Abstract
Anti-amyloid therapies (AATs) are increasingly being recognized as promising treatment options for Alzheimer’s disease (AD). Amyloid-related imaging abnormalities (ARIAs), small areas of edema and microbleeds in the brain presenting as abnormal signals in MRIs of the brain for patients with AD, are the most common side effects of AATs. While most ARIAs are asymptomatic, they can be associated with symptoms like nausea, headache, confusion, and gait instability and, less commonly, with more serious complications such as seizures and death. Cerebral amyloid angiopathy (CAA) has been found to be a major risk for ARIA development. The identification of sensitive and reliable non-invasive biomarkers for CAA has been an area of AD research over the years, but with the approval of AATs, this area has taken on a new urgency. This comprehensive review highlights several potential biomarkers, such as Aβ40, Aβ40/42, phosphorylated-tau217, neurofilament light chain, glial fibrillary acidic protein, secreted phosphoprotein 1, placental growth factor, triggering receptor expressed on myeloid cells 2, cluster of differentiation 163, proteomics, and microRNA. Identifying and staging CAA even before its consequences can be detected via neuroimaging are critical to allow clinicians to judiciously select appropriate candidates for AATs, stratify monitoring, properly manage therapeutic regimens for those experiencing symptomatic ARIAs, and optimize the treatment to achieve the best outcomes. Future studies can test potential plasma biomarkers in human beings and evaluate predictive values of individual markers for CAA severity.
1. Introduction
In the United States (U.S.), Alzheimer’s disease (AD) is the 5th leading cause of death [1] and the most common cause of dementia among older adults [2]. AD affected approximately 6.9 million Americans aged 65 and older in 2024 [1]. Considering the growing number of older adults, the prevalence and incidence of AD are expected to increase, with estimations indicating that nearly 13 million people will be affected by 2050 [1]. Anti-amyloid therapies (AATs) are approved by the U.S. Food and Drug Administration and have become promising treatment options for AD [3,4,5]. However, like other medications, AATs have side effects. Amyloid-related imaging abnormalities (ARIAs), i.e., abnormal signals seen in MRIs of the brain for patients with AD, are the most common side effect of AATs. Cerebral amyloid angiopathy (CAA) has been found to be a major risk for ARIA occurrence. The identification of sensitive and reliable non-invasive biomarkers for CAA has become a current trend of AD research following the approval of AATs. There have been great advances in the development of blood-based biomarkers that can help us to understand AD. Blood-based testing is a rapid, minimally invasive, and inexpensive method with high accuracy in identifying AD [6,7,8]. However, very few studies aiming to identify blood-based biomarkers associated with the development or severity of CAA have been conducted. In this review, we aim to explain why this is an urgent area of research, highlight potential candidate blood-based biomarkers (amyloid β, tau, glial fibrillary acidic protein, neurofilament light chain, secreted phosphoprotein 1, placental growth factor, triggering receptor expressed on myeloid cells 2, cluster of differentiation 163, proteomics, and microRNA), and outline a discovery strategy for identifying novel blood-based biomarkers for CAA.
2. AD, AATs, and ARIAs
The first sign of AD is the accumulation of amyloid β plaques in the brain. AD typically advances from preclinical AD to mild cognitive impairment (MCI) and potentially dementia due to AD (Table 1) [1,9].

Table 1.
Progressive stages of Alzheimer’s Disease [1].
Each individual spends a different amount of time in each stage. For example, influenced by age, genetics, biological sex, clinical setting, and baseline CSF t-tau levels, the total disease duration varies between 12 and 25 years, while the duration of the preclinical stage varies between 2 and 15 years, the length of the MCI stage varies between 3 to 7 years, the duration of the mild AD dementia stage varies between 2 and 6 years, and the length of the moderate AD dementia stage varies between 1 and 7 years [9]. Older people, males, ApoE Ɛ4 carriers, and those with increased CSF t-tau levels experience faster cognitive decline with a shorter duration [9]. Identifying ways to slow down cognitive decline and prolong the duration within a milder disease stage is desperately needed to increase the quality of health of older adults.
Approved and experimental monoclonal antibodies designed to target Aβ plaques and their precursor molecules have proven to be promising therapeutic tools with beneficial effects on individuals with mild cognitive impairment and mild dementia due to AD in randomized controlled trials [3,4,5]. The FDA-approved anti-amyloid drugs include lecanemab, aducanumab, and donanemab. ARIAs are the most common side effect of AATs, and they are classified into two subtypes: ARIA-E (edema/effusion) and ARIA-H (microhemorrhages, macrohemorrhages and/or superficial siderosis) [4,10]. The common clinical symptoms of ARIAs include changes in mental status, confusion, headache, visual disturbances, vomiting, nausea, gait disturbances, tremor, and even death [4,5,10,11]. Within the first 3 months for 10 mg/kg monthly and biweekly dosing regimens in a phase 2 lecanemab trial, the incidence of ARIA-E was <10% and that for symptomatic ARIA was <3% [3,10]. In a phase 3 lecanemab trial, the incidence of ARIA-E was 12.6% (vs. 1.7% with placebo) and that for ARIA-H was 17.3% (vs. 9.05% with placebo) [5]. As AATs are becoming promising treatment avenues, improved knowledge of the pathophysiology of ARIA is necessary for the early detection of those who are at greatest risk.
3. Risk Factors for ARIA Development
Several risk factors for ARIA, such as CAA; the presence of one or more microhemorrhages/superficial siderosis and white matter disease at baseline; a higher baseline amyloid load; hypertension [12]; the initial treatment period (e.g., most instances of ARIA-E occur within the first 3 months of treatment); the type of antibody used [13,14,15,16,17,18,19,20]; higher dosages [11]; an ApoE Ɛ4 genotype, with ApoE Ɛ4 homozygotes posing the greatest risk [15,21,22,23,24]; and anti-thrombin use [12], have been identified. Because CAA is highly prevalent in AD (up to 93.6%) [25] and both conditions have the same underlying mechanism, namely, impaired Aβ clearance [24], CAA is a key risk factor for ARIA development.
