogene, receptor tyrosine kinase |
| KRAS | KRAS proto-oncogene, GTPase |
| L1CAM | L1 cell adhesion molecule |
| LBX1 | Ladybird homeobox 1 |
| MAP2K1 | Mitogen-activated protein kinase kinase 1 |
| MAPK | Mitogen-activated protein kinase 1 |
| MBS | Moebius syndrome |
| MITF | Melanocyte-inducing transcription factor |
| MSX | Msh Homeobox |
| MWS | Mowat–Wilson syndrome |
| MYCN | MYCN proto-oncogene |
| MYO1H | Myosin IH |
| MYT1 | Myelin transcription factor 1 |
| NC | Neural crest |
| NCCs | Neural crest cells |
| NF1 | Neurofibromin 1 |
| NGS | Next-generation sequencing |
| NOTCH | NOTCH receptor |
| NRAS | NRAS proto-oncogene |
| NRAS | NRAS proto-oncogene, GTPase |
| NREM | Non-rapid eye movement |
| NRG1 | Neuregulin 1 |
| NSD | Nuclear receptor binding SET domain protein |
| NSD3 | Nuclear receptor binding SET domain protein 3 |
| NTN1 | Netrin 1 |
| OAVS | Oculo-auriculo-vertebral spectrum |
| OCA | Oculocutaneous albinism |
| PA | Pharyngeal arch |
| PARMs | Polyalanine repeat expansion mutations |
| PAX3/6/7 | Paired Box3/6/7 |
| PBX2 | Pre-B-cell leukemia homeobox 2 |
| PHOX2B | Paired-like homeobox 2B |
| PITX2 | Paired-like homeodomain 2 |
| PPGL | Pheochromocytoma–paraganglioma |
| PRDM5/16 | PR/SET domain 5/16 |
| PRS | Pierre Robin sequence |
| PSPN | Persephin |
| PTPN11 | Protein tyrosine phosphatase non-receptor type 11 |
| RAB | Member of the RAS oncogene family |
| RAF1 | Raf-1 proto-oncogene, serine/threonine kinase |
| RET | Rearranged during transfection |
| ROBO | Roundabout guidance receptor |
| SAMS | Short stature, auditory canal atresia, mandibular hypoplasia, and skeletal abnormalities |
| SDHB | Succinate dehydrogenase complex iron sulfur subunit B |
| SDHC | Succinate dehydrogenase complex subunit C |
| SDHD | Succinate dehydrogenase complex subunit D |
| SF3B2 | Splicing factor 3b subunit 2 |
| SHH | Sonic hedgehog |
| SHOC2 | SHOC2 leucine-rich repeat scaffold |
| SIP1 | Smad interaction protein 1 |
| SLIT | Slit guidance ligand |
| SNAI1/2 | Snail family transcriptional repressor 1/2 |
| SOS1 | Son of Sevenless Ras/Rac guanine nucleotide exchange factor 1 |
| SOX | SRY-box transcription factor |
| SP7 | Transcription factor Sp7 |
| Tak1 | Transforming growth factor beta-activated kinase 1 |
| TBX1 | T-box transcription factor 1 |
| TDO | Tricho-dento-osseous syndrome |
| TERT | Telomerase reverse transcriptase |
| TFAP2 | Transcription factor AP-2 |
| TGF-β | Transforming growth factor beta |
| TH | Tyrosine hydroxylase |
| TIMP3 | Tissue inhibitor 3 of metalloprotease-1 |
| TRIM44 | Tripartite motif-containing 44 |
| TUBB3 | Tubulin beta 3 class II |
| TWIST | Twist family bHLH transcription factor |
| USP9X | Ubiquitin specific peptidase 9 X-linked |
| UVA | Ultraviolet A |
| VHL | Von Hippel–Lindau tumor suppressor |
| VSMCs | Vascular smooth muscle cells |
| WS | Waardenburg syndrome |
| ZEB2 | Zinc finger E-box binding homeobox 2 |
| ZIC1 | Zic family member 1 |
References
- Dermitzakis, I.; Manthou, M.E.; Meditskou, S.; Miliaras, D.; Kesidou, E.; Boziki, M.; Petratos, S.; Grigoriadis, N.; Theotokis, P. Developmental Cues and Molecular Drivers in Myelinogenesis: Revisiting Early Life to Re-Evaluate the Integrity of CNS Myelin. Curr. Issues Mol. Biol. 2022, 44, 3208–3237. [Google Scholar] [CrossRef] [PubMed]
- Theotokis, P. Exploring Myelin Dynamics in Demyelinating Disorders at the Molecular Level. Curr. Issues Mol. Biol. 2024, 46, 1754–1756. [Google Scholar] [CrossRef] [PubMed]
- Simões-Costa, M.; Bronner, M.E. Establishing Neural Crest Identity: A Gene Regulatory Recipe. Development 2015, 142, 242–257. [Google Scholar] [CrossRef]
- Jacques-Fricke, B.T.; Gammill, L.S. Neural Crest Specification and Migration Independently Require NSD3-Related Lysine Methyltransferase Activity. Mol. Biol. Cell 2014, 25, 4174–4186. [Google Scholar] [CrossRef] [PubMed]
- Bronner, M.E.; LeDouarin, N.M. Development and Evolution of the Neural Crest: An Overview. Dev. Biol. 2012, 366, 2–9. [Google Scholar] [CrossRef] [PubMed]
- Taneyhill, L.A. To Adhere or Not to Adhere: The Role of Cadherins in Neural Crest Cell Development. Cell Adhes. Migr. 2008, 2, 223–230. [Google Scholar] [CrossRef]
- Bolande, R.P. Neurocristopathy: Its Growth and Development in 20 Years. Pediatr. Pathol. Lab. Med. J. Soc. Pediatr. Pathol. Affil. Int. Paediatr. Pathol. Assoc. 1997, 17, 1–25. [Google Scholar]
- Brabletz, T.; Kalluri, R.; Nieto, M.A.; Weinberg, R.A. EMT in Cancer. Nat. Rev. Cancer 2018, 18, 128–134. [Google Scholar] [CrossRef]
- Watt, K.E.N.; Trainor, P.A. Neurocristopathies; Elsevier: Amsterdam, The Netherlands, 2014; pp. 361–394. [Google Scholar]
- Meulemans, D.; Bronner-Fraser, M. Gene-Regulatory Interactions in Neural Crest Evolution and Development. Dev. Cell 2004, 7, 291–299. [Google Scholar] [CrossRef]
- Vega-Lopez, G.A.; Cerrizuela, S.; Aybar, M.J. Trunk Neural Crest Cells: Formation, Migration and Beyond. Int. J. Dev. Biol. 2017, 61, 5–15. [Google Scholar] [CrossRef]
- Groves, A.K.; LaBonne, C. Setting Appropriate Boundaries: Fate, Patterning and Competence at the Neural Plate Border. Dev. Biol. 2014, 389, 2–12. [Google Scholar] [CrossRef]
- Khudyakov, J.; Bronner-Fraser, M. Comprehensive Spatiotemporal Analysis of Early Chick Neural Crest Network Genes. Dev. Dyn. Off. Publ. Am. Assoc. Anat. 2009, 238, 716–723. [Google Scholar] [CrossRef]
- Nikitina, N.; Sauka-Spengler, T.; Bronner-Fraser, M. Dissecting Early Regulatory Relationships in the Lamprey Neural Crest Gene Network. Proc. Natl. Acad. Sci. USA 2008, 105, 20083–20088. [Google Scholar] [CrossRef]
- Garnett, A.T.; Square, T.A.; Medeiros, D.M. BMP, Wnt and FGF Signals Are Integrated through Evolutionarily Conserved Enhancers to Achieve Robust Expression of Pax3 and Zic Genes at the Zebrafish Neural Plate Border. Development 2012, 139, 4220–4231. [Google Scholar] [CrossRef]
- Bhat, N.; Kwon, H.-J.; Riley, B.B. A Gene Network That Coordinates Preplacodal Competence and Neural Crest Specification in Zebrafish. Dev. Biol. 2013, 373, 107–117. [Google Scholar] [CrossRef]
- Matsuo-Takasaki, M.; Matsumura, M.; Sasai, Y. An Essential Role of Xenopus Foxi1a for Ventral Specification of the Cephalic Ectoderm during Gastrulation. Development 2005, 132, 3885–3894. [Google Scholar] [CrossRef]
- Murdoch, B.; DelConte, C.; García-Castro, M.I. Pax7 Lineage Contributions to the Mammalian Neural Crest. PLoS ONE 2012, 7, e41089. [Google Scholar] [CrossRef]
- Barembaum, M.; Bronner, M.E. Identification and Dissection of a Key Enhancer Mediating Cranial Neural Crest Specific Expression of Transcription Factor, Ets-1. Dev. Biol. 2013, 382, 567–575. [Google Scholar] [CrossRef]
- Aybar, M.J.; Nieto, M.A.; Mayor, R. Snail Precedes Slug in the Genetic Cascade Required for the Specification and Migration of the Xenopus Neural Crest. Development 2003, 130, 483–494. [Google Scholar] [CrossRef]
- Chacon, J.; Rogers, C.D. Early Expression of Tubulin Beta-III in Avian Cranial Neural Crest Cells. Gene Expr. Patterns 2019, 34, 119067. [Google Scholar] [CrossRef]
- Karunamuni, G.H.; Ma, P.; Gu, S.; Rollins, A.M.; Jenkins, M.W.; Watanabe, M. Connecting Teratogen-Induced Congenital Heart Defects to Neural Crest Cells and Their Effect on Cardiac Function. Birth Defects Res. Part C Embryo Today Rev. 2014, 102, 227–250. [Google Scholar] [CrossRef]
- Fujita, K.; Ogawa, R.; Kawawaki, S.; Ito, K. Roles of Chromatin Remodelers in Maintenance Mechanisms of Multipotency of Mouse Trunk Neural Crest Cells in the Formation of Neural Crest-Derived Stem Cells. Mech. Dev. 2014, 133, 126–145. [Google Scholar] [CrossRef]
- Multiple Mutations in Mouse Chd7 Provide Models for CHARGE Syndrome|Human Molecular Genetics|Oxford Academic. Available online: https://academic.oup.com/hmg/article/14/22/3463/614337?login=false (accessed on 18 February 2024).
