
Sulodexide Inhibits Arterial Contraction via the Endothelium-Dependent Nitric Oxide Pathway

 , , , , ,
, , , , ,
Abstract
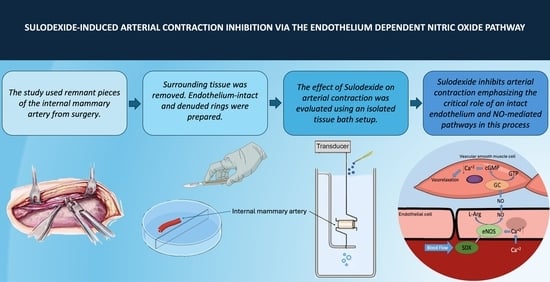
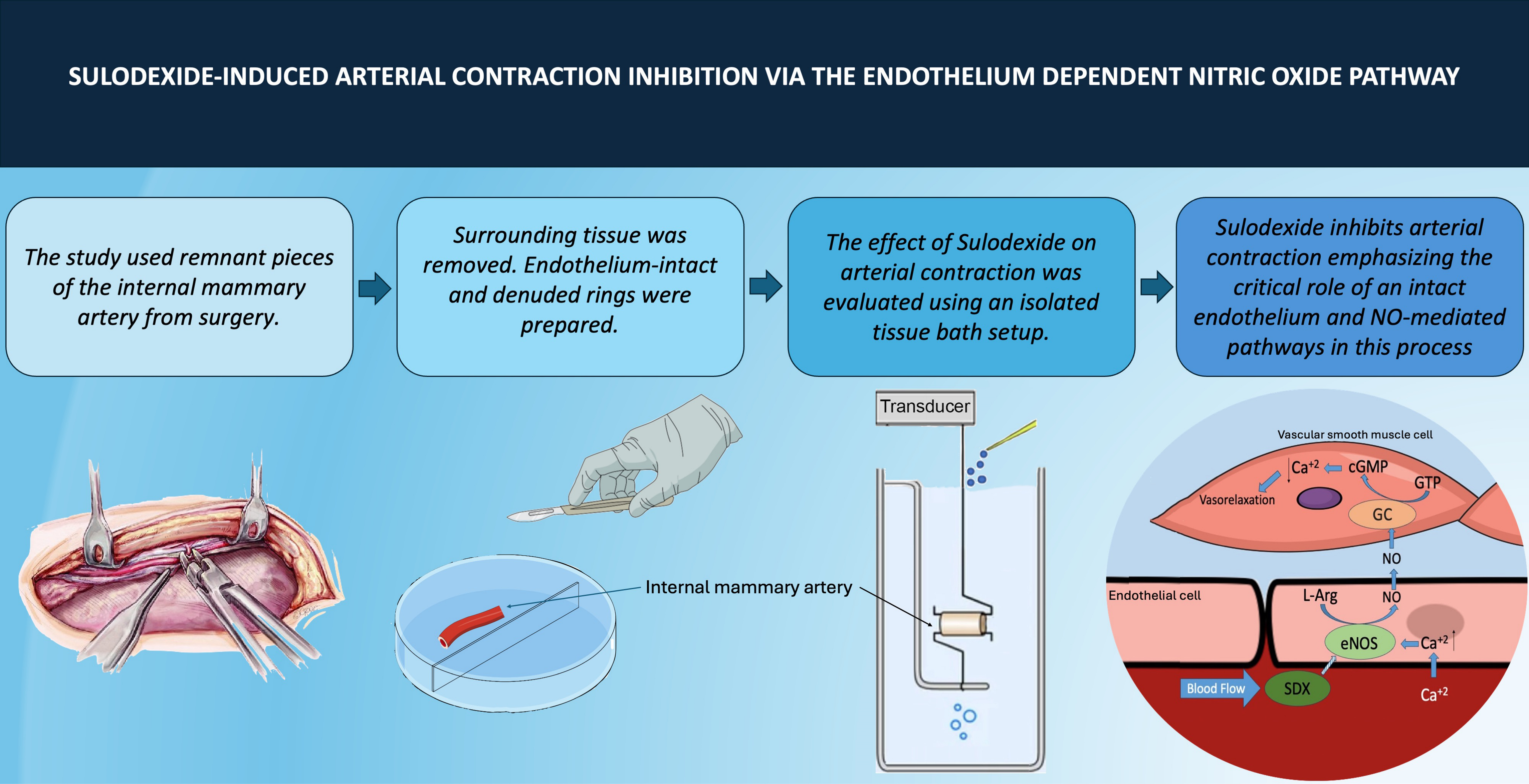
:
1. Introduction
2. Materials and Methods
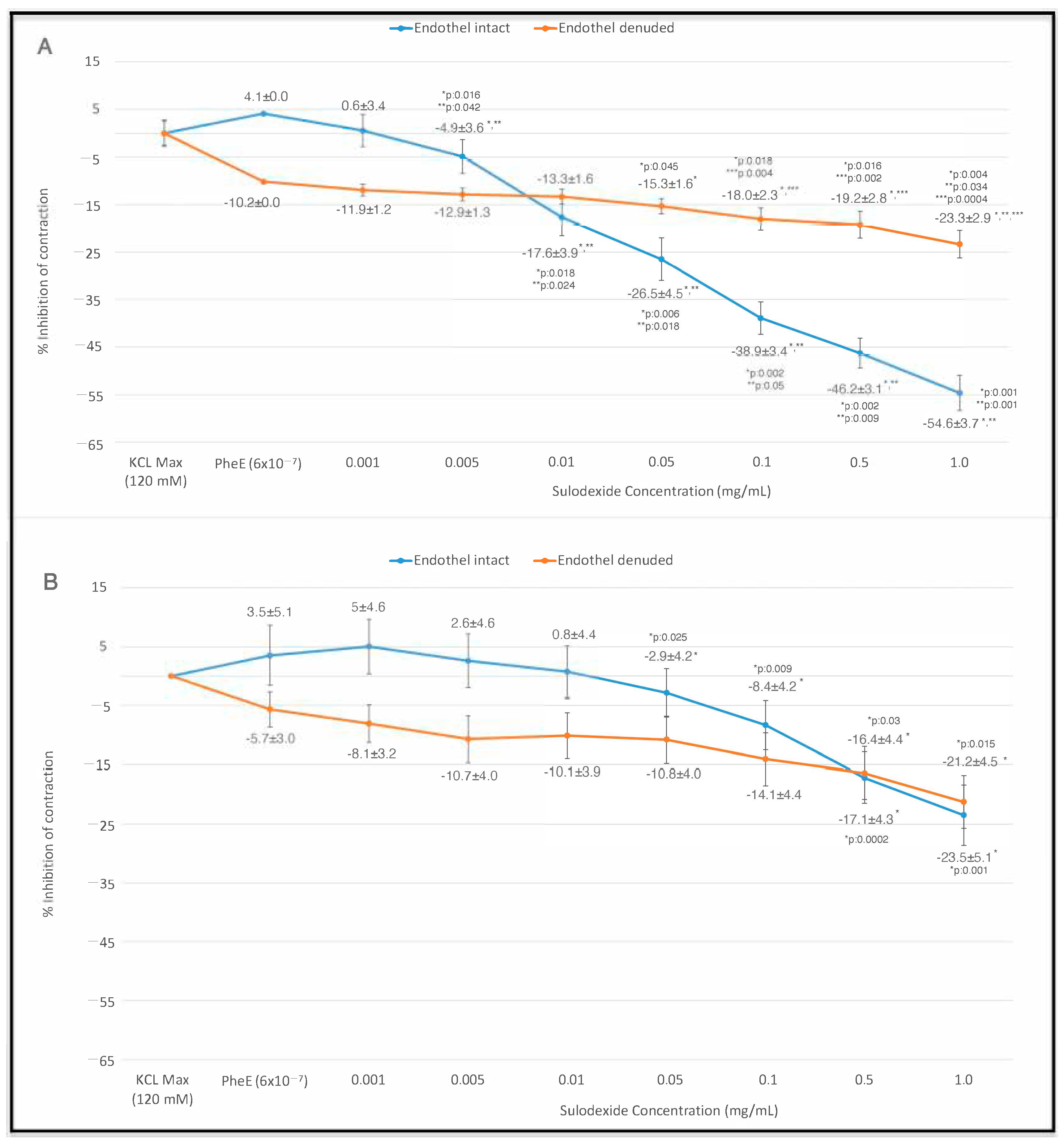
3. Results
4. Discussion
5. Conclusions
6. Clinical Relevance
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Takano, H.; Dora, K.A.; Garland, C.J. Spreading Vasodilatation in Resistance Arteries. J. Smooth Muscle Res. 2005, 41, 303–311. [Google Scholar] [CrossRef] [PubMed]
- Masola, V.; Zaza, G.; Onisto, M.; Lupo, A.; Gambaro, G. Glycosaminoglycans, Proteoglycans and Sulodexide and the endothelium: Biological Roles and Pharmacological Effects. Int. Angiol. 2014, 33, 243–254. [Google Scholar] [PubMed]
- Sosińska, P.; Baum, E.; Maćkowiak, B.; Maj, M.; Sumińska-Jasińska, K.; Staniszewski, R.; Brȩborowicz, A. Sulodexide Reduces the Proinflammatory Effect of Serum from Patients with Peripheral Artery Disease in Human Arterial Endothelial Cells. Cell Physiol. Biochem. 2016, 40, 1005–1012. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Liu, X.; Zhao, Z.; Ni, L.; Liu, C. Sulodexide Recovers Endothelial Function through Reconstructing Glycocalyx in the Balloon-Injury Rat Carotid Artery Model. Oncotarget 2017, 8, 91350–91361. [Google Scholar] [CrossRef] [PubMed]
- Mangiafico, R.; Fiore, C. Current Management of Intermittent Claudication: The Role of Pharmacological and Nonpharmacological Symptom-Directed Therapies. Curr. Vasc. Pharmacol. 2009, 7, 394–413. [Google Scholar] [CrossRef] [PubMed]
- Karino, T.; Motomiya, M. Flow through a Venous Valve and Its Implication for Thrombus Formation. Thromb. Res. 1984, 36, 245–257. [Google Scholar] [CrossRef] [PubMed]
- Blann, A.D.; Khoo, C.W. The Prevention and Treatment of Venous Thromboembolism with LMWHs and New Anticoagulants. Vasc. Health Risk Manag. 2009, 5, 693–704. [Google Scholar] [CrossRef] [PubMed]
- Nenci, G.G. Dermatan Sulphate as an Antithrombotic Drug. Pathophysiol. Haemost. Thromb. 2002, 32, 303–307. [Google Scholar] [CrossRef] [PubMed]