The high prevalence of CAA in AD places many patients at risk for ARIA-associated morbidity. ARIAs can be serious (e.g., leading to hospitalization, disability, etc.) and life-threatening for approximately 1% of patients [11,26,27]. The morbidity and cost of care can be substantial. This highlights the importance of detecting and staging CAA in its “preclinical” stage, even before its consequences can be detected on neuroimaging. However, the ability to identify people with preclinical CAA is severely limited due to a lack of a reliable, specific, and sensitive screening tool.
Histopathological confirmation via brain biopsy or autopsy, which is not clinically feasible, is the gold standard for CAA diagnosis [28]. Among in vivo tools, CT and MRI are most reliable in identifying CAA according to the modified Boston criteria [29], but these approaches detect only the secondary consequences of CAA [30]. CAA is often asymptomatic. Its presence may be detected in vivo through surrogate telltale signs in MRIs, such as edema or hemorrhaging, which can present as micro- or macro-hemorrhages or superficial siderosis [3,5,10,31]. The modified Boston criteria offer one schema for identifying CAA [32] but only in the presence of bleeding. In addition, the imaging markers lack specificity because they may be partially induced by arteriosclerotic small-vessel disease [33]. Amyloid PET imaging has limited diagnostic accuracy for CAA because it is unable to differentiate between vascular and parenchymal amyloid β (Aβ) [34].
4. Diagnosis of CAA
The identification of sensitive and reliable non-invasive biomarkers for CAA has been an area of AD research for some time, but with the approval of AAT, research in this area has taken on a new urgency. The Foundation of the NIH Biomarkers Consortium reported that plasma Aβ is highly correlated with amyloid PET or CSF amyloid β (Aβ) levels [35]. However, plasma tests have mainly been used to detect AD, not CAA. A recent study conducted on animals found reduced Aβ40 levels in CSF and plasma in early-stage CAA prior to the onset of cerebral microbleeds as assessed via MRI and histological staining; then, Aβ40 levels abruptly dropped at the early onset of CAA and continued to decrease as CAA progressed [36]. Considering the advantages of using plasma tests and the promising study findings, further studies measuring Aβ peptide levels in humans for CAA diagnosis are needed to advance AD management. However, there is a need to explore the relationship between CAA and parenchymal amyloid plaque, and this endeavor requires the use of a multi-biomarker panel. Thus, in this review, we propose to investigate several potential biomarkers for CAA identification, such as Aβ40, Aβ40/42, phosphorylated-tau217 (p-tau217), neurofilament light chain (NfL), glial fibrillary acidic protein (GFAP), secreted phosphoprotein 1 (SPP1), placental growth factor (PIGF), triggering receptor expressed on myeloid cells 2 (TREM2), cluster of differentiation (CD)163, proteomics, and microRNA. Figure 1 depicts the pathways of the potential biomarkers to CAA via neurodegeneration, neuroinflammation, and pathological angiogenesis.
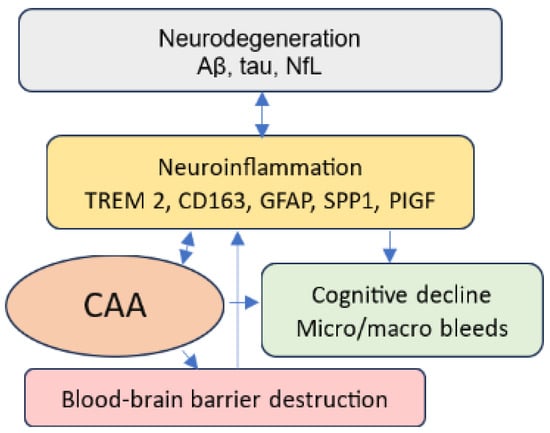
Figure 1.
Neurodegeneration, neuroinflammation, and CAA.
Neurodegeneration, neuroinflammation, and pathological angiogenesis are direct consequences of excess Aβ in the brain. They are frequently seen in AD brains and mutually reinforcing [37,38]. The presence of Aβ and tau in neurodegenerative disease triggers and exacerbates neuroinflammation by activating glial cells, such as microglia and astrocytes, and immune cells, such as macrophages. Both microglia and perivascular macrophages play important roles in Aβ clearance in parenchyma and CAA [39]. Activated microglia, macrophages, and neuroinflammation can also exacerbate Aβ deposition, resulting in a vicious cycle. The presence of Aβ in the blood vessels is related to neuroinflammation and vessel inflammation, contributing to blood–brain barrier destruction, which further leads to neuroinflammation and cognitive impairment.
Aβ, made up of 39–43-residue amyloidogenic peptides (including Aβ1-40 and Aβ1-42), are produced from amyloid precursor protein [40,41]. Soluble Aβ is produced throughout one’s life, but some of it is pathologically converted into insoluble Aβ, which is the major component of the neuritic plaques in the brain parenchyma and vessel Aβ [42]. Neuritic plaques, the result of Aβ accumulation in the brain parenchyma, are a pathological hallmark of AD [40,43]. In contrast, CAA is the accumulation of Aβ in cerebral vessels, primarily in the medium-sized arteries and arterioles in the leptomeningeal and cortical regions [42,44,45,46,47,48]. Advanced CAA can render vessels fragile and prone to rupturing, resulting in intracerebral hemorrhages in lobar regions, cortical superficial siderosis, and white matter hyperintensities [36,49,50,51,52,53]. Cerebral microhemorrhages, cortical superficial siderosis, and a history of intracranial hemorrhage are well-established surrogate markers of CAA severity [54]. While Aβ42 is thought to be the foundational seed for both parenchymal plaques and CAA formation, higher cerebral Aβ40 levels and Aβ40/42 ratios are more likely related to CAA formation, whereas higher cerebral Aβ42 and Aβ42/40 levels are more likely related to parenchymal plaques [44,45,46].