- Barriga, E.H.; Theveneau, E. In Vivo Neural Crest Cell Migration Is Controlled by “Mixotaxis”. Front. Physiol. 2020, 11, 586432. [Google Scholar] [CrossRef]
- Gallik, K.L.; Treffy, R.W.; Nacke, L.M.; Ahsan, K.; Rocha, M.; Green-Saxena, A.; Saxena, A. Neural Crest and Cancer: Divergent Travelers on Similar Paths. Mech. Dev. 2017, 148, 89–99. [Google Scholar] [CrossRef]
- Taneyhill, L.A.; Schiffmacher, A.T. Cadherin Dynamics during Neural Crest Cell Ontogeny. Prog. Mol. Biol. Transl. Sci. 2013, 116, 291–315. [Google Scholar]
- Ji, Y.; Hao, H.; Reynolds, K.; McMahon, M.; Zhou, C.J. Wnt Signaling in Neural Crest Ontogenesis and Oncogenesis. Cells 2019, 8, 1173. [Google Scholar] [CrossRef]
- Kumar, D.; Nitzan, E.; Kalcheim, C. YAP Promotes Neural Crest Emigration through Interactions with BMP and Wnt Activities. Cell Commun. Signal. 2019, 17, 69. [Google Scholar] [CrossRef]
- Shoval, I.; Ludwig, A.; Kalcheim, C. Antagonistic Roles of Full-Length N-Cadherin and Its Soluble BMP Cleavage Product in Neural Crest Delamination. Development 2007, 134, 491–501. [Google Scholar] [CrossRef]
- Ahmad, M.H.; Ghosh, B.; Rizvi, M.A.; Ali, M.; Kaur, L.; Mondal, A.C. Neural Crest Cells Development and Neuroblastoma Progression: Role of Wnt Signaling. J. Cell. Physiol. 2023, 238, 306–328. [Google Scholar] [CrossRef]
- Ille, F.; Atanasoski, S.; Falk, S.; Ittner, L.M.; Märki, D.; Büchmann-Møller, S.; Wurdak, H.; Suter, U.; Taketo, M.M.; Sommer, L. Wnt/BMP Signal Integration Regulates the Balance between Proliferation and Differentiation of Neuroepithelial Cells in the Dorsal Spinal Cord. Dev. Biol. 2007, 304, 394–408. [Google Scholar] [CrossRef]
- Hong, L.; Li, N.; Gasque, V.; Mehta, S.; Ye, L.; Wu, Y.; Li, J.; Gewies, A.; Ruland, J.; Hirschi, K.K.; et al. Prdm6 Controls Heart Development by Regulating Neural Crest Cell Differentiation and Migration. JCI Insight 2022, 7, e156046. [Google Scholar] [CrossRef]
- Basch, M.L.; Bronner-Fraser, M. Neural Crest Inducing Signals. Neural Crest Induction Differ. 2006, 589, 24–31. [Google Scholar]
- Strobl-Mazzulla, P.H.; Sauka-Spengler, T.; Bronner-Fraser, M. Histone Demethylase JmjD2A Regulates Neural Crest Specification. Dev. Cell 2010, 19, 460–468. [Google Scholar] [CrossRef]
- Beleza-Meireles, A.; Clayton-Smith, J.; Saraiva, J.M.; Tassabehji, M. Oculo-Auriculo-Vertebral Spectrum: A Review of the Literature and Genetic Update. J. Med. Genet. 2014, 51, 635–645. [Google Scholar] [CrossRef]
- Wieczorek, D.; Ludwig, M.; Boehringer, S.; Jongbloet, P.H.; Gillessen-Kaesbach, G.; Horsthemke, B. Reproduction Abnormalities and Twin Pregnancies in Parents of Sporadic Patients with Oculo-Auriculo-Vertebral Spectrum/Goldenhar Syndrome. Hum. Genet. 2007, 121, 369–376. [Google Scholar] [CrossRef]
- Barisic, I.; Odak, L.; Loane, M.; Garne, E.; Wellesley, D.; Calzolari, E.; Dolk, H.; Addor, M.-C.; Arriola, L.; Bergman, J.; et al. Prevalence, Prenatal Diagnosis and Clinical Features of Oculo-Auriculo-Vertebral Spectrum: A Registry-Based Study in Europe. Eur. J. Hum. Genet. 2014, 22, 1026–1033. [Google Scholar] [CrossRef]
- Singhal, D.; Tripathy, K. Oculo Auriculo Vertebral Spectrum. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2024. [Google Scholar]
- Szabo-Rogers, H.L.; Smithers, L.E.; Yakob, W.; Liu, K.J. New Directions in Craniofacial Morphogenesis. Dev. Biol. 2010, 341, 84–94. [Google Scholar] [CrossRef]
- Berenguer, M.; Tingaud-Sequeira, A.; Colovati, M.; Melaragno, M.I.; Bragagnolo, S.; Perez, A.B.A.; Arveiler, B.; Lacombe, D.; Rooryck, C. A Novel de Novo Mutation in MYT1, the Unique OAVS Gene Identified so Far. Eur. J. Hum. Genet. EJHG 2017, 25, 1083–1086. [Google Scholar] [CrossRef]
- Varma, A.R.; Meshram, R.J.; Varma, A.R.; Dixit, A.S.; Zabak, S.S.; Kulkarni, C.A. Multicorrection Goldenhar Syndrome (Facio-Auriculo-Vertebral Dysplasia): A Rare Follow-up Case of 12-Year-Old Female. Pan Afr. Med. J. 2021, 39, 96. [Google Scholar] [CrossRef]
- Foerst-Potts, L.; Sadler, T.W. Disruption of Msx-1 and Msx-2 Reveals Roles for These Genes in Craniofacial, Eye, and Axial Development. Dev. Dyn. 1997, 209, 70–84. [Google Scholar] [CrossRef]
- Lopes, M.; Goupille, O.; Cloment, C.S.; Lallemand, Y.; Cumano, A.; Robert, B. Msx Genes Define a Population of Mural Cell Precursors Required for Head Blood Vessel Maturation. Development 2011, 138, 3055–3066. [Google Scholar] [CrossRef]
- Stoll, C.; Viville, B.; Treisser, A.; Gasser, B. A family with dominant oculoauriculovertebral spectrum. Am. J. Med. Genet. 1998, 78, 345–349. [Google Scholar] [CrossRef]
- Zhang, Y.; Bi, S.; Dai, L.; Zhao, Y.; Liu, Y.; Shi, Z. Clinical Report and Genetic Analysis of a Chinese Neonate with Craniofacial Microsomia Caused by a Splicing Variant of the Splicing Factor 3b Subunit 2 Gene. Mol. Genet. Genomic Med. 2023, 11, e2268. [Google Scholar] [CrossRef]
- Rosa, R.F.M.; Graziadio, C.; Lenhardt, R.; Alves, R.P.M.; Paskulin, G.A.; Zen, P.R.G. Central Nervous System Abnormalities in Patients with Oculo-Auriculo-Vertebral Spectrum (Goldenhar Syndrome). Arq. Neuropsiquiatr. 2010, 68, 98–102. [Google Scholar] [CrossRef]
- Seifi, M.; Walter, M.A. Axenfeld-Rieger Syndrome. Clin. Genet. 2018, 93, 1123–1130. [Google Scholar] [CrossRef]
- Zamora, E.A.; Salini, B. Axenfeld-Rieger Syndrome. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2024. [Google Scholar]
- Reis, L.M.; Amor, D.J.; Haddad, R.A.; Nowak, C.B.; Keppler-Noreuil, K.M.; Chisholm, S.A.; Semina, E.V. Alternative Genetic Diagnoses in Axenfeld–Rieger Syndrome Spectrum. Genes 2023, 14, 1948. [Google Scholar] [CrossRef]
- Taneyhill, L.A.; Bronner-Fraser, M. Recycling Signals in the Neural Crest. J. Biol. 2006, 4, 10. [Google Scholar] [CrossRef]
- Ittner, L.M.; Wurdak, H.; Schwerdtfeger, K.; Kunz, T.; Ille, F.; Leveen, P.; Hjalt, T.A.; Suter, U.; Karlsson, S.; Hafezi, F.; et al. Compound Developmental Eye Disorders Following Inactivation of TGFbeta Signaling in Neural-Crest Stem Cells. J. Biol. 2005, 4, 11. [Google Scholar] [CrossRef]
- Vega-Lopez, G.A.; Cerrizuela, S.; Tribulo, C.; Aybar, M.J. Neurocristopathies: New Insights 150 Years after the Neural Crest Discovery. Dev. Biol. 2018, 444, S110–S143. [Google Scholar] [CrossRef]
- Sauka-Spengler, T.; Bronner-Fraser, M. A Gene Regulatory Network Orchestrates Neural Crest Formation. Nat. Rev. Mol. Cell Biol. 2008, 9, 557–568. [Google Scholar] [CrossRef]
- Katouni, K.; Nikolaou, A.; Mariolis, T.; Protogerou, V.; Chrysikos, D.; Theofilopoulou, S.; Filippou, D. Syndromic Craniosynostosis: A Comprehensive Review. Cureus 2023, 15, e50448. [Google Scholar] [CrossRef]
- Piacentino, M.L.; Li, Y.; Bronner, M.E. Epithelial-to-Mesenchymal Transition and Different Migration Strategies as Viewed from the Neural Crest. Curr. Opin. Cell Biol. 2020, 66, 43–50. [Google Scholar] [CrossRef]
- Mellott, D.O.; Burke, R.D. Divergent Roles for Eph and Ephrin in Avian Cranial Neural Crest. BMC Dev. Biol. 2008, 8, 56. [Google Scholar] [CrossRef] [PubMed]
- Bush, J.O. Chapter Five—Cellular and Molecular Mechanisms of EPH/EPHRIN Signaling in Evolution and Development. In Current Topics in Developmental Biology; Soriano, P.M., Ed.; Cell Signaling Pathways in Development; Academic Press: Cambridge, MA, USA, 2022; Volume 149, pp. 153–201. [Google Scholar]
- Gad, A.; Laurino, M.; Maravilla, K.R.; Matsushita, M.; Raskind, W.H. Sensorineural Deafness, Distinctive Facial Features, and Abnormal Cranial Bones: A New Variant of Waardenburg Syndrome? Am. J. Med. Genet. Part A 2008, 146, 1880–1885. [Google Scholar] [CrossRef]
- Wang, Q.; Fang, W.-H.; Krupinski, J.; Kumar, S.; Slevin, M.; Kumar, P. Pax Genes in Embryogenesis and Oncogenesis. J. Cell. Mol. Med. 2008, 12, 2281–2294. [Google Scholar] [CrossRef]
- Fazel, M.; Afshari, E.; Jarrahi, N. Dental Management of Tricho-Dento-Osseous Syndrome in Adolescent Patients: Literature Review and Case Presentation. Dent. Res. J. 2021, 18, 98. [Google Scholar] [CrossRef]
- Jin, H.-S.; Son, J.; Seo, Y.-J.; Choi, S.-R.; Ahn, H.-B.; Go, Y.; Lim, J.; Oh, K.-I.; Ryu, K.-S.; Lee, J.-H. Salt Dependence of DNA Binding Activity of Human Transcription Factor Dlx3. Int. J. Mol. Sci. 2022, 23, 9497. [Google Scholar] [CrossRef]
- Miletich, I.; Sharpe, P.T. Neural Crest Contribution to Mammalian Tooth Formation. Birth Defects Res. Part C Embryo Today Rev. 2004, 72, 200–212. [Google Scholar] [CrossRef]
- Viale-Bouroncle, S.; Felthaus, O.; Schmalz, G.; Brockhoff, G.; Reichert, T.E.; Morsczeck, C. The Transcription Factor DLX3 Regulates the Osteogenic Differentiation of Human Dental Follicle Precursor Cells. Stem Cells Dev. 2012, 21, 1936–1947. [Google Scholar] [CrossRef]
- Jain, P.; Kaul, R.; Saha, S.; Sarkar, S. Tricho-Dento-Osseous Syndrome and Precocious Eruption. J. Clin. Exp. Dent. 2017, 9, e494–e497. [Google Scholar] [CrossRef]
- Whitehouse, L.L.E.; Smith, C.E.L.; Poulter, J.A.; Brown, C.J.; Patel, A.; Lamb, T.; Brown, L.R.; O’Sullivan, E.A.; Mitchell, R.E.; Berry, I.R.; et al. Novel DLX3 Variants in Amelogenesis Imperfecta with Attenuated Tricho-dento-osseous Syndrome. Oral Dis. 2019, 25, 182–191. [Google Scholar] [CrossRef] [PubMed]
- Bhandari, R.; Ferri, S.; Whittaker, B.; Liu, M.; Lazzaro, D.R. Peters Anomaly: Review of the Literature. Cornea 2011, 30, 939. [Google Scholar] [CrossRef] [PubMed]
- Iseri, S.U.; Osborne, R.J.; Farrall, M.; Wyatt, A.W.; Mirza, G.; Nürnberg, G.; Kluck, C.; Herbert, H.; Martin, A.; Hussain, M.S.; et al. Seeing Clearly: The Dominant and Recessive Nature of FOXE3 in Eye Developmental Anomalies. Hum. Mutat. 2009, 30, 1378–1386. [Google Scholar] [CrossRef] [PubMed]
- Halder, G.; Callaerts, P.; Gehring, W.J. Induction of Ectopic Eyes by Targeted Expression of the Eyeless Gene in Drosophila. Science 1995, 267, 1788–1792. [Google Scholar] [CrossRef] [PubMed]
- Vincent, A. Further support of the role of CYP1B1 in patients with Peters anomaly. Molecular Vision. 2006. Available online: http://www.molvis.org/molvis/v12/a59/ (accessed on 20 February 2024).