- Lasierra-Cirujeda, J.; Coronel, P.; Aza, M.J.; Gimeno, M. Use of Sulodexide in Patients with Peripheral Vascular Disease. J. Blood Med. 2010, 1, 105. [Google Scholar] [CrossRef]
- Carroll, B.J.; Piazza, G.; Goldhaber, S.Z. Sulodexide in Venous Disease. J. Thromb. Haemost. 2019, 17, 31–38. [Google Scholar] [CrossRef]
- Afratis, N.; Gialeli, C.; Nikitovic, D.; Tsegenidis, T.; Karousou, E.; Theocharis, A.D.; Pavão, M.S.; Tzanakakis, G.N.; Karamanos, N.K. Glycosaminoglycans: Key Players in Cancer Cell Biology and Treatment. FEBS J. 2012, 279, 1177–1197. [Google Scholar] [CrossRef]
- Veraldi, N.; Guerrini, M.; Urso, E.; Risi, G.; Bertini, S.; Bensi, D.; Bisio, A. Fine Structural Characterization of Sulodexide. J. Pharm. Biomed. Anal. 2018, 156, 67–79. [Google Scholar] [CrossRef] [PubMed]
- Silvestro, L.; Lanzarotti, E.; Marchi, E.; Gori, M.; Pescador, R.; Ferro, L.; Milani, M.R.; Da Col, R.; Coppini, A. Human Pharmacokinetics of Glycosaminoglycans Using Deuterium-Labeled and Unlabeled Substances: Evidence for Oral Absorption. Semin. Thromb. Hemost. 1994, 20, 281–292. [Google Scholar] [CrossRef]
- Coccheri, S.; Mannello, F. Development and Use of Sulodexide in Vascular Diseases: Implications for Treatment. Drug Des. Devel Ther. 2013, 8, 49–65. [Google Scholar] [CrossRef] [PubMed]
- Ofosu, F.A. Pharmacological Actions of Sulodexide. Semin. Thromb. Hemost. 1998, 24, 127–138. [Google Scholar] [CrossRef]
- Marchi, E.; Barbanti, M.; Milani, R.; Breccia, A.; Fini, A.; Gattavecchia, E. Organ Glycosaminoglycan Distribution after Intravenous and Oral Administration in Rats. Semin. Thromb. Hemost. 1994, 20, 297–300. [Google Scholar] [CrossRef]
- Raffetto, J.D.; Calanni, F.; Mattana, P.; Khalil, R.A. Sulodexide Promotes Arterial Relaxation via Endothelium-Dependent Nitric Oxide-Mediated Pathway. Biochem. Pharmacol. 2019, 166, 347–356. [Google Scholar] [CrossRef]
- Raffetto, J.D.; Yu, W.; Wang, X.; Calanni, F.; Mattana, P.; Khalil, R.A. Sulodexide Improves Contraction and Decreases Matrix Metalloproteinase-2 and -9 in Veins under Prolonged Stretch. J. Cardiovasc. Pharmacol. 2020, 75, 211–221. [Google Scholar] [CrossRef] [PubMed]