Aβ40 and Aβ42 reflect amyloid precursor protein metabolism and p-tau for tangle pathology [28]. Since CAA is the result of impaired clearance of Aβ from interstitial cerebral fluid, CSF contains biomarkers reflecting this process [34]. Several studies have tested CSF biomarkers such as Aβ40 and Aβ42 to identify early-stage CAA [33,34,55,56,57]. Table 2 shows CSF Aβ40, Aβ42, and Aβ40/42 levels with respect to individuals with CAA and AD [30]. All these studies agreed that people with CAA had lower CSF Aß40 and Aß42 levels than both the control and AD groups, but they were inconclusive on the ability to distinguish CAA from AD [56]. None of these studies included the use of a multi-biomarker panel to aid in the differentiation of CAA from AD. P-tau refers to tau proteins that have undergone phosphorylation [58]. Plasma p-tau 217 is one of the most promising biomarkers for diagnosing AD [59,60]. For example, plasma p-tau 217 was found to offer high accuracy in identifying elevated Aβ levels (area under the curve [AUC]: 0.92–0.96; 95% CI, 0.89–0.99) and tau pathology (AUC, 0.93–0.97; 95% CI, 0.84–0.99) in 786 individuals from three single-center observational cohorts [59]. In another study, plasma p-tau 217 was found to be offer high accuracy (AUC, 0.80–0.91) in identifying Aβ pathology, determined either via Aβ positron emission tomography or CSF Aβ42/40 ratio [60]. A meta-analysis reported that the core CSF biomarkers (Aβ40, Aβ42, and p-tau) can serve as molecular biomarkers of CAA [28]. However, the relationship between plasma Aβ and p-tau in the context of CAA has not been extensively studied.
Table 2.
Results of CSF Aβ42, Aβ40, and Aβ40/42.
Glial fibrillary acidic protein (GFAP), an astrocytic marker, is a Food and Drug Administration-cleared marker used to treat mild traumatic brain injuries/concussions [61]. The GFAP blood test helps clinicians determine the need for computerized tomography for patients with a suspected head injury and helps prevent unnecessary neuroimaging [61]. GFAP has recently been reported to be a potential biomarker for AD [62,63,64,65]. Studies have found that patients with AD have significantly higher GFAP levels than healthy controls [62,63,64]. In addition, elevated GFAP levels have been associated with increased neuritic (p < 0.001) and diffuse plaque density or counts (p = 0.001) as well as with Braak stages and tangle counts (p < 0.05) [63]. Plasma GFAP was found to be able to detect AD more accurately than CSF GFAP [66,67]. In addition, both serum and CSF GFAP were found to be biomarkers for neuroaxonal damage and astrocytosis in CAA [68].
Neurofilament light chain (NfL) is a protein and considered to be a biomarker for several neurodegenerative diseases. NfL is mainly found in large-caliber myelinated axons, and it is essential for the transmission of electrical impulses [69]. A low amount of NfL is present in normal physiological conditions such as brain development, maturation, and aging, but a high amount of NfL is present in the cerebrospinal fluid and blood when axonal damage or neurodegeneration occurs [69]. NfL is a reliable biomarker of neuronal damage, dementia, stroke, and traumatic brain injuries, and it is released into the bloodstream and CSF when brain cells are damaged [70,71,72]. NfL can be a powerful tool for measuring and predicting AD progression but not diagnosing AD because brain cell damage can be caused by other factors [70,72]. Elevated plasma NfL levels were found to have an association with changes in global cognition, attention, and amyloid PET longitudinally [70]. Studying the severity of AD can give insights into how the severity of AD is related to or exacerbates CAA. A recent study found serum NfL to be a biomarker for advanced CAA [68], but more studies are needed to confirm this finding.
Secreted phosphoprotein 1 (SPP1), also known as osteopontin, plays a role in cell adhesion, migration, and inflammation [73]. Osteopontin functions as a “bridge builder” in the central nervous system (CNS). It is expressed in nearly all cells in the CNS and interacts with several different types of receptors and proteins [74]. The longitudinal Northern Manhattan Study (NOMAS) conducted on three groups of adults aged ≥ 40 with dementia and cerebral small vessel disease (CSVD), dementia with no CSVD, and no dementia and no CSVD reported that the highest levels of plasma oesteoponin were in the group with dementia and CSVD, supporting a significant association between osteoponin levels and dementia with evidence of CSVD [75]. In addition, the NOMAS reported there was a strong correlation between osteoponin and white matter hyperintensity volumes [75]. Studies have identified that elevated levels of SPP1 are associated with increased neuroinflammation and neurodegeneration; thus, SPP1 is upregulated in several neuro-disorders, including multiple sclerosis, AD, and age-related macular degeneration [75,76]. A study based on the ROSMAP reported SPP1 expression was associated with faster cognitive decline, greater odds of AD and CAA, and activated microglia and neuroinflammatory disorders [77].
Placental growth factor (PIGF) is part of the vascular endothelial growth factor family. It is released in the highest amounts in the placenta and decidual tissues, but it can also be detected in endothelial tissues and bone marrow erythroblasts as well as a variety of tissues such as lung and heart tissue, adipose tissues, and skeletal muscle [78,79]. PIGF is either present in low amounts or nonexistent in healthy tissues, but it is present in high concentrations in diseased conditions. PIGF potentially plays roles in wound healing, tumor growth, and collateral vessel formation when ischemia occurs [80]. PIGF plays a role in angiogenesis, especially in pathological angiogenesis and inflammation [78,79]. In addition, PIGF can help distinguish whether cognitive impairment is the result of AD or an issue involving other vascular issues [79]. A multisite observational cohort study reported that there is a significant association between PIGF and cognitive impairment associated with white matter injury and dementia [81]. Others have also found elevated PIGF levels in people with a higher burden of white matter injury and cerebral microbleeds associated with AD [82]. A pilot study indicated that Aβ regulates angiogenesis through PIGF expression [38]. Endothelial dysfunction caused by PIGF is a common condition in CAA, highlighting the complex relationship between CAA and PIGF in vascular pathology. GFAP, SPP1, and PIGF are emerging biomarkers for AD research, and understanding their relationships with CAA might provide insights into the mechanisms underlying vascular changes in CAA.