- Weh, E.; Reis, L.M.; Tyler, R.C.; Bick, D.; Rhead, W.J.; Wallace, S.; McGregor, T.L.; Dills, S.K.; Chao, M.C.; Murray, J.C.; et al. Novel B3GALTL Mutations in Classic Peters plus Syndrome and Lack of Mutations in a Large Cohort of Patients with Similar Phenotypes. Clin. Genet. 2014, 86, 142–148. [Google Scholar] [CrossRef] [PubMed]
- Sandal, G.; Pirgon, O.; Ormeci, A.R. Bamforth Syndrome: Is Porencephaly a New Finding? Genet. Couns. 2013, 24, 279–282. [Google Scholar]
- Venza, I.; Visalli, M.; Parrillo, L.; De Felice, M.; Teti, D.; Venza, M. MSX1 and TGF-Β3 Are Novel Target Genes Functionally Regulated by FOXE1. Hum. Mol. Genet. 2011, 20, 1016–1025. [Google Scholar] [CrossRef] [PubMed]
- Brancaccio, A.; Minichiello, A.; Grachtchouk, M.; Antonini, D.; Sheng, H.; Parlato, R.; Dathan, N.; Dlugosz, A.A.; Missero, C. Requirement of the Forkhead Gene Foxe1, a Target of Sonic Hedgehog Signaling, in Hair Follicle Morphogenesis. Hum. Mol. Genet. 2004, 13, 2595–2606. [Google Scholar] [CrossRef] [PubMed]
- Nakada, C.; Iida, A.; Tabata, Y.; Watanabe, S. Forkhead Transcription Factor Foxe1 Regulates Chondrogenesis in Zebrafish. J. Exp. Zoolog. Part B Mol. Dev. Evol. 2009, 312, 827–840. [Google Scholar] [CrossRef]
- A Mouse Model for Hereditary Thyroid Dysgenesis and Cleft Palate|Nature Genetics. Available online: https://www.nature.com/articles/ng0898_395 (accessed on 19 February 2024).
- De Bono, C.; Liu, Y.; Ferrena, A.; Valentine, A.; Zheng, D.; Morrow, B.E. Single-Cell Transcriptomics Uncovers a Non-Autonomous Tbx1-Dependent Genetic Program Controlling Cardiac Neural Crest Cell Development. Nat. Commun. 2023, 14, 1551. [Google Scholar] [CrossRef]
- Raveh, E.; Papsin, B.C.; Forte, V. Branchio-Oculo-Facial Syndrome. Int. J. Pediatr. Otorhinolaryngol. 2000, 53, 149–156. [Google Scholar] [CrossRef] [PubMed]
- Craniofacial Defects in AP-2 Null Mutant Mice–Morriss-Kay. BioEssays-Wiley Online Library. 1996. Available online: https://onlinelibrary.wiley.com/doi/abs/10.1002/bies.950181004?casa_token=O6qzygTSCA0AAAAA:pvNHU655PPfieeFglu1hOIrq5v85mVvPjgk9rW2H6Oq297OGf-RI4zX8JBky9CAm0v4QCteZWmgCgm0 (accessed on 20 February 2024).
- Schorle, H.; Meier, P.; Buchert, M.; Jaenisch, R.; Mitchell, P.J. Transcription Factor AP-2 Essential for Cranial Closure and Craniofacial Development. Nature 1996, 381, 235–238. [Google Scholar] [CrossRef] [PubMed]
- West-Mays, J.A.; Zhang, J.; Nottoli, T.; Hagopian-Donaldson, S.; Libby, D.; Strissel, K.J.; Williams, T. AP-2αTranscription Factor Is Required for Early Morphogenesis of the Lens Vesicle. Dev. Biol. 1999, 206, 46–62. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Cornell, R.A. Redundant Activities of Tfap2a and Tfap2c Are Required for Neural Crest Induction and Development of Other Non-Neural Ectoderm Derivatives in Zebrafish Embryos. Dev. Biol. 2007, 304, 338–354. [Google Scholar] [CrossRef] [PubMed]
- Yi, S.; Albino, F.P.; Wood, B.C.; Sauerhammer, T.M.; Rogers, G.F.; Oh, A.K. An Unconventional Presentation of Branchio-Oculo-Facial Syndrome. J. Craniofac. Surg. 2016, 27, 1412. [Google Scholar] [CrossRef] [PubMed]
- Balikova, I.; Devriendt, K.; Fryns, J.-P.; Vermeesch, J.R. FOXD1 Duplication Causes Branchial Defects and Interacts with the TFAP2A Gene Implicated in the Branchio-Oculo-Facial Syndrome in Causing Eye Effects in Zebrafish. Mol. Syndromol. 2011, 1, 255–261. [Google Scholar] [CrossRef]
- Mizuno, T.; Mizuta, I.; Watanabe-Hosomi, A.; Mukai, M.; Koizumi, T. Clinical and Genetic Aspects of CADASIL. Front. Aging Neurosci. 2020, 12, 91. [Google Scholar] [CrossRef] [PubMed]
- Joutel, A.; Andreux, F.; Gaulis, S.; Domenga, V.; Cecillon, M.; Battail, N.; Piga, N.; Chapon, F.; Godfrain, C.; Tournier-Lasserve, E. The Ectodomain of the Notch3 Receptor Accumulates within the Cerebrovasculature of CADASIL Patients. J. Clin. Investig. 2000, 105, 597–605. [Google Scholar] [CrossRef] [PubMed]
- The Developmental Biology of Genetic Notch Disorders|Development|The Company of Biologists. Available online: https://journals.biologists.com/dev/article/144/10/1743/47916/The-developmental-biology-of-genetic-Notch (accessed on 20 February 2024).
- Pericytes as a New Target for Pathological Processes in CADASIL-Dziewulska. Neuropathology-Wiley Online Library. 2012. Available online: https://onlinelibrary.wiley.com/doi/abs/10.1111/j.1440-1789.2011.01290.x?casa_token=SpjS1YB0kP8AAAAA:XRDYcQMER26N94doc-DjuuCABGaKI5EshSPMkI4he232bPTEUc4Z3HYA-jrogVrUdVCxZOeScOxftI0 (accessed on 20 February 2024).
- Angiogenesis in Health and Disease|Nature Medicine. Available online: https://www.nature.com/articles/nm0603-653 (accessed on 20 February 2024).
- Baeten, J.T.; Lilly, B. Differential Regulation of NOTCH2 and NOTCH3 Contribute to Their Unique Functions in Vascular Smooth Muscle Cells. J. Biol. Chem. 2015, 290, 16226–16237. [Google Scholar] [CrossRef]
- Notch3 Is Necessary for Blood Vessel Integrity in the Central Nervous System|Arteriosclerosis, Thrombosis, and Vascular Biology. Available online: https://www.ahajournals.org/doi/full/10.1161/ATVBAHA.114.304849 (accessed on 20 February 2024).
- Notch3 Directs Differentiation of Brain Mural Cells from Human Pluripotent Stem Cell–Derived Neural Crest. Available online: https://www.science.org/doi/10.1126/sciadv.adi1737 (accessed on 20 February 2024).
- Ando, K.; Wang, W.; Peng, D.; Chiba, A.; Lagendijk, A.K.; Barske, L.; Crump, J.G.; Stainier, D.Y.R.; Lendahl, U.; Koltowska, K.; et al. Peri-Arterial Specification of Vascular Mural Cells from Naïve Mesenchyme Requires Notch Signaling. Development 2019, 146, dev165589. [Google Scholar] [CrossRef]
- French, C.R.; Seshadri, S.; Destefano, A.L.; Fornage, M.; Arnold, C.R.; Gage, P.J.; Skarie, J.M.; Dobyns, W.B.; Millen, K.J.; Liu, T.; et al. Mutation of FOXC1 and PITX2 Induces Cerebral Small-Vessel Disease. J. Clin. Investig. 2014, 124, 4877–4881. [Google Scholar] [CrossRef] [PubMed]
- Capone, C.; Cognat, E.; Ghezali, L.; Baron-Menguy, C.; Aubin, D.; Mesnard, L.; Stöhr, H.; Domenga-Denier, V.; Nelson, M.T.; Joutel, A. Reducing Timp3 or Vitronectin Ameliorates Disease Manifestations in CADASIL Mice. Ann. Neurol. 2016, 79, 387–403. [Google Scholar] [CrossRef] [PubMed]
- Daruich, A.; Duncan, M.; Robert, M.P.; Lagali, N.; Semina, E.V.; Aberdam, D.; Ferrari, S.; Romano, V.; des Roziers, C.B.; Benkortebi, R.; et al. Congenital Aniridia beyond Black Eyes: From Phenotype and Novel Genetic Mechanisms to Innovative Therapeutic Approaches. Prog. Retin. Eye Res. 2023, 95, 101133. [Google Scholar] [CrossRef] [PubMed]
- Landsend, E.C.S.; Lagali, N.; Utheim, T.P. Congenital Aniridia—A Comprehensive Review of Clinical Features and Therapeutic Approaches. Surv. Ophthalmol. 2021, 66, 1031–1050. [Google Scholar] [CrossRef] [PubMed]
- Takamiya, M.; Stegmaier, J.; Kobitski, A.Y.; Schott, B.; Weger, B.D.; Margariti, D.; Cereceda Delgado, A.R.; Gourain, V.; Scherr, T.; Yang, L.; et al. Pax6 Organizes the Anterior Eye Segment by Guiding Two Distinct Neural Crest Waves. PLoS Genet. 2020, 16, e1008774. [Google Scholar] [CrossRef]
- Samant, M.; Chauhan, B.K.; Lathrop, K.L.; Nischal, K.K. Congenital Aniridia: Etiology, Manifestations and Management. Expert Rev. Ophthalmol. 2016, 11, 135–144. [Google Scholar] [CrossRef] [PubMed]
- Tibrewal, S.; Ratna, R.; Gour, A.; Agarkar, S.; Dubey, S.; Ganesh, S.; Kekunnaya, R.; Sangwan, V.; Liu, Y.; Vanita, V. Clinical and Molecular Aspects of Congenital Aniridia—A Review of Current Concepts. Indian J. Ophthalmol. 2022, 70, 2280–2292. [Google Scholar] [CrossRef] [PubMed]
- Farlie, P.G.; Baker, N.L.; Yap, P.; Tan, T.Y. Frontonasal Dysplasia: Towards an Understanding of Molecular and Developmental Aetiology. Mol. Syndromol. 2016, 7, 312–321. [Google Scholar] [CrossRef]
- Guion-Almeida, M.L.; Richieri-Costa, A.; Saavedra, D.; Cohen, M.M. Frontonasal Dysplasia: Analysis of 21 Cases and Literature Review. Int. J. Oral Maxillofac. Surg. 1996, 25, 91–97. [Google Scholar] [CrossRef]
- Twigg, S.R.; Versnel, S.L.; Nürnberg, G.; Lees, M.M.; Bhat, M.; Hammond, P.; Hennekam, R.C.; Hoogeboom, A.J.M.; Hurst, J.A.; Johnson, D.; et al. Frontorhiny, a Distinctive Presentation of Frontonasal Dysplasia Caused by Recessive Mutations in the ALX3 Homeobox Gene. Am. J. Hum. Genet. 2009, 84, 698–705. [Google Scholar] [CrossRef]
- Kariminejad, A.; Bozorgmehr, B.; Alizadeh, H.; Ghaderi-Sohi, S.; Toksoy, G.; Uyguner, Z.O.; Kayserili, H. Skull Defects, Alopecia, Hypertelorism, and Notched Alae Nasi Caused by Homozygous ALX4 Gene Mutation. Am. J. Med. Genet. Part A 2014, 164, 1322–1327. [Google Scholar] [CrossRef]