- Palareti, G.; Legnani, C.; Antonucci, E.; Zorzi, S.; Bignamini, A.A.; Lodigiani, C.; Tosetto, A.; Bertù, L.; Pengo, V.; Testa, S.; et al. Design and Rationale of a Randomized, Placebo-Controlled Trial on the Efficacy and Safety of Sulodexide for Extended Treatment in Elderly Patients after a First Venous Thromboembolism. Intern. Emerg. Med. 2021, 16, 359–368. [Google Scholar] [CrossRef]
- Elleuch, N.; Zidi, H.; Bellamine, Z.; Hamdane, A.; Guerchi, M.; Jellazi, N.; On behalf of the CVD Study Investigators; Nizar, E.; Jamila, R.; Habib, G.M.; et al. Sulodexide in Patients with Chronic Venous Disease of the Lower Limbs: Clinical Efficacy and Impact on Quality of Life. Adv. Ther. 2016, 33, 1536–1549. [Google Scholar] [CrossRef]
- Andreozzi, G.M.; Bignamini, A.A.; Davì, G.; Palareti, G.; Matuška, J.; Holý, M.; Pawlaczyk-Gabriel, K.; Džupina, A.; Sokurenko, G.Y.; Didenko, Y.P.; et al. Sulodexide for the Prevention of Recurrent Venous Thromboembolism: The Sulodexide in Secondary Prevention of Recurrent Deep Vein Thrombosis (SURVET) Study: A Multicenter, Randomized, Double-Blind, Placebo-Controlled Trial. Circulation 2015, 132, 1891–1897. [Google Scholar] [CrossRef] [PubMed]
- Dobiaš, L.; Petrová, M.; Vojtko, R.; Uličná, O.; Vančová, O.; Kristová, V. Effect of Sulodexide on Vascular Responses and Liver Mitochondrial Function in Diabetic Rats. Physiol. Res. 2015, 64, S497–S505. [Google Scholar] [CrossRef] [PubMed]
- Kristová, V.; Líšková, S.; Sotníková, R.; Vojtko, R.; Kurtanský, A. Sulodexide Improves Endothelial Dysfunction in Streptozotocin-Induced Diabetes in Rats. Physiol. Res. 2008, 57, 491–494. [Google Scholar] [CrossRef] [PubMed]
- Serra, R.; Gallelli, L.; Conti, A.; De Caridi, G.; Massara, M.; Spinelli, F.; Buffone, G.; Caliò, F.G.; Amato, B.; Ceglia, S.; et al. The Effects of Sulodexide on Both Clinical and Molecular Parameters in Patients with Mixed Arterial and Venous Ulcers of Lower Limbs. Drug Des. Devel Ther. 2014, 8, 519–527. [Google Scholar] [CrossRef] [PubMed]