The central nervous system (CNS) contains numerous myeloid cells and defensive barriers such as the meninges, the perivascular space, and the choroid plexus [83]. The central myeloid cells include parenchymal microglia as well as non-parenchymal macrophages such as perivascular macrophages, meningeal macrophages, choroid plexus macrophages, and monocytes [83,84]. Microglia are glial cells and considered the tissue-resident macrophages of the CNS located in the parenchyma [77,85]. Microglia constitute 10–12% of CNS cells and perform three essential functions: sensing, housekeeping, and protection against injurious self- and foreign stimuli (host defense) [86]. Microglia sense and scan the surrounding area every few hours and rapidly polarize toward a focal injury. The housekeeping functions of microglia include engaging in synaptic remodeling, phagocytosing dead or dying cells by moving to the site of neuronal death, and maintaining myelin homeostasis. Microglia mediate the host’s defenses against pathogens by initiating neuroinflammatory responses (e.g., producing cytokines and chemokines, recruiting additional cells and inducing their activity to clear pathogens, and maintaining brain homeostats (a neuroprotective role)) [83,85]. These neuroinflammatory responses help CNS neural networks mature, leading to healthy CNS function [76]. Healthy brains are capable of engaging in these responses, but dysregulation of any of the three responses (e.g., via persistent neuroinflammation) can lead to imbalance and neurodegeneration. The emergence of disease-associated microglia (DAMs), along with unique gene expression signatures, is the consequence of neuroinflammation and neurodegeneration [76]. Microglial activation plays an important role in the pathophysiology of AD [87,88] and CAA-related inflammation [89,90], eventually leading to ARIA. Identifying the crucial factors influencing the shift from healthy microglia to DAMs might help in developing microglial-cell-based therapeutic approaches [91].
Microglia may have a protective effect on AD by limiting the spread of Aβ and tau pathologies, but they also have a detrimental effect when phagocytosing Aβ, as they further damage unaffected brain tissue [92]. Understanding the relationship between microglia and AD may hold the key to AD management.
Triggering receptor expressed on myeloid cells 2 (TREM2) is a major microglial activation marker and plays a crucial role in microglial survival, activation, phagocytosis, and the maintenance of brain homeostasis and inflammatory responses to injuries or neurodegeneration [93]. Several studies have found that TREM2 is a reliable biomarker of AD and a contributor to neuroinflammation [94,95,96,97]. In an AD mouse model study, the loss of TREM2 was associated with a remarkable increase in Aβ load in the brain but also a dramatic decrease in CAA, showing the different effects of TREM2 in parenchymal and vascular Aβ pathologies [97]. A study reported a positive association between the concentration of p-tau in the CSF and TREM2 levels [95]. Others have proposed a potential connection of TREM2 with ARIA [98]. TREM2 is expressed in microglia and macrophages, and thus CSF levels and blood levels may have unique characters. Measuring TREM2 as a blood-based biomarker may provide unique evidence for CAA, wherein the macrophages may have a more dominant role.
Cluster of differentiation 163 (CD163) is a macrophage activation marker and has anti-inflammatory properties [99]. CD163 was found to be correlated with small-vessel injury in early AD in a mouse model [99]. Both microglia and macrophage activation markers have been gaining attention, especially when the identification of CAA biomarkers is urgently needed for ARIA management [98]. An animal model study reported that increased perivascular macrophage (e.g., CD163) turnover promotes vascular Aβ clearance, suggesting that activating perivascular macrophage could be a potential therapeutic strategy for clearing vascular amyloids [100]. Macrophages are white blood cells that play a crucial role in immune surveillance (immune cells) and the phagocytosis of pathogens and cellular debris [39]. CNS-associated macrophages are found in the meninges (leptomeninges and dura) [85]. Activated by pathogen invasion, macrophages produce inflammatory cytokines and activate signaling pathways leading to tissue repair [101]. Both microglia and the phagocytic activity of perivascular macrophages have been found to play a significant role in Aβ clearance not only in parenchyma but also in cerebral vessels [39]. Many potential microglial and macrophage molecules have been identified and are currently being tested in preclinical models, and much more effort is needed to speed up the transition to the clinical setting. Table 3 below provides the results of our literature search for potential biomarkers.
Table 3.
Summary of the literature on potential biomarkers.
Proteomics has remarkable potential with respect to advance clinical research and biomarker discovery. Studies have identified several proteins that can indicate amyloid pathology [104,105,106,107]. For example, a study identified specific groups of proteins differentially expressed in patients with CAA pathology and CAA rat models (e.g., rTg-DI) in the CSF [106]. They found that the cathepsin protein family (CTSB, CTSD, and CTSS) and its main inhibitor (CST3) in rats and synaptic proteins (e.g., VGF, NPTX1, and NRXN2) and several members of the granin family (SCG1, SCG2, SCG3, and SCG5) were differentially expressed in humans in comparison to the controls. Another proteomics study involving a laser dissection microscopy-assisted mass spectrometry analysis of post-mortem human brain tissue identified potentially highly selective markers of CAA (e.g., clusterin, apolipoprotein E (APOE), serum amyloid P-component, norrin, collagen and alpha-2(VI)) [105]. Considering the remarkable promise of proteomics in identifying amyloid pathology, further studies are needed to identify CAA in humans [91].
MicroRNAs are short (~22 nt-long) and constitute a class of non-coding RNAs [108] that play a role in gene expression [109]. MicroRNAs can be used for diagnostic or therapeutic purposes because of the ability to detect circulating microRNA (c-microRNA) [109]. C-microRNA is released when cells are injured, inflamed, necrotic, or apoptotic [109]. Several studies have reported associations between dysregulated microRNA expression and neurodegenerative diseases such as AD [110,111,112]. An RNA-sequencing study reported that microRNAs are most abundant in human plasma, making up over 42.34% of all raw reads and 76.20% of all mappable reads [113]. Since these microRNAs are transported throughout the body through “insulation”—unlike free-floating mRNAs, which are quickly degraded by RNases—they have significantly longer half-lives (which can be 5 days or longer in some cases), making these RNA transcripts great biomarker candidates for disease [109]. MicroRNAs function as modulators of both neuronal and immune processes (termed “neurimmiRs”) in the CNS [114]. MicroR-124, microR-132, and microR-146 are prototypes of microRNAs and play important roles in neurodegeneration, neurogenesis, and synaptic plasticity [115]. MicroRNAs have emerged as promising markers for understanding the underlying complex pathology of AD based on its involvement in Aβ metabolism, neuroinflammation, and synaptic function [108,116]. Because of its role in Aβ metabolism, microRNA may be a biomarker for CAA. More clinical studies are necessary.
5. Discussion
The advent of the use of AATs for the prevention and treatment of AD and the recognition that ARIAs might limit their usefulness have prompted a new line of research exploring how to better predict the AAT-related risk posed by ARIAs. Because CAA is a major risk factor posed by ARIAs, the identification of CAA and risk stratification based on CAA may help optimize AAT use. As discussed above, there are many potential biomarkers that can be used to identify and stage CAA even before its consequences can be detected via neuroimaging.