- Beverdam, A.; Brouwer, A.; Reijnen, M.; Korving, J.; Meijlink, F. Severe Nasal Clefting and Abnormal Embryonic Apoptosis in Alx3/Alx4 Double Mutant Mice. Development 2001, 128, 3975–3986. [Google Scholar] [CrossRef]
- Dee, C.T.; Szymoniuk, C.R.; Mills, P.E.D.; Takahashi, T. Defective Neural Crest Migration Revealed by a Zebrafish Model of Alx1-Related Frontonasal Dysplasia. Hum. Mol. Genet. 2013, 22, 239–251. [Google Scholar] [CrossRef]
- Minoux, M.; Rijli, F.M. Molecular Mechanisms of Cranial Neural Crest Cell Migration and Patterning in Craniofacial Development. Development 2010, 137, 2605–2621. [Google Scholar] [CrossRef]
- Hu, D.; Marcucio, R.S.; Helms, J.A. A Zone of Frontonasal Ectoderm Regulates Patterning and Growth in the Face. Development 2003, 130, 1749–1758. [Google Scholar] [CrossRef]
- Hu, D.; Young, N.M.; Li, X.; Xu, Y.; Hallgrímsson, B.; Marcucio, R.S. A Dynamic Shh Expression Pattern, Regulated by SHH and BMP Signaling, Coordinates Fusion of Primordia in the Amniote Face. Development 2015, 142, 567–574. [Google Scholar] [CrossRef]
- Hu, D.; Marcucio, R.S. A SHH-Responsive Signaling Center in the Forebrain Regulates Craniofacial Morphogenesis via the Facial Ectoderm. Development 2009, 136, 107–116. [Google Scholar] [CrossRef]
- van den Elzen, M.E.P.; Twigg, S.R.F.; Goos, J.A.C.; Hoogeboom, A.J.M.; van den Ouweland, A.M.W.; Wilkie, A.O.M.; Mathijssen, I.M.J. Phenotypes of Craniofrontonasal Syndrome in Patients with a Pathogenic Mutation in EFNB1. Eur. J. Hum. Genet. 2014, 22, 995–1001. [Google Scholar] [CrossRef]
- Brugmann, S.A.; Allen, N.C.; James, A.W.; Mekonnen, Z.; Madan, E.; Helms, J.A. A Primary Cilia-Dependent Etiology for Midline Facial Disorders. Hum. Mol. Genet. 2010, 19, 1577–1592. [Google Scholar] [CrossRef]
- Canalis, E.; Zanotti, S. Hajdu-Cheney Syndrome: A Review. Orphanet J. Rare Dis. 2014, 9, 200. [Google Scholar] [CrossRef]
- Majewski, J.; Schwartzentruber, J.A.; Caqueret, A.; Patry, L.; Marcadier, J.; Fryns, J.-P.; Boycott, K.M.; Ste-Marie, L.-G.; McKiernan, F.E.; Marik, I.; et al. Mutations in NOTCH2 in Families with Hajdu-Cheney Syndrome. Hum. Mutat. 2011, 32, 1114–1117. [Google Scholar] [CrossRef]
- Manderfield, L.J.; High, F.A.; Engleka, K.A.; Liu, F.; Li, L.; Rentschler, S.; Epstein, J.A. Notch Activation of Jagged1 Contributes to the Assembly of the Arterial Wall. Circulation 2012, 125, 314–323. [Google Scholar] [CrossRef]
- High, F.A.; Zhang, M.; Proweller, A.; Tu, L.; Parmacek, M.S.; Pear, W.S.; Epstein, J.A. An Essential Role for Notch in Neural Crest during Cardiovascular Development and Smooth Muscle Differentiation. J. Clin. Investig. 2007, 117, 353–363. [Google Scholar] [CrossRef]
- Keyte, A.; Hutson, M.R. The Neural Crest in Cardiac Congenital Anomalies. Differentiation 2012, 84, 25–40. [Google Scholar] [CrossRef]
- Hilton, M.J.; Tu, X.; Wu, X.; Bai, S.; Zhao, H.; Kobayashi, T.; Kronenberg, H.M.; Teitelbaum, S.L.; Ross, F.P.; Kopan, R.; et al. Notch Signaling Maintains Bone Marrow Mesenchymal Progenitors by Suppressing Osteoblast Differentiation. Nat. Med. 2008, 14, 306–314. [Google Scholar] [CrossRef]
- Zanotti, S.; Smerdel-Ramoya, A.; Stadmeyer, L.; Durant, D.; Radtke, F.; Canalis, E. Notch Inhibits Osteoblast Differentiation and Causes Osteopenia. Endocrinology 2008, 149, 3890–3899. [Google Scholar] [CrossRef]
- Sekine, C.; Koyanagi, A.; Koyama, N.; Hozumi, K.; Chiba, S.; Yagita, H. Differential Regulation of Osteoclastogenesis by Notch2/Delta-like 1 and Notch1/Jagged1 Axes. Arthritis Res. Ther. 2012, 14, R45. [Google Scholar] [CrossRef]
- Jheon, A.H.; Prochazkova, M.; Meng, B.; Wen, T.; Lim, Y.-J.; Naveau, A.; Espinoza, R.; Cox, T.C.; Sone, E.D.; Ganss, B.; et al. Inhibition of Notch Signaling During Mouse Incisor Renewal Leads to Enamel Defects. J. Bone Miner. Res. 2016, 31, 152–162. [Google Scholar] [CrossRef]
- Felszeghy, S.; Suomalainen, M.; Thesleff, I. Notch Signalling Is Required for the Survival of Epithelial Stem Cells in the Continuously Growing Mouse Incisor. Differentiation 2010, 80, 241–248. [Google Scholar] [CrossRef] [PubMed]
- Kumar, D. Moebius Syndrome. J. Med. Genet. 1990, 27, 122–126. [Google Scholar] [CrossRef]
- Terzis, J.K.; Noah, E.M. Möbius and Möbius-like Patients: Etiology, Diagnosis, and Treatment Options. Clin. Plast. Surg. 2002, 29, 497–514. [Google Scholar] [CrossRef]
- Verzijl, H.T.F.M.; van der Zwaag, B.; Cruysberg, J.R.M.; Padberg, G.W. Möbius Syndrome Redefined. Neurology 2003, 61, 327–333. [Google Scholar] [CrossRef]
- Van Der Zwaag, B.; Verzijl, H.; De Bernabe, D.B.-V.; Schuster, V.; Van Bokhoven, H.; Kremer, H.; Van Reen, M.; Wichers, G.; Brunner, H.; Padberg, G. Mutation Analysis in the Candidate Möbius Syndrome Genes PGT and GATA2 on Chromosome 3 and EGR2 on Chromosome 10. J. Med. Genet. 2002, 39, e30. [Google Scholar] [CrossRef]
- Kadakia, S.; Helman, S.N.; Schwedhelm, T.; Saman, M.; Azizzadeh, B. Examining the Genetics of Congenital Facial Paralysis—A Closer Look at Moebius Syndrome. Oral Maxillofac. Surg. 2015, 19, 109–116. [Google Scholar] [CrossRef] [PubMed]
- Patel, R.M.; Liu, D.; Gonzaga-Jauregui, C.; Jhangiani, S.; Lu, J.T.; Sutton, V.R.; Fernbach, S.D.; Azamian, M.; White, L.; Edmond, J.C.; et al. An Exome Sequencing Study of Moebius Syndrome Including Atypical Cases Reveals an Individual with CFEOM3A and a TUBB3 Mutation. Mol. Case Stud. 2017, 3, a000984. [Google Scholar] [CrossRef]
- Kurosaka, H.; Trainor, P.A.; Leroux-Berger, M.; Iulianella, A. Cranial Nerve Development Requires Co-Ordinated Shh and Canonical Wnt Signaling. PLoS ONE 2015, 10, e0120821. [Google Scholar] [CrossRef]
- Chiang, C.; Litingtung, Y.; Lee, E.; Young, K.E.; Corden, J.L.; Westphal, H.; Beachy, P.A. Cyclopia and Defective Axial Patterning in Mice Lacking Sonic Hedgehog Gene Function. Nature 1996, 383, 407–413. [Google Scholar] [CrossRef]
- Goddard, J.M.; Rossel, M.; Manley, N.R.; Capecchi, M.R. Mice with Targeted Disruption of Hoxb-1 Fail to Form the Motor Nucleus of the VIIth Nerve. Development 1996, 122, 3217–3228. [Google Scholar] [CrossRef]
- Perrone, L.; Daltoé, F.; Araújo, L.; Nunes, F.; Gallottini, M. Others Clinical and Genetics Findings in Möbius Syndrome: Role of Hoxa1 and Hoxb1 Mutations. Int. J. Pathol. Clin. Res. 2019, 5, 085. [Google Scholar]
- Kremer, H.; Kuyt, L.P.; van den Helm, B.; van Reen, M.; Leunissen, J.A.M.; Hamel, B.C.J.; Jansen, C.; Mariman, E.C.M.; Frants, R.R.; Padberg, G.W. Localization of a Gene for Möbius Syndrome to Chromosome 3q by Linkage Analysis in a Dutch Family. Hum. Mol. Genet. 1996, 5, 1367–1371. [Google Scholar] [CrossRef] [PubMed]
- Uzumcu, A.; Karaman, B.; Toksoy, G.; Uyguner, Z.O.; Candan, S.; Eris, H.; Tatli, B.; Geckinli, B.; Yuksel, A.; Kayserili, H.; et al. Molecular Genetic Screening of MBS1 Locus on Chromosome 13 for Microdeletions and Exclusion of FGF9, GSH1 and CDX2 as Causative Genes in Patients with Moebius Syndrome. Eur. J. Med. Genet. 2009, 52, 315–320. [Google Scholar] [CrossRef]
- Webb, B.D.; Shaaban, S.; Gaspar, H.; Cunha, L.F.; Iacovelli, A.J.; Ye, X.; Camminady, A.; Engle, E.C.; Jabs, E.W. Moebius Syndromes, Its Genetics Remain Undefined. 10, 11 Sorting out Its Genetics Has Been Complicated, in Part, by the Not-Infrequent Misdiagnosis of Moebius Syndrome in Children Who Have CFP but Do Not Have Limited Abduction of the Eye. Am. J. Hum. Genet. 2012, 179, 91–171. [Google Scholar]
- Baxter, D.; Shanks, A.L. Pierre Robin Syndrome. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2024. [Google Scholar]
- Varadarajan, S.; Balaji, T.M.; Raj, A.T.; Gupta, A.A.; Patil, S.; Alhazmi, T.H.; Alaqi, H.A.A.; Al Omar, N.E.M.; Almutaher, S.A.B.A.; Jafer, A.A.; et al. Genetic Mutations Associated with Pierre Robin Syndrome/Sequence: A Systematic Review. Mol. Syndromol. 2021, 12, 69–86. [Google Scholar] [CrossRef]
- Yamashita, S.; Kataoka, K.; Yamamoto, H.; Kato, T.; Hara, S.; Yamaguchi, K.; Renard-Guillet, C.; Katou, Y.; Shirahige, K.; Ochi, H.; et al. Comparative Analysis Demonstrates Cell Type-Specific Conservation of SOX9 Targets between Mouse and Chicken. Sci. Rep. 2019, 9, 12560. [Google Scholar] [CrossRef]
- Bi, W.; Deng, J.M.; Zhang, Z.; Behringer, R.R.; de Crombrugghe, B. Sox9 Is Required for Cartilage Formation. Nat. Genet. 1999, 22, 85–89. [Google Scholar] [CrossRef] [PubMed]
- Bjork, B.C.; Turbe-Doan, A.; Prysak, M.; Herron, B.J.; Beier, D.R. Prdm16 Is Required for Normal Palatogenesis in Mice. Hum. Mol. Genet. 2010, 19, 774–789. [Google Scholar] [CrossRef]