- Bikdeli, B.; Chatterjee, S.; Kirtane, A.J.; Parikh, S.A.; Andreozzi, G.M.; Desai, N.R.; Francese, D.P.; Gibson, C.M.; Piazza, G.; Goldhaber, S.Z.; et al. Sulodexide versus Control and the Risk of Thrombotic and Hemorrhagic Events: Meta-Analysis of Randomized Trials. Semin. Thromb. Hemost. 2020, 46, 908–918. [Google Scholar] [CrossRef] [PubMed]
- Cospite, M.; Ferrara, F.; Cospite, V.; Palazzini, E. Sulodexide and the Microcirculatory Component in Microphlebopathies. Curr. Med. Res. Opin. 1992, 13, 56–60. [Google Scholar] [CrossRef] [PubMed]
- Gianesini, S.; Rimondi, E.; Raffetto, J.D.; Melloni, E.; Pellati, A.; Menegatti, E.; Avruscio, G.P.; Bassetto, F.; Costa, A.L.; Rockson, S. Human Collecting Lymphatic Glycocalyx Identification by Electron Microscopy and Immunohistochemistry. Sci. Rep. 2023, 13, 3022. [Google Scholar] [CrossRef] [PubMed]
- Borawski, J.; Dubowski, M.; Pawlak, K.; Mysliwiec, M. Sulodexide Induces Hepatocyte Growth Factor Release in Humans. Eur. J. Pharmacol. 2007, 558, 167–171. [Google Scholar] [CrossRef] [PubMed]
- Correction Arosio, E.; Ferrari, G.; Santoro, L.; Gianese, F.; Coccheri, S.; Zamboni, P.; Cisno, L.; Marchetti, F.; Anna, S.; Paroni, G.; et al. A Placebo-Controlled, Double-Blind Study of Mesoglycan in the Treatment of Chronic Venous Ulcers. Eur. J. Vasc. Endovasc. Surg. 2001, 22, 365–372. [Google Scholar] [CrossRef] [PubMed]
- Coccheri, S.; Scondotto, G.; Agnelli, G.; Palazzini, E.; Zamboni, V. Sulodexide in the Treatment of Intermittent Claudication. Results of a Randomized, Double-Blind, Multicentre, Placebo-Controlled Study. Eur. Heart J. 2002, 23, 1057–1065. [Google Scholar] [CrossRef]
- Luzzi, R.; Belcaro, G.; Dugall, M.; Hu, S.; Arpaia, G.; Ledda, A.; Ippolito, E.; Corsi, M.; Ricci, A.; Cotellese, R.; et al. The Efficacy of Sulodexide in the Prevention of Postthrombotic Syndrome. Clin. Appl. Thromb. Hemost. 2014, 20, 594–599. [Google Scholar] [CrossRef]
- Olde Engberink, R.H.G.; Heerspink, H.J.L.; de Zeeuw, D.; Vogt, L. Blood Pressure-Lowering Effects of Sulodexide Depend on Albuminuria Severity: Post Hoc Analysis of the Sulodexide Microalbuminuria and Macroalbuminuria Studies. Br. J. Clin. Pharmacol. 2016, 82, 1351–1357. [Google Scholar] [CrossRef] [PubMed]