Identifying specific and sensitive diagnostic markers that can be used to identify patients with AD who are at greater risk of ARIA development prior to treatment with an AAT is of substantial importance. Even their use within AAT clinical trials is an urgent priority for maximizing patient safety and improving the risk-vs.-benefit trade-off of therapies. AATs cause serious (e.g., leading to hospitalization and disability) and life-threatening ARIAs in approximately 1% of patients [11,26,27]. The morbidity and cost of care for ARIAs can be substantial. In 2024, about 7 million Americans were living with AD [1]. If as few as one million received AAT, one can expect serious complications in 10,000 cases. The average adjusted cost of hospitalization per inpatient stay in 2019 was USD 14,101 [117]. The 65-and-older population is estimated to reach 82 million by 2050 [118], while cases of AD are expected to reach approximately 13 million by 2050 [1]. The healthcare costs for ARIA-related complications will escalate with the increasing number of older adults and the growing number of patients with AD starting AATs. In addition, there is no alternative to AATs with respect to treatment. Therefore, it is of upmost importance to study which patients will be prone to these side effects before deciding which patients to subject to a treatment protocol.
The review of the literature given above demonstrates that while plasma biomarkers have led to great progress in AD research, their application for identifying CAA has been limited but constitutes a promising arena. There is a need to study the existing long list of biomarkers and identify novel biomarkers that may help identify CAA. This approach should rely on samples well characterized via gold-standard quantitative assessments made using digital neuropathology along with antemortem data assessed using the Boston Criteria 2.0. The biomarker candidates identified need to be validated in a second cohort for which there are only antemortem imaging data assessed using the Boston criteria 2.0.
6. Limitations and Suggestions for Future Studies
Autopsy-based CAA identification is a gold standard, but it is not practical. The modified Boston criteria are helpful only in the presence of bleeding. To date, little research has been conducted to identify blood-based biomarkers for CAA. Detecting and staging CAA even before its consequences can be detected via neuroimaging are critical to allow clinicians to judiciously select appropriate candidates for AATs, stratify monitoring, properly manage therapeutic regimens for those experiencing symptomatic ARIAs, and optimize treatments to achieve the best outcomes. In our opinion, p-tau 217 and Aβ40 may have the greatest potential as CAA biomarkers because of the strong associations with Aβ accumulation and tau pathology, whereas PIGF may have the least potential as a CAA biomarker because of its indirect involvement in AD pathophysiology. However, more studies are necessary to establish the roles of the potential promising biomarkers we have reviewed in this paper. Future studies can test potential plasma biomarkers in human beings and evaluate the predictive value of individual markers for CAA severity.
7. Conclusions
CAA is a major risk factor for ARIA development. Identifying biomarkers for CAA is necessary for the proper management of AD. Post-mortem diagnosis is a definite means of diagnosing CAA, but it is not practical. The MRI-based Boston criteria are useful for diagnosing CAA, but they are only helpful after a hemorrhage has occurred. The current research on CAA biomarkers is centered on Aβ in CSF. The use of plasma-based biomarkers is a convenient and non-invasive strategy for detecting and staging CAA in its “preclinical” stage before its consequences are detected via neuroimaging. We propose further exploring the list of existing biomarkers and identifying novel biomarkers for the better prediction and early detection of CAA to minimize the risk of ARIAs in patients receiving AAT for the treatment and prevention of AD, an urgent global public health issue.
Author Contributions
M.-K.S.—Original draft, conceptualization, review & editing; J.L.D., K.N., N.M.D., N.T.S., D.A.B., A.I.L.: review & editing, A.A.: review & editing, supervision. All authors have read and agreed to the published version of the manuscript.
Funding
M.-K.S. was in part supported by the National Institute on Aging of the National Institutes of Health under Award Numbers 1R03AG072110-01A1 and 1R03AG070579-01. A.A. was in part supported by the National Institute on Aging of the National Institutes of Health under Award Numbers 1RF1AG069121 and 1R01AG073474.
Conflicts of Interest
The authors have no conflicts of interest to report.
References
- Alzheimer’s Association. Alzheimer’s Facts and Figures Report. Available online: https://www.alz.org/alzheimers-dementia/facts-figures (accessed on 28 January 2025).
- National Institute on Aging. Alzheimer’s Disease Fact Sheet. Available online: https://www.nia.nih.gov/health/alzheimers-and-dementia/alzheimers-disease-fact-sheet (accessed on 28 January 2025).