- Song, Z.; Liu, C.; Iwata, J.; Gu, S.; Suzuki, A.; Sun, C.; He, W.; Shu, R.; Li, L.; Chai, Y.; et al. Mice with Tak1 Deficiency in Neural Crest Lineage Exhibit Cleft Palate Associated with Abnormal Tongue Development. J. Biol. Chem. 2013, 288, 10440–10450. [Google Scholar] [CrossRef]
- Parada, C.; Han, D.; Grimaldi, A.; Sarrión, P.; Park, S.S.; Pelikan, R.; Sanchez-Lara, P.A.; Chai, Y. Disruption of the ERK/MAPK Pathway in Neural Crest Cells as a Potential Cause of Pierre Robin Sequence. Development 2015, 142, 3734–3745. [Google Scholar] [CrossRef]
- Adam, M.P.; Conta, J.; Bean, L.J. Mowat-Wilson Syndrome. In GeneReviews®; Adam, M.P., Feldman, J., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J., Gripp, K.W., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Tronina, A.; Świerczyńska, M.; Filipek, E. First Case Report of Developmental Bilateral Cataract with a Novel Mutation in the ZEB2 Gene Observed in Mowat-Wilson Syndrome. Medicina 2023, 59, 101. [Google Scholar] [CrossRef] [PubMed]
- Ivanovski, I.; Djuric, O.; Caraffi, S.G.; Santodirocco, D.; Pollazzon, M.; Rosato, S.; Cordelli, D.M.; Abdalla, E.; Accorsi, P.; Adam, M.P.; et al. Phenotype and Genotype of 87 Patients with Mowat–Wilson Syndrome and Recommendations for Care. Genet. Med. 2018, 20, 965–975. [Google Scholar] [CrossRef] [PubMed]
- Charney, R.M.; Prasad, M.S.; Juan-Sing, C.; Patel, L.J.; Hernandez, J.C.; Wu, J.; García-Castro, M.I. Mowat-Wilson Syndrome Factor ZEB2 Controls Early Formation of Human Neural Crest through BMP Signaling Modulation. Stem Cell Rep. 2023, 18, 2254–2267. [Google Scholar] [CrossRef] [PubMed]
- Iwafuchi-Doi, M.; Matsuda, K.; Murakami, K.; Niwa, H.; Tesar, P.J.; Aruga, J.; Matsuo, I.; Kondoh, H. Transcriptional Regulatory Networks in Epiblast Cells and during Anterior Neural Plate Development as Modeled in Epiblast Stem Cells. Development 2012, 139, 3926–3937. [Google Scholar] [CrossRef] [PubMed]
- Van de Putte, T.; Maruhashi, M.; Francis, A.; Nelles, L.; Kondoh, H.; Huylebroeck, D.; Higashi, Y. Mice Lacking Zfhx1b, the Gene That Codes for Smad-Interacting Protein-1, Reveal a Role for Multiple Neural Crest Cell Defects in the Etiology of Hirschsprung Disease–Mental Retardation Syndrome. Am. J. Hum. Genet. 2003, 72, 465–470. [Google Scholar] [CrossRef] [PubMed]
- Parry, D.A.; Logan, C.V.; Stegmann, A.P.; Abdelhamed, Z.A.; Calder, A.; Khan, S.; Bonthron, D.T.; Clowes, V.; Sheridan, E.; Ghali, N.; et al. SAMS, a Syndrome of Short Stature, Auditory-Canal Atresia, Mandibular Hypoplasia, and Skeletal Abnormalities Is a Unique Neurocristopathy Caused by Mutations in Goosecoid. Am. J. Hum. Genet. 2013, 93, 1135–1142. [Google Scholar] [CrossRef] [PubMed]
- Twigg, S.R.F.; Wilkie, A.O.M. New Insights into Craniofacial Malformations. Hum. Mol. Genet. 2015, 24, R50–R59. [Google Scholar] [CrossRef] [PubMed]
- Gaunt, S.J.; Blum, M.; De Robertis, E.M. Expression of the Mouse Goosecoid Gene during Mid-Embryogenesis May Mark Mesenchymal Cell Lineages in the Developing Head, Limbs and Body Wall. Development 1993, 117, 769–778. [Google Scholar] [CrossRef] [PubMed]
- Yamada, G.; Mansouri, A.; Torres, M.; Stuart, E.T.; Blum, M.; Schultz, M.; Robertis, E.M.D.; Gruss, P. Targeted Mutation of the Murine Goosecoid Gene Results in Craniofacial Defects and Neonatal Death. Development 1995, 121, 2917–2922. [Google Scholar] [CrossRef] [PubMed]
- Belo, J.A.; Leyns, L.; Yamada, G.; De Robertis, E.M. The Prechordal Midline of the Chondrocranium Is Defective in Goosecoid-1 Mouse Mutants. Mech. Dev. 1998, 72, 15–25. [Google Scholar] [CrossRef] [PubMed]
- Clouthier, D.E.; Garcia, E.; Schilling, T.F. Regulation of Facial Morphogenesis by Endothelin Signaling: Insights from Mice and Fish. Am. J. Med. Genet. Part A 2010, 152, 2962–2973. [Google Scholar] [CrossRef]
- Nair, S.; Li, W.; Cornell, R.; Schilling, T.F. Requirements for Endothelin Type-A Receptors and Endothelin-1 Signaling in the Facial Ectoderm for the Patterning of Skeletogenic Neural Crest Cells in Zebrafish. Development 2007, 134, 335–345. [Google Scholar] [CrossRef]
- Karpinski, B.A.; Maynard, T.M.; Fralish, M.S.; Nuwayhid, S.; Zohn, I.E.; Moody, S.A.; LaMantia, A.-S. Dysphagia and Disrupted Cranial Nerve Development in a Mouse Model of DiGeorge (22q11) Deletion Syndrome. Dis. Model. Mech. 2014, 7, 245–257. [Google Scholar] [CrossRef] [PubMed]
- Calmont, A.; Ivins, S.; Van Bueren, K.L.; Papangeli, I.; Kyriakopoulou, V.; Andrews, W.D.; Martin, J.F.; Moon, A.M.; Illingworth, E.A.; Basson, M.A.; et al. Tbx1 Controls Cardiac Neural Crest Cell Migration during Arch Artery Development by Regulating Gbx2 Expression in the Pharyngeal Ectoderm. Development 2009, 136, 3173–3183. [Google Scholar] [CrossRef] [PubMed]
- Escot, S.; Blavet, C.; Faure, E.; Zaffran, S.; Duband, J.-L.; Fournier-Thibault, C. Disruption of CXCR4 Signaling in Pharyngeal Neural Crest Cells Causes DiGeorge Syndrome-like Malformations. Development 2016, 143, 582–588. [Google Scholar] [CrossRef] [PubMed]
- Pathologic Features of the CHARGE Association: Support for Involvement of the Neural Crest–Siebert. Teratology-Wiley Online Library. 1985. Available online: https://onlinelibrary.wiley.com/doi/abs/10.1002/tera.1420310303?casa_token=bAHTKYxP0ykAAAAA:oWhuTX_PrEEl269-Hr3_V7wDESo74fnX95vSpg1gZ3EfwMHRuV4_IbYcSbMg0qIru6i_P_VrobE5UgA (accessed on 18 February 2024).
- Pauli, S.; Bajpai, R.; Borchers, A. CHARGEd with Neural Crest Defects. Am. J. Med. Genet. Part C Semin. Med. Genet. 2017, 175, 478–486. [Google Scholar] [CrossRef] [PubMed]
- Randall, V.; McCue, K.; Roberts, C.; Kyriakopoulou, V.; Beddow, S.; Barrett, A.N.; Vitelli, F.; Prescott, K.; Shaw-Smith, C.; Devriendt, K.; et al. Great Vessel Development Requires Biallelic Expression of Chd7 and Tbx1 in Pharyngeal Ectoderm in Mice. J. Clin. Investig. 2009, 119, 3301–3310. [Google Scholar] [CrossRef] [PubMed]
- Kvigne, V.L.; Leonardson, G.R.; Neff-Smith, M.; Brock, E.; Borzelleca, J.; Welty, T.K. Characteristics of Children Who Have Full or Incomplete Fetal Alcohol Syndrome. J. Pediatr. 2004, 145, 635–640. [Google Scholar] [CrossRef] [PubMed]
- Smith, S.M.; Garic, A.; Flentke, G.R.; Berres, M.E. Neural Crest Development in Fetal Alcohol Syndrome. Birth Defects Res. Part C Embryo Today Rev. 2014, 102, 210–220. [Google Scholar] [CrossRef]
- Washington Smoak, I.; Byrd, N.A.; Abu-Issa, R.; Goddeeris, M.M.; Anderson, R.; Morris, J.; Yamamura, K.; Klingensmith, J.; Meyers, E.N. Sonic Hedgehog Is Required for Cardiac Outflow Tract and Neural Crest Cell Development. Dev. Biol. 2005, 283, 357–372. [Google Scholar] [CrossRef]
- Dworkin, S.; Boglev, Y.; Owens, H.; Goldie, S.J. The Role of Sonic Hedgehog in Craniofacial Patterning, Morphogenesis and Cranial Neural Crest Survival. J. Dev. Biol. 2016, 4, 24. [Google Scholar] [CrossRef]
- Ormestad, M.; Astorga, J.; Landgren, H.; Wang, T.; Johansson, B.R.; Miura, N.; Carlsson, P. Foxf1 and Foxf2 Control Murine Gut Development by Limiting Mesenchymal Wnt Signaling and Promoting Extracellular Matrix Production. Development 2006, 133, 833–843. [Google Scholar] [CrossRef]
- Connor, F.L.; Lorenzo, C.D. Chronic Intestinal Pseudo-Obstruction: Assessment and Management. Gastroenterology 2006, 130, S29–S36. [Google Scholar] [CrossRef] [PubMed]
- Kirby, M.L.; Hutson, M.R. Factors Controlling Cardiac Neural Crest Cell Migration. Cell Adhes. Migr. 2010, 4, 609–621. [Google Scholar] [CrossRef] [PubMed]
- Kamath, B.M.; Spinner, N.B.; Emerick, K.M.; Chudley, A.E.; Booth, C.; Piccoli, D.A.; Krantz, I.D. Vascular Anomalies in Alagille Syndrome. Circulation 2004, 109, 1354–1358. [Google Scholar] [CrossRef] [PubMed]
- Roberts, A.E.; Allanson, J.E.; Tartaglia, M.; Gelb, B.D. Noonan Syndrome. Lancet 2013, 381, 333–342. [Google Scholar] [CrossRef] [PubMed]
- Tartaglia, M.; Pennacchio, L.A.; Zhao, C.; Yadav, K.K.; Fodale, V.; Sarkozy, A.; Pandit, B.; Oishi, K.; Martinelli, S.; Schackwitz, W.; et al. Gain-of-Function SOS1 Mutations Cause a Distinctive Form of Noonan Syndrome. Nat. Genet. 2007, 39, 75–79. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, T.; Gulick, J.; Pratt, R.; Robbins, J. Noonan Syndrome Is Associated with Enhanced pERK Activity, the Repression of Which Can Prevent Craniofacial Malformations. Proc. Natl. Acad. Sci. USA 2009, 106, 15436–15441. [Google Scholar] [CrossRef] [PubMed]
- Mattina, T.; Perrotta, C.S.; Grossfeld, P. Jacobsen Syndrome. Orphanet J. Rare Dis. 2009, 4, 9. [Google Scholar] [CrossRef]