- Engberink, R.H.G.O.; Rorije, N.M.G.; Lambers Heerspink, H.J.; De Zeeuw, D.; Van Den Born, B.J.H.; Vogt, L. The Blood Pressure Lowering Potential of Sulodexide—A Systematic Review and Meta-Analysis. Br. J. Clin. Pharmacol. 2015, 80, 1245–1253. [Google Scholar] [CrossRef]
- Ligi, D.; Benitez, S.; Croce, L.; Rivas-Urbina, A.; Puig, N.; Ordóñez-Llanos, J.; Mannello, F.; Sanchez-Quesada, J.L. Electronegative LDL Induces MMP-9 and TIMP-1 Release in Monocytes through CD14 Activation: Inhibitory Effect of Glycosaminoglycan Sulodexide. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 3559–3567. [Google Scholar] [CrossRef]
- Liu, Y.N.; Zhou, J.; Li, T.; Wu, J.; Xie, S.H.; Liu, H.F.; Liu, Z.; Park, T.S.; Wang, Y.; Liu, W.J. Sulodexide Protects Renal Tubular Epithelial Cells from Oxidative Stress-Induced Injury via Upregulating Klotho Expression at an Early Stage of Diabetic Kidney Disease. J. Diabetes Res. 2017, 2017, 4989847. [Google Scholar] [CrossRef]
- Pletinck, A.; Van Landschoot, M.; Steppan, S.; Laukens, D.; Passlick-Deetjen, J.; Vanholder, R.; Van Biesen, W. Oral Supplementation with Sulodexide Inhibits Neo-Angiogenesis in a Rat Model of Peritoneal Perfusion. Nephrol. Dial. Transplant. 2012, 27, 548–556. [Google Scholar] [CrossRef]
- Ciszewicz, M.; PoLubinska, A.; Antoniewicz, A.; Suminska-Jasinska, K.; BrEborowicz, A. Sulodexide Suppresses Inflammation in Human Endothelial Cells and Prevents Glucose Cytotoxicity. Transl. Res. 2009, 153, 118–123. [Google Scholar] [CrossRef]
- Mannello, F.; Raffetto, J.D. Matrix Metalloproteinase Activity and Glycosaminoglycans in Chronic Venous Disease: The Linkage among Cell Biology, Pathology and Translational Research. Am. J. Transl. Res. 2011, 3, 149. [Google Scholar] [PubMed]
- Mannello, F.; Medda, V.; Ligi, D.; Raffetto, J.D. Glycosaminoglycan Sulodexide Inhibition of MMP-9 Gelatinase Secretion and Activity: Possible Pharmacological Role against Collagen Degradation in Vascular Chronic Diseases. Curr. Vasc. Pharmacol. 2013, 11, 354–365. [Google Scholar] [CrossRef] [PubMed]