- Swanson, C.J.; Zhang, Y.; Dhadda, S.; Wang, J.; Kaplow, J.; Lai, R.Y.K.; Lannfelt, L.; Bradley, H.; Rabe, M.; Koyama, A.; et al. A randomized, double-blind, phase 2b proof-of-concept clinical trial in early Alzheimer’s disease with lecanemab, an anti-Aβ protofibril antibody. Alzheimers Res. Ther. 2021, 13, 80. [Google Scholar] [CrossRef] [PubMed]
- Withington, C.G.; Turner, R.S. Amyloid-Related Imaging Abnormalities With Anti-amyloid Antibodies for the Treatment of Dementia Due to Alzheimer’s Disease. Front. Neurol. 2022, 13, 862369. [Google Scholar] [CrossRef] [PubMed]
- van Dyck, C.H. Anti-Amyloid-β Monoclonal Antibodies for Alzheimer’s Disease: Pitfalls and Promise. Biol. Psychiatry 2018, 83, 311–319. [Google Scholar] [CrossRef]
- Li, Y.; Schindler, S.E.; Bollinger, J.G.; Ovod, V.; Mawuenyega, K.G.; Weiner, M.W.; Shaw, L.M.; Masters, C.L.; Fowler, C.J.; Trojanowski, J.Q.; et al. Validation of Plasma Amyloid-β 42/40 for Detecting Alzheimer Disease Amyloid Plaques. Neurology 2022, 98, e688–e699. [Google Scholar] [CrossRef]
- Brand, A.L.; Lawler, P.E.; Bollinger, J.G.; Li, Y.; Schindler, S.E.; Li, M.; Lopez, S.; Ovod, V.; Nakamura, A.; Shaw, L.M.; et al. The performance of plasma amyloid beta measurements in identifying amyloid plaques in Alzheimer’s disease: A literature review. Alzheimers Res. Ther. 2022, 14, 195. [Google Scholar] [CrossRef]
- Nakamura, T.; Kawarabayashi, T.; Seino, Y.; Hirohata, M.; Nakahata, N.; Narita, S.; Itoh, K.; Nakaji, S.; Shoji, M. Aging and APOE-ε4 are determinative factors of plasma Aβ42 levels. Ann. Clin. Transl. Neurol. 2018, 5, 1184–1191. [Google Scholar] [CrossRef]
- Vermunt, L.; Sikkes, S.A.M.; van den Hout, A.; Handels, R.; Bos, I.; van der Flier, W.M.; Kern, S.; Ousset, P.-J.; Maruff, P.; Skoog, I.; et al. Duration of preclinical, prodromal, and dementia stages of Alzheimer’s disease in relation to age, sex, and APOE genotype. Alzheimers Dement. 2019, 15, 888–898. [Google Scholar] [CrossRef]
- Honig, L.S.; Barakos, J.; Dhadda, S.; Kanekiyo, M.; Reyderman, L.; Irizarry, M.; Kramer, L.D.; Swanson, C.J.; Sabbagh, M. ARIA in patients treated with lecanemab (BAN2401) in a phase 2 study in early Alzheimer’s disease. Alzheimers Dement. 2023, 9, e12377. [Google Scholar] [CrossRef]
- Salloway, S.; Chalkias, S.; Barkhof, F.; Burkett, P.; Barakos, J.; Purcell, D.; Suhy, J.; Forrestal, F.; Tian, Y.; Umans, K.; et al. Amyloid-Related Imaging Abnormalities in 2 Phase 3 Studies Evaluating Aducanumab in Patients with Early Alzheimer Disease. JAMA Neurol. 2022, 79, 13–21. [Google Scholar] [CrossRef]
- Doran, S.J.; Sawyer, R.P. Risk factors in developing amyloid related imaging abnormalities (ARIA) and clinical implications. Front. Neurosci. 2024, 18, 1326784. [Google Scholar] [CrossRef]
- Budd Haeberlein, S.; O’Gorman, J.; Chiao, P.; Bussière, T.; von Rosenstiel, P.; Tian, Y.; Zhu, Y.; von Hehn, C.; Gheuens, S.; Skordos, L.; et al. Clinical Development of Aducanumab, an Anti-Aβ Human Monoclonal Antibody Being Investigated for the Treatment of Early Alzheimer’s Disease. J. Prev. Alzheimers Dis. 2017, 4, 255–263. [Google Scholar] [CrossRef] [PubMed]
- Carlson, C.; Siemers, E.; Hake, A.; Case, M.; Hayduk, R.; Suhy, J.; Oh, J.; Barakos, J. Amyloid-related imaging abnormalities from trials of solanezumab for Alzheimer’s disease. Alzheimers Dement. 2016, 2, 75–85. [Google Scholar] [CrossRef] [PubMed]
- Cummings, J.L.; Cohen, S.; van Dyck, C.H.; Brody, M.; Curtis, C.; Cho, W.; Ward, M.; Friesenhahn, M.; Rabe, C.; Brunstein, F.; et al. ABBY: A phase 2 randomized trial of crenezumab in mild to moderate Alzheimer disease. Neurology 2018, 90, e1889–e1897. [Google Scholar] [CrossRef] [PubMed]
- Doody, R.S.; Thomas, R.G.; Farlow, M.; Iwatsubo, T.; Vellas, B.; Joffe, S.; Kieburtz, K.; Raman, R.; Sun, X.; Aisen, P.S.; et al. Phase 3 trials of solanezumab for mild-to-moderate Alzheimer’s disease. N. Engl. J. Med. 2014, 370, 311–321. [Google Scholar] [CrossRef] [PubMed]
- Greenberg, S.M.; Bacskai, B.J.; Hernandez-Guillamon, M.; Pruzin, J.; Sperling, R.; van Veluw, S.J. Cerebral amyloid angiopathy and Alzheimer disease—One peptide, two pathways. Nat. Rev. Neurol. 2020, 16, 30–42. [Google Scholar] [CrossRef]
- Landen, J.W.; Andreasen, N.; Cronenberger, C.L.; Schwartz, P.F.; Börjesson-Hanson, A.; Östlund, H.; Sattler, C.A.; Binneman, B.; Bednar, M.M. Ponezumab in mild-to-moderate Alzheimer’s disease: Randomized phase II PET-PIB study. Alzheimers Dement. 2017, 3, 393–401. [Google Scholar] [CrossRef]
- Leurent, C.; Goodman, J.A.; Zhang, Y.; He, P.; Polimeni, J.R.; Gurol, M.E.; Lindsay, M.; Frattura, L.; Sohur, U.S.; Viswanathan, A.; et al. Immunotherapy with ponezumab for probable cerebral amyloid angiopathy. Ann. Clin. Transl. Neurol. 2019, 6, 795–806. [Google Scholar] [CrossRef]
- Mintun, M.A.; Lo, A.C.; Duggan Evans, C.; Wessels, A.M.; Ardayfio, P.A.; Andersen, S.W.; Shcherbinin, S.; Sparks, J.; Sims, J.R.; Brys, M.; et al. Donanemab in Early Alzheimer’s Disease. N. Engl. J. Med. 2021, 384, 1691–1704. [Google Scholar] [CrossRef]
- Barakos, J.; Purcell, D.; Suhy, J.; Chalkias, S.; Burkett, P.; Marsica Grassi, C.; Castrillo-Viguera, C.; Rubino, I.; Vijverberg, E. Detection and Management of Amyloid-Related Imaging Abnormalities in Patients with Alzheimer’s Disease Treated with Anti-Amyloid Beta Therapy. J. Prev. Alzheimers Dis. 2022, 9, 211–220. [Google Scholar] [CrossRef]
- Chiong, W.; Tolchin, B.D.; Bonnie, R.J.; Busl, K.; Cruz-Flores, S.; Epstein, L.G.; Greene, E.P.; Illes, J.; Kirschen, M.; Larriviere, D.G.; et al. Decisions With Patients and Families Regarding Aducanumab in Alzheimer Disease, With Recommendations for Consent: AAN Position Statement. Neurology 2022, 98, 154–159. [Google Scholar] [CrossRef]
- Decourt, B.; Boumelhem, F.; Pope, E.D.; Shi, J.; Mari, Z.; Sabbagh, M.N. Critical Appraisal of Amyloid Lowering Agents in AD. Curr. Neurol. Neurosci. Rep. 2021, 21, 39. [Google Scholar] [CrossRef] [PubMed]
- Sveikata, L.; Charidimou, A.; Viswanathan, A. Vessels Sing Their ARIAs: The Role of Vascular Amyloid in the Age of Aducanumab. Stroke 2022, 53, 298–302. [Google Scholar] [CrossRef] [PubMed]
- Walker, K.A.; Chawla, S.; Nogueras-Ortiz, C.; Coresh, J.; Sharrett, A.R.; Wong, D.F.; Jack, C.R.J.; Spychalla, A.J.; Gottesman, R.F.; Kapogiannis, D. Neuronal insulin signaling and brain structure in nondemented older adults: The Atherosclerosis Risk in Communities Study. Neurobiol. Aging 2021, 97, 65–72. [Google Scholar] [CrossRef] [PubMed]
- Eli Lilly and Company. Lilly’s Donanemab Significantly Slowed Cognitive and Functional Decline in Phase 3 Study of Early Alzheimer’s Disease. Available online: https://investor.lilly.com/news-releases/news-release-details/lillys-donanemab-significantly-slowed-cognitive-and-functional (accessed on 28 January 2025).