- Favier, R.; Akshoomoff, N.; Mattson, S.; Grossfeld, P. Jacobsen Syndrome: Advances in Our Knowledge of Phenotype and Genotype. Am. J. Med. Genet. Part C Semin. Med. Genet. 2015, 169, 239–250. [Google Scholar] [CrossRef] [PubMed]
- Ye, M.; Coldren, C.; Liang, X.; Mattina, T.; Goldmuntz, E.; Benson, D.W.; Ivy, D.; Perryman, M.B.; Garrett-Sinha, L.A.; Grossfeld, P. Deletion of ETS-1, a Gene in the Jacobsen Syndrome Critical Region, Causes Ventricular Septal Defects and Abnormal Ventricular Morphology in Mice. Hum. Mol. Genet. 2010, 19, 648–656. [Google Scholar] [CrossRef]
- Ahmed, J.N.; Mui, R.K.; Masood, S. Waardenburg Syndrome. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2024. [Google Scholar]
- Yang, S.-Z.; Hou, L.; Qi, X.; Wang, G.-J.; Huang, S.-S.; Zhang, S.-S.; Huang, B.-Q.; Yang, Y.; Li, B.-C.; Liu, S.; et al. A Gross Deletion of the PAX3 Gene in a Large Chinese Family with Waardenburg Syndrome Type I. World J. Pediatr. 2023, 19, 1203–1207. [Google Scholar] [CrossRef]
- Hou, L.; Pavan, W.J. Transcriptional and Signaling Regulation in Neural Crest Stem Cell-Derived Melanocyte Development: Do All Roads Lead to Mitf? Cell Res. 2008, 18, 1163–1176. [Google Scholar] [CrossRef] [PubMed]
- Kapur, R.P. Early Death of Neural Crest Cells Is Responsible for Total Enteric Aganglionosis in Sox10(Dom)/Sox10(Dom) Mouse Embryos. Pediatr. Dev. Pathol. Off. J. Soc. Pediatr. Pathol. Paediatr. Pathol. Soc. 1999, 2, 559–569. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Lo, L.; Dormand, E.; Anderson, D.J. SOX10 Maintains Multipotency and Inhibits Neuronal Differentiation of Neural Crest Stem Cells. Neuron 2003, 38, 17–31. [Google Scholar] [CrossRef] [PubMed]
- Elworthy, S.; Lister, J.A.; Carney, T.J.; Raible, D.W.; Kelsh, R.N. Transcriptional Regulation of Mitfa Accounts for the Sox10 Requirement in Zebrafish Melanophore Development. Development 2003, 130, 2809–2818. [Google Scholar] [CrossRef] [PubMed]
- Bondurand, N.; Natarajan, D.; Barlow, A.; Thapar, N.; Pachnis, V. Maintenance of Mammalian Enteric Nervous System Progenitors by SOX10 and Endothelin 3 Signalling. Development 2006, 133, 2075–2086. [Google Scholar] [CrossRef] [PubMed]
- Steingrímsson, E.; Copeland, N.G.; Jenkins, N.A. Melanocytes and the Microphthalmia Transcription Factor Network. Annu. Rev. Genet. 2004, 38, 365–411. [Google Scholar] [CrossRef]
- Nagy, N.; Goldstein, A.M. Endothelin-3 Regulates Neural Crest Cell Proliferation and Differentiation in the Hindgut Enteric Nervous System. Dev. Biol. 2006, 293, 203–217. [Google Scholar] [CrossRef]
- Cobaleda, C.; Pérez-Caro, M.; Vicente-Dueñas, C.; Sánchez-García, I. Function of the Zinc-Finger Transcription Factor SNAI2 in Cancer and Development. Annu. Rev. Genet. 2007, 41, 41–61. [Google Scholar] [CrossRef]
- Bondurand, N.; Dastot-Le Moal, F.; Stanchina, L.; Collot, N.; Baral, V.; Marlin, S.; Attie-Bitach, T.; Giurgea, I.; Skopinski, L.; Reardon, W.; et al. Deletions at the SOX10 Gene Locus Cause Waardenburg Syndrome Types 2 and 4. Am. J. Hum. Genet. 2007, 81, 1169–1185. [Google Scholar] [CrossRef]
- Pingault, V.; Ente, D.; Dastot-Le Moal, F.; Goossens, M.; Marlin, S.; Bondurand, N. Review and Update of Mutations Causing Waardenburg Syndrome. Hum. Mutat. 2010, 31, 391–406. [Google Scholar] [CrossRef]
- Shah, M.; Patton, E.; Zedek, D. Piebaldism. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2024. [Google Scholar]
- Agarwal, S.; Ojha, A. Piebaldism: A Brief Report and Review of the Literature. Indian Dermatol. Online J. 2012, 3, 144–147. [Google Scholar] [CrossRef]
- Perez-Losada, J.; Sanchez-Martin, M.; Perez, M.; Pérez-Mancera, P.; Sánchez-García, I.; Perez-Losada, J.; Sanchez-Martin, M.; Perez-Caro, M.; Perez-Mancera, P.A.; Sanchez-Garcia, I. The Radioresistance Biological Function of the SCF/Kit Signaling Pathway Is Mediated by the Zinc-Finger Transcription Factor Slug. Oncogene 2003, 22, 4205–4211. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Perfetto, M.; Materna, C.; Li, R.; Thi Tran, H.; Vleminckx, K.; Duncan, M.K.; Wei, S. A New Transgenic Reporter Line Reveals Wnt-Dependent Snai2 Re-Expression and Cranial Neural Crest Differentiation in Xenopus. Sci. Rep. 2019, 9, 11191. [Google Scholar] [CrossRef]
- Ma, E.Z.; Zhou, A.E.; Hoegler, K.M.; Khachemoune, A. Oculocutaneous Albinism: Epidemiology, Genetics, Skin Manifestation, and Psychosocial Issues. Arch. Dermatol. Res. 2023, 315, 107–116. [Google Scholar] [CrossRef]
- Marçon, C.R.; Maia, M. Albinism: Epidemiology, Genetics, Cutaneous Characterization, Psychosocial Factors. An. Bras. Dermatol. 2019, 94, 503–520. [Google Scholar] [CrossRef] [PubMed]
- Cichorek, M.; Wachulska, M.; Stasiewicz, A.; Tymińska, A. Skin Melanocytes: Biology and Development. Adv. Dermatol. Allergol. Dermatol. Alergol. 2013, 30, 30–41. [Google Scholar] [CrossRef] [PubMed]
- Hirobe, T. How Are Proliferation and Differentiation of Melanocytes Regulated? Pigment Cell Melanoma Res. 2011, 24, 462–478. [Google Scholar] [CrossRef]
- Hari, L.; Miescher, I.; Shakhova, O.; Suter, U.; Chin, L.; Taketo, M.; Richardson, W.D.; Kessaris, N.; Sommer, L. Temporal Control of Neural Crest Lineage Generation by Wnt/β-Catenin Signaling. Development 2012, 139, 2107–2117. [Google Scholar] [CrossRef]
- Schouwey, K.; Beermann, F. The Notch Pathway: Hair Graying and Pigment Cell Homeostasis. Histol. Histopathol. 2008, 23, 609–619. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, L.; Chen, X.; Liu, S.; Han, W.; Yu, X.; Cao, X.; Liu, X.; Wang, J. Congenital Central Hypoventilation Syndrome in Chinese Population: Analysis of Three New Cases and Review of the Literature. Mol. Genet. Genomic Med. 2023, 11, e2267. [Google Scholar] [CrossRef]
- Di Lascio, S.; Benfante, R.; Cardani, S.; Fornasari, D. Research Advances on Therapeutic Approaches to Congenital Central Hypoventilation Syndrome (CCHS). Front. Neurosci. 2021, 14, 615666. [Google Scholar] [CrossRef]
- Morikawa, Y.; Zehir, A.; Maska, E.; Deng, C.; Schneider, M.D.; Mishina, Y.; Cserjesi, P. BMP Signaling Regulates Sympathetic Nervous System Development through Smad4-Dependent and -Independent Pathways. Development 2009, 136, 3575–3584. [Google Scholar] [CrossRef] [PubMed]
- Ditmer, M.; Turkiewicz, S.; Gabryelska, A.; Sochal, M.; Białasiewicz, P. Adolescent Congenital Central Hypoventilation Syndrome: An Easily Overlooked Diagnosis. Int. J. Environ. Res. Public. Health 2021, 18, 13402. [Google Scholar] [CrossRef] [PubMed]
- Pattyn, A.; Morin, X.; Cremer, H.; Goridis, C.; Brunet, J.-F. Expression and Interactions of the Two Closely Related Homeobox Genes Phox2a and Phox2b during Neurogenesis. Development 1997, 124, 4065–4075. [Google Scholar] [CrossRef]
- Pla, P.; Hirsch, M.-R.; Le Crom, S.; Reiprich, S.; Harley, V.R.; Goridis, C. Identification of Phox2b-Regulated Genes by Expression Profiling of Cranial Motoneuron Precursors. Neural Develop. 2008, 3, 14. [Google Scholar] [CrossRef]
- Pattyn, A.; Morin, X.; Cremer, H.; Goridis, C.; Brunet, J.-F. The Homeobox Gene Phox2b Is Essential for the Development of Autonomic Neural Crest Derivatives. Nature 1999, 399, 366–370. [Google Scholar] [CrossRef] [PubMed]
- Gaultier, C.; Trang, H.; Dauger, S.; Gallego, J. Pediatric Disorders with Autonomic Dysfunction: What Role for PHOX2B? Pediatr. Res. 2005, 58, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Lo, L.; Morin, X.; Brunet, J.-F.; Anderson, D.J. Specification of Neurotransmitter Identity by Phox2 Proteins in Neural Crest Stem Cells. Neuron 1999, 22, 693–705. [Google Scholar] [CrossRef] [PubMed]
- Kanai, M.; Numakura, C.; Sasaki, A.; Shirahata, E.; Akaba, K.; Hashimoto, M.; Hasegawa, H.; Shirasawa, S.; Hayasaka, K. Congenital Central Hypoventilation Syndrome: A Novel Mutation of the RET Gene in an Isolated Case. Tohoku J. Exp. Med. 2002, 196, 241–246. [Google Scholar] [CrossRef]
- Spielmann, M.; Hernandez-Miranda, L.R.; Ceccherini, I.; Weese-Mayer, D.E.; Kragesteen, B.K.; Harabula, I.; Krawitz, P.; Birchmeier, C.; Leonard, N.; Mundlos, S. Mutations in MYO1H Cause a Recessive Form of Central Hypoventilation with Autonomic Dysfunction. J. Med. Genet. 2017, 54, 754–761. [Google Scholar] [CrossRef]
- Wang, Q.; Pu, S.; Xiang, B.; Chen, J. A Novel Structural Variant of RET Causes Familial Hirschsprung’s Disease via Nonsense-Mediated mRNA Decay. Genes Dis. 2024, 11, 7–10. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, S.; Fries, L.E.; Yaacov, O.; Hu, N.; Berk-Rauch, H.E.; Chakravarti, A. RET Enhancer Haplotype-Dependent Remodeling of the Human Fetal Gut Development Program. PLoS Genet. 2023, 19, e1011030. [Google Scholar] [CrossRef] [PubMed]