- Połubińska, A.; Staniszewski, R.; Baum, E.; Sumińska-Jasińska, K.; Brȩborowicz, A. Sulodexide Modifies Intravascular Homeostasis What Affects Function of the Endothelium. Adv. Med. Sci. 2013, 58, 304–310. [Google Scholar] [CrossRef]
- Harenberg, J. Review of Pharmacodynamics, Pharmacokinetics, and Therapeutic Properites of Sulodexide. Med. Res. Rev. 1998, 18, 1–20. [Google Scholar] [CrossRef]
- Doganci, S.; Ince, M.E.; Demeli, M.; Ors Yildirim, N.; Pehlivanoglu, B.; Yildirim, A.K.; Gianesini, S.; Chi, Y.W.; Yildirim, V. Sulodexide Develops Contraction in Human Saphenous Vein via Endothelium-Dependent Nitric Oxide Pathway. J. Clin. Med. 2023, 12, 1019. [Google Scholar] [CrossRef] [PubMed]
- Busse, R.; Fleming, I. Nitric Oxide, Nitric Oxide Synthase, and Hypertensive Vascular Disease. Curr. Hypertens. Rep. 1999, 1, 88–95. [Google Scholar] [CrossRef] [PubMed]
- Parkington, H.C.; Coleman, H.A.; Tare, M. Prostacyclin and Endothelium-Dependent Hyperpolarization. Pharmacol. Res. 2004, 49, 509–514. [Google Scholar] [CrossRef] [PubMed]
- Klinger, J.R.; Kadowitz, P.J. The Nitric Oxide Pathway in Pulmonary Vascular Disease. Am. J. Cardiol. 2017, 120, S71–S79. [Google Scholar] [CrossRef] [PubMed]
- Félétou, M.; Vanhoutte, P.M. Endothelium-Derived Hyperpolarizing Factor: Where Are We Now? Arterioscler. Thromb. Vasc. Biol. 2006, 26, 1215–1225. [Google Scholar] [CrossRef] [PubMed]
- Lincoln, T.M.; Cornwell, T.L.; Taylor, A.E. CGMP-Dependent Protein Kinase Mediates the Reduction of Ca2+ by CAMP in Vascular Smooth Muscle Cells. Am. J. Physiol. 1990, 258, C399–C407. [Google Scholar] [CrossRef] [PubMed]
- Raffetto, J.D.; Qiao, X.; Koledova, V.V.; Khalil, R.A. Prolonged Increases in Vein Wall Tension Increase Matrix Metalloproteinases and Decrease Constriction in Rat Vena Cava: Potential Implications in Varicose Veins. J. Vasc. Surg. 2008, 48, 447–456. [Google Scholar] [CrossRef] [PubMed]
- Raffetto, J.D.; Yu, P.; Reslan, O.M.; Xia, Y.; Khalil, R.A. Endothelium-Dependent Nitric Oxide and Hyperpolarization-Mediated Venous Relaxation Pathways in Rat Inferior Vena Cava. J. Vasc. Surg. 2012, 55, 1716–1725. [Google Scholar] [CrossRef]