- Highlights of Prescribing Information. Available online: https://us.eisai.com/-/media/files/useisai/792710051-2021-08-11 (accessed on 28 January 2025).
- Charidimou, A.; Boulouis, G.; Frosch, M.P.; Baron, J.-C.; Pasi, M.; Albucher, J.F.; Banerjee, G.; Barbato, C.; Bonneville, F.; Brandner, S.; et al. The Boston criteria version 2.0 for cerebral amyloid angiopathy: A multicentre, retrospective, MRI-neuropathology diagnostic accuracy study. Lancet Neurol. 2022, 21, 714–725. [Google Scholar] [CrossRef] [PubMed]
- Knudsen, K.A.; Rosand, J.; Karluk, D.; Greenberg, S.M. Clinical diagnosis of cerebral amyloid angiopathy: Validation of the Boston criteria. Neurology 2001, 56, 537–539. [Google Scholar] [CrossRef]
- Verbeek, M.M.; Kremer, B.P.H.; Rikkert, M.O.; Van Domburg, P.H.M.F.; Skehan, M.E.; Greenberg, S.M. Cerebrospinal fluid amyloid beta(40) is decreased in cerebral amyloid angiopathy. Ann. Neurol. 2009, 66, 245–249. [Google Scholar] [CrossRef]
- Agarwal, A.; Gupta, V.; Brahmbhatt, P.; Desai, A.; Vibhute, P.; Joseph-Mathurin, N.; Bathla, G. Amyloid-related Imaging Abnormalities in Alzheimer Disease Treated with Anti-Amyloid-β Therapy. RadioGraphics 2023, 43, e230009. [Google Scholar] [CrossRef]
- Greenberg, S.M.; Charidimou, A. Diagnosis of Cerebral Amyloid Angiopathy: Evolution of the Boston Criteria. Stroke 2018, 49, 491–497. [Google Scholar] [CrossRef]
- De Kort, A.M.; Kuiperij, H.B.; Marques, T.M.; Jäkel, L.; van den Berg, E.; Kersten, I.; van Berckel-Smit, H.E.P.; Duering, M.; Stoops, E.; Abdo, W.F.; et al. Decreased Cerebrospinal Fluid Amyloid β 38, 40, 42, and 43 Levels in Sporadic and Hereditary Cerebral Amyloid Angiopathy. Ann. Neurol. 2023, 93, 1173–1186. [Google Scholar] [CrossRef]
- Banerjee, G.; Carare, R.; Cordonnier, C.; Greenberg, S.M.; Schneider, J.A.; Smith, E.E.; van Buchem, M.; van der Grond, J.; Verbeek, M.M.; Werring, D.J. The increasing impact of cerebral amyloid angiopathy: Essential new insights for clinical practice. J. Neurol. Neurosurg. Psychiatry 2017, 88, 982–994. [Google Scholar] [CrossRef]
- Zicha, S.; Bateman, R.J.; Shaw, L.M.; Zetterberg, H.; Bannon, A.W.; Horton, W.A.; Baratta, M.; Kolb, H.C.; Dobler, I.; Mordashova, Y.; et al. Comparative analytical performance of multiple plasma Aβ42 and Aβ40 assays and their ability to predict positron emission tomography amyloid positivity. Alzheimers Dement. 2023, 19, 956–966. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Xu, F.; Hoos, M.D.; Lee, H.; Benveniste, H.; Nostrand, W.E.V. Reduced Levels of Cerebrospinal Fluid/Plasma Aβ40 as an Early Biomarker for Cerebral Amyloid Angiopathy in RTg-DI Rats. Int. J. Mol. Sci. 2020, 21, 303. [Google Scholar] [CrossRef] [PubMed]
- Jefferies, W.A.; Price, K.A.; Biron, K.E.; Fenninger, F.; Pfeifer, C.G.; Dickstein, D.L. Adjusting the compass: New insights into the role of angiogenesis in Alzheimer’s disease. Alzheimers Res. Ther. 2013, 5, 64. [Google Scholar] [CrossRef] [PubMed]
- Sheikh, A.M.; Yano, S.; Tabassum, S.; Mitaki, S.; Michikawa, M.; Nagai, A. Alzheimer’s Amyloid β Peptide Induces Angiogenesis in an Alzheimer’s Disease Model Mouse through Placental Growth Factor and Angiopoietin 2 Expressions. Int. J. Mol. Sci. 2023, 24, 4510. [Google Scholar] [CrossRef]
- Lai, A.Y.; McLaurin, J. Clearance of amyloid-β peptides by microglia and macrophages: The issue of what, when and where. Future Neurol. 2012, 7, 165–176. [Google Scholar] [CrossRef]
- Gu, L.; Guo, Z. Alzheimer’s Aβ42 and Aβ40 peptides form interlaced amyloid fibrils. J. Neurochem. 2013, 126, 305–311. [Google Scholar] [CrossRef]
- Zhang, Y.W.; Xu, H. Molecular and cellular mechanisms for Alzheimer’s disease: Understanding APP metabolism. Curr. Mol. Med. 2007, 7, 687–696. [Google Scholar] [CrossRef]
- Han, B.H.; Zhou, M.L.; Vellimana, A.K.; Milner, E.; Kim, D.H.; Greenberg, J.K.; Chu, W.; Mach, R.H.; Zipfel, G.J. Resorufin analogs preferentially bind cerebrovascular amyloid: Potential use as imaging ligands for cerebral amyloid angiopathy. Mol. Neurodegener. 2011, 6, 86. [Google Scholar] [CrossRef]