- Chalazonitis, A.; D’Autréaux, F.; Pham, T.D.; Kessler, J.A.; Gershon, M.D. Bone Morphogenetic Proteins Regulate Enteric Gliogenesis by Modulating ErbB3 Signaling. Dev. Biol. 2011, 350, 64–79. [Google Scholar] [CrossRef] [PubMed]
- Chalazonitis, A.; Pham, T.D.; Li, Z.; Roman, D.; Guha, U.; Gomes, W.; Kan, L.; Kessler, J.A.; Gershon, M.D. Bone Morphogenetic Protein Regulation of Enteric Neuronal Phenotypic Diversity: Relationship to Timing of Cell Cycle Exit. J. Comp. Neurol. 2008, 509, 474–492. [Google Scholar] [CrossRef] [PubMed]
- Kovács, T.; Halasy, V.; Pethő, C.; Szőcs, E.; Soós, Á.; Dóra, D.; de Santa Barbara, P.; Faure, S.; Stavely, R.; Goldstein, A.M.; et al. Essential Role of BMP4 Signaling in the Avian Ceca in Colorectal Enteric Nervous System Development. Int. J. Mol. Sci. 2023, 24, 15664. [Google Scholar] [CrossRef] [PubMed]
- Lang, D.; Chen, F.; Milewski, R.; Li, J.; Lu, M.M.; Epstein, J.A. Pax3 Is Required for Enteric Ganglia Formation and Functions with Sox10 to Modulate Expression of c-ret. J. Clin. Investig. 2000, 106, 963–971. [Google Scholar] [CrossRef] [PubMed]
- Honoré, S.M.; Aybar, M.J.; Mayor, R. Sox10 Is Required for the Early Development of the Prospective Neural Crest in Xenopus Embryos. Dev. Biol. 2003, 260, 79–96. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Zhu, Y.; Xie, X.; Yao, Y.; Zhang, J.; Zhang, R.; Huang, L.; Cheng, J.; Xia, H.; He, J.; et al. Pleiotropic Effect of Common PHOX2B Variants in Hirschsprung Disease and Neuroblastoma. Aging 2019, 11, 1252–1261. [Google Scholar] [CrossRef]
- Okamoto, N.; Del Maestro, R.; Valero, R.; Monros, E.; Poo, P.; Kanemura, Y.; Yamasaki, M. Hydrocephalus and Hirschsprung’s Disease with a Mutation of L1CAM. J. Hum. Genet. 2004, 49, 334–337. [Google Scholar] [CrossRef]
- Gunadi; Budi, N.Y.P.; Sethi, R.; Fauzi, A.R.; Kalim, A.S.; Indrawan, T.; Iskandar, K.; Makhmudi, A.; Adrianto, I.; San, L.P. NRG1 Variant Effects in Patients with Hirschsprung Disease. BMC Pediatr. 2018, 18, 292. [Google Scholar] [CrossRef]
- Epifanova, E.; Babaev, A.; Newman, A.G.; Tarabykin, V. Role of Zeb2/Sip1 in Neuronal Development. Brain Res. 2019, 1705, 24–31. [Google Scholar] [CrossRef] [PubMed]
- Porokuokka, L.L.; Virtanen, H.T.; Lindén, J.; Sidorova, Y.; Danilova, T.; Lindahl, M.; Saarma, M.; Andressoo, J.-O. Gfra1 Underexpression Causes Hirschsprung’s Disease and Associated Enterocolitis in Mice. Cell. Mol. Gastroenterol. Hepatol. 2019, 7, 655–678. [Google Scholar] [CrossRef] [PubMed]
- PRIME PubMed|Susceptibility of ECE1 Polymorphisms to Hirschsprung’s Disease in Southern Chinese Children. Available online: https://www.unboundmedicine.com/medline/citation/36619519/Susceptibility_of_ECE1_polymorphisms_to_Hirschsprung’s_disease_in_southern_Chinese_children_ (accessed on 21 February 2024).
- Fernandez, R.M.; Ruiz-Ferrer, M.; Lopez-Alonso, M.; Antiñolo, G.; Borrego, S. Polymorphisms in the Genes Encoding the 4 RET Ligands, GDNF, NTN, ARTN, PSPN, and Susceptibility to Hirschsprung Disease. J. Pediatr. Surg. 2008, 43, 2042–2047. [Google Scholar] [CrossRef] [PubMed]
- Katkat, E.; Demirci, Y.; Heger, G.; Karagulle, D.; Papatheodorou, I.; Brazma, A.; Ozhan, G. Canonical Wnt and TGF-β/BMP Signaling Enhance Melanocyte Regeneration but Suppress Invasiveness, Migration, and Proliferation of Melanoma Cells. Front. Cell Dev. Biol. 2023, 11, 1297910. [Google Scholar] [CrossRef]
- Differential Requirements of BMP and Wnt Signalling during Gastrulation and Neurulation Define Two Steps in Neural Crest Induction|Development|The Company of Biologists. Available online: https://journals.biologists.com/dev/article/136/5/771/65570/Differential-requirements-of-BMP-and-Wnt (accessed on 22 February 2024).
- García-Castro, M.I.; Marcelle, C.; Bronner-Fraser, M. Ectodermal Wnt Function as a Neural Crest Inducer. Science 2002, 297, 848–851. [Google Scholar] [CrossRef]
- Meng, D.; Carvajal, R.D. KIT as an Oncogenic Driver in Melanoma: An Update on Clinical Development. Am. J. Clin. Dermatol. 2019, 20, 315–323. [Google Scholar] [CrossRef] [PubMed]
- Gong, H.Z.; Zheng, H.Y.; Li, J. The Clinical Significance of KIT Mutations in Melanoma: A Meta-Analysis. Melanoma Res. 2018, 28, 259–270. [Google Scholar] [CrossRef]
- Bhattacharya, D.; Azambuja, A.P.; Simoes-Costa, M. Metabolic Reprogramming Promotes Neural Crest Migration via Yap/Tead Signaling. Dev. Cell 2020, 53, 199–211.e6. [Google Scholar] [CrossRef]
- Haq, R.; Fisher, D.E. Biology and Clinical Relevance of the Micropthalmia Family of Transcription Factors in Human Cancer. J. Clin. Oncol. 2011, 29, 3474–3482. [Google Scholar] [CrossRef]
- Hartman, M.L.; Czyz, M. MITF in Melanoma: Mechanisms behind Its Expression and Activity. Cell. Mol. Life Sci. CMLS 2015, 72, 1249–1260. [Google Scholar] [CrossRef]
- Toma, J.G.; McKenzie, I.A.; Bagli, D.; Miller, F.D. Isolation and Characterization of Multipotent Skin-Derived Precursors from Human Skin. Stem Cells 2005, 23, 727–737. [Google Scholar] [CrossRef]
- Lang, D.; Lu, M.M.; Huang, L.; Engleka, K.A.; Zhang, M.; Chu, E.Y.; Lipner, S.; Skoultchi, A.; Millar, S.E.; Epstein, J.A. Pax3 Functions at a Nodal Point in Melanocyte Stem Cell Differentiation. Nature 2005, 433, 884–887. [Google Scholar] [CrossRef]
- Muratovska, A.; Zhou, C.; He, S.; Goodyer, P.; Eccles, M.R. Paired-Box Genes Are Frequently Expressed in Cancer and Often Required for Cancer Cell Survival. Oncogene 2003, 22, 7989–7997. [Google Scholar] [CrossRef]
- Plummer, R.S.; Shea, C.R.; Nelson, M.; Powell, S.K.; Freeman, D.M.; Dan, C.P.; Lang, D. PAX3 Expression in Primary Melanomas and Nevi. Mod. Pathol. 2008, 21, 525–530. [Google Scholar] [CrossRef]
- Muñoz-Couselo, E.; Adelantado, E.Z.; Ortiz, C.; García, J.S.; Perez-Garcia, J. NRAS-Mutant Melanoma: Current Challenges and Future Prospect. OncoTargets Ther. 2017, 10, 3941–3947. [Google Scholar] [CrossRef]
- Kiuru, M.; Busam, K.J. The NF1 Gene in Tumor Syndromes and Melanoma. Lab. Investig. J. Tech. Methods Pathol. 2017, 97, 146–157. [Google Scholar] [CrossRef]
- Helbing, D.-L.; Schulz, A.; Morrison, H. Pathomechanisms in Schwannoma Development and Progression. Oncogene 2020, 39, 5421–5429. [Google Scholar] [CrossRef]
- Li, W.; You, L.; Cooper, J.; Schiavon, G.; Pepe-Caprio, A.; Zhou, L.; Ishii, R.; Giovannini, M.; Hanemann, C.O.; Long, S.B.; et al. Merlin/NF2 Suppresses Tumorigenesis by Inhibiting the E3 Ubiquitin Ligase CRL4DCAF1 in the Nucleus. Cell 2010, 140, 477–490. [Google Scholar] [CrossRef]
- Morrison, H.; Sherman, L.S.; Legg, J.; Banine, F.; Isacke, C.; Haipek, C.A.; Gutmann, D.H.; Ponta, H.; Herrlich, P. The NF2 Tumor Suppressor Gene Product, Merlin, Mediates Contact Inhibition of Growth through Interactions with CD44. Genes Dev. 2001, 15, 968–980. [Google Scholar] [CrossRef]
- Li, Z.; Ngan, E.S.-W. New Insights Empowered by Single-Cell Sequencing: From Neural Crest to Enteric Nervous System. Comput. Struct. Biotechnol. J. 2022, 20, 2464–2472. [Google Scholar] [CrossRef] [PubMed]
- Britsch, S.; Goerich, D.E.; Riethmacher, D.; Peirano, R.I.; Rossner, M.; Nave, K.-A.; Birchmeier, C.; Wegner, M. The Transcription Factor Sox10 Is a Key Regulator of Peripheral Glial Development. Genes Dev. 2001, 15, 66–78. [Google Scholar] [CrossRef] [PubMed]
- Terry, M.; Gupta, R.; Ravindranathan, A.; Wu, J.; Chan, E.; Bollen, A.W.; Chang, S.M.; Berger, M.S.; Jacques, L.; Solomon, D.A. Somatic Mosaic SOX10 Indel Mutations Underlie a Form of Segmental Schwannomatosis. Acta Neuropathol. 2023, 146, 857–860. [Google Scholar] [CrossRef] [PubMed]
- Williams, E.A.; Ravindranathan, A.; Gupta, R.; Stevers, N.O.; Suwala, A.K.; Hong, C.; Kim, S.; Yuan, J.B.; Wu, J.; Barreto, J.; et al. Novel SOX10 Indel Mutations Drive Schwannomas through Impaired Transactivation of Myelination Gene Programs. Neuro-Oncology 2023, 25, 2221–2236. [Google Scholar] [CrossRef] [PubMed]
- Bahmad, H.F.; Thiravialingam, A.; Sriganeshan, K.; Gonzalez, J.; Alvarez, V.; Ocejo, S.; Abreu, A.R.; Avellan, R.; Arzola, A.H.; Hachem, S.; et al. Clinical Significance of SOX10 Expression in Human Pathology. Curr. Issues Mol. Biol. 2023, 45, 10131–10158. [Google Scholar] [CrossRef] [PubMed]
- Brodeur, G.M. Neuroblastoma: Biological Insights into a Clinical Enigma. Nat. Rev. Cancer 2003, 3, 203–216. [Google Scholar] [CrossRef] [PubMed]
- Raabe, E.H.; Laudenslager, M.; Winter, C.; Wasserman, N.; Cole, K.; LaQuaglia, M.; Maris, D.J.; Mosse, Y.P.; Maris, J.M. Prevalence and Functional Consequence of PHOX2B Mutations in Neuroblastoma. Oncogene 2008, 27, 469–476. [Google Scholar] [CrossRef]