- Li, J.; Parovian, C.; Li, J.; Hampton, T.G.; Metais, C.; Tkachenko, E.; Sellke, F.W.; Simons, M. Modulation of Microvascular Signaling by Heparan Sulfate Matrix: Studies in Syndecan-4 Transgenic Mice. Microvasc. Res. 2002, 64, 38–46. [Google Scholar] [CrossRef]
- Ahmed, I.; Yandrapalli, S. Internal Mammary Artery Bypass; StatPearls: Treasure Island, FL, USA, 2023. [Google Scholar]
- Yildiz, O.; Seyrek, M.; Gul, H.; Un, I.; Yildirim, V.; Ozal, E.; Uzun, M.; Bolu, E. Testosterone Relaxes Human Internal Mammary Artery in Vitro. J. Cardiovasc. Pharmacol. 2005, 45, 580–585. [Google Scholar] [CrossRef] [PubMed]
- Gaudino, M.; Benedetto, U.; Fremes, S.; Hare, D.L.; Hayward, P.; Moat, N.; Moscarelli, M.; Di Franco, A.; Nasso, G.; Peric, M.; et al. Effect of Calcium-Channel Blocker Therapy on Radial Artery Grafts after Coronary Bypass Surgery. J. Am. Coll. Cardiol. 2019, 73, 2299–2306. [Google Scholar] [CrossRef] [PubMed]
- Florian, J.A.; Kosky, J.R.; Ainslie, K.; Pang, Z.; Dull, R.O.; Tarbell, J.M. Heparan Sulfate Proteoglycan Is a Mechanosensor on Endothelial Cells. Circ. Res. 2003, 93, e136–e142. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
| Endothelium Integrity | Intact | Denuded | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Patient Number | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 | 14 | 15 | 16 | |
| Age | 71 | 52 | 68 | 71 | 57 | 68 | 50 | 62 | 70 | 46 | 72 | 62 | 61 | 56 | 71 | 56 | |
| Sex | M | F | M | M | M | M | M | M | M | M | F | M | M | M | M | M | |
| Hypertension | + | − | + | + | − | + | + | + | + | − | + | + | + | + | + | − | |
| Hyperlipidemia | + | − | − | − | − | − | + | − | + | − | − | − | − | + | − | − | |
| Diabetes | + | − | − | + | + | + | + | + | + | + | + | + | − | + | − | + | |
| Use of | Beta blockers | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + |
| ACE-I | − | − | − | + | − | + | + | − | + | − | − | − | + | − | + | − | |
| ARNI | + | − | − | − | − | − | − | + | − | − | + | − | − | − | − | − | |
| Diuretics | + | − | + | + | − | − | − | + | − | − | + | − | − | − | − | − | |
| Force of Contraction (g/100 mg Wet Tissue Weight) | ||||||
|---|---|---|---|---|---|---|
| Groups | Control | L-NAME Pre-Incubated | ||||
| Basal Tonus | KCl-Induced | PheE-Induced | Basal Tonus | KCl-Induced | PheE-Induced | |
| Endothelium-intact | 3.05 ± 0.37 | 6.05 ± 0.54 | 6.33 ± 0.52 | 3.32 ± 0.12 | 7.84 ± 0.56 # | 7.32 ± 0.57 # |
| Endothelium-denuded | 4.91 ± 0.41 * | 8.31 ± 0.68 * | 7.55 ± 0.61 ** | 4.09 ± 0.25 | 8.32 ± 0.30 | 7.15 ± 0.4 |
| PheE-Induced Only | Response to SDX | |||||||
|---|---|---|---|---|---|---|---|---|
| Endothelium Integrity | Intact | Denuded | Intact | Denuded | ||||
| Groups | Control | L-NAME Incubated | Control | L-NAME Incubated | Control | L-NAME Incubated | Control | L-NAME Incubated |
| Emax | 102.4 ± 2.8 | 110.9 ± 2.1 | 98.4 ± 1.4 | 101.2 ± 6.5 | 101.2 ± 8.3 | 103.2 ± 6.3 | 97.5 ± 5.8 | 101.1 ± 7.1 |
| pEC50 | 6.31 ± 0.16 | 7.31 ± 0.13 * | 7.11 ± 0.13 ** | 7.05 ± 0.18 ** | 5.92 ± 0.2 | 4.80 ± 0.14 * | 4.62 ± 0.21 ** | 4.83 ± 0.19 ** |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ors Yildirim, N.; Yildirim, A.K.; Demeli Ertus, M.; Dastan, A.O.; Pehlivanoglu, B.; Chi, Y.-W.; Gianesini, S.; Doganci, S.; Yildirim, V. Sulodexide Inhibits Arterial Contraction via the Endothelium-Dependent Nitric Oxide Pathway. J. Clin. Med. 2024, 13, 2332. https://doi.org/10.3390/jcm13082332
Ors Yildirim N, Yildirim AK, Demeli Ertus M, Dastan AO, Pehlivanoglu B, Chi Y-W, Gianesini S, Doganci S, Yildirim V. Sulodexide Inhibits Arterial Contraction via the Endothelium-Dependent Nitric Oxide Pathway. Journal of Clinical Medicine. 2024; 13(8):2332. https://doi.org/10.3390/jcm13082332
Chicago/Turabian StyleOrs Yildirim, Nadide, Alperen Kutay Yildirim, Meric Demeli Ertus, Ahmet Onur Dastan, Bilge Pehlivanoglu, Yung-Wei Chi, Sergio Gianesini, Suat Doganci, and Vedat Yildirim. 2024. "Sulodexide Inhibits Arterial Contraction via the Endothelium-Dependent Nitric Oxide Pathway" Journal of Clinical Medicine 13, no. 8: 2332. https://doi.org/10.3390/jcm13082332