- Rohrer, H. Transcriptional Control of Differentiation and Neurogenesis in Autonomic Ganglia. Eur. J. Neurosci. 2011, 34, 1563–1573. [Google Scholar] [CrossRef]
- Wang, W.; Zhong, Q.; Teng, L.; Bhatnagar, N.; Sharma, B.; Zhang, X.; Luther, W.; Haynes, L.P.; Burgoyne, R.D.; Vidal, M.; et al. Mutations That Disrupt PHOXB Interaction with the Neuronal Calcium Sensor HPCAL1 Impede Cellular Differentiation in Neuroblastoma. Oncogene 2014, 33, 3316–3324. [Google Scholar] [CrossRef]
- Ponzoni, M.; Bachetti, T.; Corrias, M.V.; Brignole, C.; Pastorino, F.; Calarco, E.; Bensa, V.; Giusto, E.; Ceccherini, I.; Perri, P. Recent Advances in the Developmental Origin of Neuroblastoma: An Overview. J. Exp. Clin. Cancer Res. 2022, 41, 92. [Google Scholar] [CrossRef]
- Lider Burciulescu, S.M.; Randon, C.; Duprez, F.; Huvenne, W.; Creytens, D.; Claes, K.B.M.; de Putter, R.; T’Sjoen, G.; Badiu, C.; Lapauw, B. Clinical Presentation of Sporadic and Hereditary Pheochromocytoma/Paraganglioma. Endocr. Oncol. 2023, 3, e220040. [Google Scholar] [CrossRef]
- Elder, E.E.; Elder, G.; Larsson, C. Pheochromocytoma and Functional Paraganglioma Syndrome: No Longer the 10% Tumor. J. Surg. Oncol. 2005, 89, 193–201. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.P.; Hung, Y.P.; O’Dorisio, T.M.; Howe, J.R.; Hornick, J.L.; Bellizzi, A.M. Examination of PHOX2B in Adult Neuroendocrine Neoplasms Reveals Relatively Frequent Expression in Pheochromocytomas and Paragangliomas. Histopathology 2017, 71, 503–510. [Google Scholar] [CrossRef] [PubMed]
- Chatzikyriakou, P.; Brempou, D.; Quinn, M.; Fishbein, L.; Noberini, R.; Anastopoulos, I.N.; Tufton, N.; Lim, E.S.; Obholzer, R.; Hubbard, J.G.; et al. A Comprehensive Characterisation of Phaeochromocytoma and Paraganglioma Tumours through Histone Protein Profiling, DNA Methylation and Transcriptomic Analysis Genome Wide. Clin. Epigenetics 2023, 15, 196. [Google Scholar] [CrossRef] [PubMed]
- Williams, S.T.; Chatzikyriakou, P.; Carroll, P.V.; McGowan, B.M.; Velusamy, A.; White, G.; Obholzer, R.; Akker, S.; Tufton, N.; Casey, R.T.; et al. SDHC Phaeochromocytoma and Paraganglioma: A UK-Wide Case Series. Clin. Endocrinol. 2022, 96, 499–512. [Google Scholar] [CrossRef] [PubMed]
- Maliszewska, A.; Leandro-Garcia, L.J.; Castelblanco, E.; Macià, A.; de Cubas, A.; Goméz-López, G.; Inglada-Pérez, L.; Álvarez-Escolá, C.; De la Vega, L.; Letón, R.; et al. Differential Gene Expression of Medullary Thyroid Carcinoma Reveals Specific Markers Associated with Genetic Conditions. Am. J. Pathol. 2013, 182, 350–362. [Google Scholar] [CrossRef]
- Romei, C.; Ciampi, R.; Elisei, R. A Comprehensive Overview of the Role of the RET Proto-Oncogene in Thyroid Carcinoma. Nat. Rev. Endocrinol. 2016, 12, 192–202. [Google Scholar] [CrossRef] [PubMed]
- Mulligan, L.M. RET Revisited: Expanding the Oncogenic Portfolio. Nat. Rev. Cancer 2014, 14, 173–186. [Google Scholar] [CrossRef]
- Nakamura, T.; Ishizaka, Y.; Nagao, M.; Hara, M.; Ishikawa, T. Expression of the Ret Proto-Oncogene Product in Human Normal and Neoplastic Tissues of Neural Crest Origin. J. Pathol. 1994, 172, 255–260. [Google Scholar] [CrossRef] [PubMed]
- Tan, L.; Jiang, T.; Tan, L.; Yu, J.-T. Toward Precision Medicine in Neurological Diseases. Ann. Transl. Med. 2016, 4, 104. [Google Scholar] [CrossRef]
- Kumar, K.R.; Cowley, M.J.; Davis, R.L. Next-Generation Sequencing and Emerging Technologies. In Proceedings of the Seminars in Thrombosis and Hemostasis, Madrid, Spain, 29–31 March 2019; Thieme Medical Publishers: Stuttgart, Germany, 2019; Volume 45, pp. 661–673. [Google Scholar]
- Dermitzakis, I.; Manthou, M.E.; Meditskou, S.; Tremblay, M.-È.; Petratos, S.; Zoupi, L.; Boziki, M.; Kesidou, E.; Simeonidou, C.; Theotokis, P. Origin and Emergence of Microglia in the CNS—An Interesting (Hi)Story of an Eccentric Cell. Curr. Issues Mol. Biol. 2023, 45, 2609–2628. [Google Scholar] [CrossRef]
- Dermitzakis, I.; Theotokis, P.; Evangelidis, P.; Delilampou, E.; Evangelidis, N.; Chatzisavvidou, A.; Avramidou, E.; Manthou, M.E. CNS Border-Associated Macrophages: Ontogeny and Potential Implication in Disease. Curr. Issues Mol. Biol. 2023, 45, 4285–4300. [Google Scholar] [CrossRef] [PubMed]
- Okuno, H.; Okano, H. Modeling Human Congenital Disorders with Neural Crest Developmental Defects Using Patient-Derived Induced Pluripotent Stem Cells. Regen. Ther. 2021, 18, 275–280. [Google Scholar] [CrossRef] [PubMed]
- Pilon, N. Treatment and Prevention of Neurocristopathies. Trends Mol. Med. 2021, 27, 451–468. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
| Affected Regions | Accepted Examples | Genes | References |
|---|---|---|---|
| Cranial–Facial Regions | Goldenhar syndrome | MSX1, MYT1, SF3B2 | [41,42,46] |
| Axenfeld–Rieger syndrome | FOXC1, FOXC2, PITX2, CYP1B1, PRDM5, JAG1, USP9X, CDK13, HCCS, AMELX, BCOR | [49,50,53] | |
| Craniosynostosis | FGFR1, FGFR2, FGFR3, TWIST1, EFNB1 | [55] | |
| Craniofacial–deafness–hand syndrome | PAX3 | [60] | |
| Tricho-dento-osseus syndrome | DLX3 | [65] | |
| Peter’s anomaly | PITX2, PAX6, FOXE3, FOXC1, CYP1B1 | [63,64,68,70] | |
| Bamforth–Lazarus syndrome | FOXE1, MSX1, TGF-b3 | [53,73] | |
| Branchio-oculo-facial syndrome | TFAP2A | [79] | |
| CADASIL | NOTCH3, FOXC1, PITX2, TIMP3, VTN | [86,94,95] | |
| Congenital aniridia | PAX6, FOXC1, PITX2, TRIM44, ELP4, DCDC1, CYP1B1 | [96,99,100] | |
| Frontonasal dysplasia | ALX3, ALX4 ALX1, EFNB1, KIF3A | [101,103,104,111,112] | |
| Hajdu–Cheney syndrome | NOTCH2 | [113] | |
| Moebius syndrome | PLXND1, REV3L, TUBB3, HOXA1, HOXB1, GSH1, CDX2, CRBP1, PBX2, EGR2, SOX14 | [21,126,127,129,131,132,133,134,135] | |
| Pierre Robin sequence | SOX9, BMP2 | [137] | |
| Mowat–Wilson syndrome | ZEB2 | [146] | |
| SAMS disorder | GSC, EDNRA | [150,154] | |
| Heart and Outflow Tract | DiGeorge syndrome | TBX1, GBX2, CXCR4, CXCL12 | [77,158] |
| CHARGE syndrome | CHD7, TBX1 | [117] | |
| Velocardiofacial syndrome | TBX1 | [77] | |
| Fetal alcohol spectrum disorders | SHH | [163] | |
| Alagille syndrome | JAG1, NOTCH2 | [117] | |
| Noonan–LEOPARD syndrome | PTPN11, SOS1, KRAS, NRAS, RAF1, BRAF, SHOC2, MAP2K1, CBL | [53,171] | |
| Jacobsen syndrome | ETS1 | [174] | |
| Melanocytes and Neuronal Ganglia | Piebaldism | KIT, SNAI2 | [188,189,190] |
| Waardenburg syndrome | PAX3, EDN3, EDNRB, SOX10, MITF, SNAI2 | [178,181,182,183,186,187] | |
| Oculocutaneous albinism | TYR, KIT | [194,195] | |
| Congenital central hypoventilation syndrome | PHOX2A, PHOX2B, GDNF, RET, EDN3, BMP2, MYO1H, LBX1 | [199,207,208,209] | |
| Enteric–Sacral Nervous System | Hirschsprung disease | RET, EDNRB, BMP4, PAX3, L1CAM, NRG1, SOX10, SIP1, PHOX2B, GFRA1, HASH1, HAND2, ECE1, GDNF, NTN, ARTN, PSPN | [210,211,214,215,216,217,218,219,220,221,222] |
| Affected Structures | Types of Cancer | Genes | References |
|---|---|---|---|
| Cranial–Facial Regions | - | - | - |
| Heart And Outflow Tract | Familial medullary thyroid carcinomas | RET, PITX2 | [255,256] |
| Melanocytes, Neuronal Ganglia, and Nerves | Malignant melanoma | KIT, PAX3, MITF, BRAF, NRAS, NF1, CDKN2A, CDK4 | [60,178,226,227,229,233,235,236] |
| Schwannoma | SOX10, NF2 | [237,238,239] | |
| Neuroblastoma | PHOX2B | [246,249] | |
| Pheochromocytoma–Paraganglioma | PHOX2B, SDHB, SDHC, SDHD, VHL | [248,252,254] | |
| Enteric Nervous System | - | - | |
| Sacral | - | - |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chatzi, D.; Kyriakoudi, S.A.; Dermitzakis, I.; Manthou, M.E.; Meditskou, S.; Theotokis, P. Clinical and Genetic Correlation in Neurocristopathies: Bridging a Precision Medicine Gap. J. Clin. Med. 2024, 13, 2223. https://doi.org/10.3390/jcm13082223
Chatzi D, Kyriakoudi SA, Dermitzakis I, Manthou ME, Meditskou S, Theotokis P. Clinical and Genetic Correlation in Neurocristopathies: Bridging a Precision Medicine Gap. Journal of Clinical Medicine. 2024; 13(8):2223. https://doi.org/10.3390/jcm13082223
Chicago/Turabian StyleChatzi, Despoina, Stella Aikaterini Kyriakoudi, Iasonas Dermitzakis, Maria Eleni Manthou, Soultana Meditskou, and Paschalis Theotokis. 2024. "Clinical and Genetic Correlation in Neurocristopathies: Bridging a Precision Medicine Gap" Journal of Clinical Medicine 13, no. 8: 2223. https://doi.org/10.3390/jcm13082223
APA StyleChatzi, D., Kyriakoudi, S. A., Dermitzakis, I., Manthou, M. E., Meditskou, S., & Theotokis, P. (2024). Clinical and Genetic Correlation in Neurocristopathies: Bridging a Precision Medicine Gap. Journal of Clinical Medicine, 13(8), 2223. https://doi.org/10.3390/jcm13082223