- Qiu, T.; Liu, Q.; Chen, Y.-X.; Zhao, Y.-F.; Li, Y.-M. Aβ42 and Aβ40: Similarities and differences. J. Pept. Sci. 2015, 21, 522–529. [Google Scholar] [CrossRef]
- Gravina, S.A.; Ho, L.; Eckman, C.B.; Long, K.E.; Otvos, L.; Younkin, L.H.; Suzuki, N.; Younkin, S.G. Amyloid beta protein (A beta) in Alzheimer’s disease brain. Biochemical and immunocytochemical analysis with antibodies specific for forms ending at A beta 40 or A beta 42(43). J. Biol. Chem. 1995, 270, 7013–7016. [Google Scholar] [CrossRef]
- Kakuda, N.; Miyasaka, T.; Iwasaki, N.; Nirasawa, T.; Wada-Kakuda, S.; Takahashi-Fujigasaki, J.; Murayama, S.; Ihara, Y.; Ikegawa, M. Distinct deposition of amyloid-β species in brains with Alzheimer’s disease pathology visualized with MALDI imaging mass spectrometry. Acta Neuropathol. Commun. 2017, 5, 73. [Google Scholar] [CrossRef] [PubMed]
- Miller, D.L.; Papayannopoulos, I.A.; Styles, J.; Bobin, S.A.; Lin, Y.Y.; Biemann, K.; Iqbal, K. Peptide compositions of the cerebrovascular and senile plaque core amyloid deposits of Alzheimer’s disease. Arch. Biochem. Biophys. 1993, 301, 41–52. [Google Scholar] [CrossRef] [PubMed]
- DeSimone, C.V.; Graff-Radford, J.; El-Harasis, M.A.; Rabinstein, A.A.; Asirvatham, S.J.; Holmes, D.R. Cerebral Amyloid Angiopathy: Diagnosis, Clinical Implications, and Management Strategies in Atrial Fibrillation. J. Am. Coll. Cardiol. 2017, 70, 1173–1182. [Google Scholar] [CrossRef] [PubMed]
- Wilcock, D.M.; Gordon, M.N.; Morgan, D. Quantification of cerebral amyloid angiopathy and parenchymal amyloid plaques with Congo red histochemical stain. Nat. Protoc. 2006, 1, 1591–1595. [Google Scholar] [CrossRef]
- Alban, S.L.; Lynch, K.M.; Ringman, J.M.; Toga, A.W.; Chui, H.C.; Sepehrband, F.; Choupan, J. Alzheimer’s Disease Neuroimaging Initiative The association between white matter hyperintensities and amyloid and tau deposition. NeuroImage Clin. 2023, 38, 103383. [Google Scholar] [CrossRef]
- Graff-Radford, J.; Arenaza-Urquijo, E.M.; Knopman, D.S.; Schwarz, C.G.; Brown, R.D.; Rabinstein, A.A.; Gunter, J.L.; Senjem, M.L.; Przybelski, S.A.; Lesnick, T.; et al. White matter hyperintensities: Relationship to amyloid and tau burden. Brain J. Neurol. 2019, 142, 2483–2491. [Google Scholar] [CrossRef]
- Smith, E.E.; Gurol, M.E.; Eng, J.A.; Engel, C.R.; Nguyen, T.N.; Rosand, J.; Greenberg, S.M. White matter lesions, cognition, and recurrent hemorrhage in lobar intracerebral hemorrhage. Neurology 2004, 63, 1606–1612. [Google Scholar] [CrossRef]
- Thanprasertsuk, S.; Martinez-Ramirez, S.; Pontes-Neto, O.M.; Ni, J.; Ayres, A.; Reed, A.; Swords, K.; Gurol, M.E.; Greenberg, S.M.; Viswanathan, A. Posterior white matter disease distribution as a predictor of amyloid angiopathy. Neurology 2014, 83, 794–800. [Google Scholar] [CrossRef]
- Walsh, P.; Sudre, C.H.; Fiford, C.M.; Ryan, N.S.; Lashley, T.; Frost, C.; Barnes, J. ADNI Investigators CSF amyloid is a consistent predictor of white matter hyperintensities across the disease course from aging to Alzheimer’s disease. Neurobiol. Aging 2020, 91, 5–14. [Google Scholar] [CrossRef]
- Antolini, L.; DiFrancesco, J.C.; Zedde, M.; Basso, G.; Arighi, A.; Shima, A.; Cagnin, A.; Caulo, M.; Carare, R.O.; Charidimou, A.; et al. Spontaneous ARIA-like Events in Cerebral Amyloid Angiopathy-Related Inflammation: A Multicenter Prospective Longitudinal Cohort Study. Neurology 2021, 97, e1809–e1822. [Google Scholar] [CrossRef]
- Grangeon, L.; Paquet, C.; Guey, S.; Zarea, A.; Martinaud, O.; Rotharmel, M.; Maltête, D.; Quillard-Muraine, M.; Nicolas, G.; Charbonnier, C.; et al. Cerebrospinal Fluid Profile of Tau, Phosphorylated Tau, Aβ42, and Aβ40 in Probable Cerebral Amyloid Angiopathy. J. Alzheimers Dis. 2022, 87, 791–802. [Google Scholar] [CrossRef] [PubMed]
- Margraf, N.G.; Jensen-Kondering, U.; Weiler, C.; Leypoldt, F.; Maetzler, W.; Philippen, S.; Bartsch, T.; Flüh, C.; Röcken, C.; Möller, B.; et al. Cerebrospinal Fluid Biomarkers in Cerebral Amyloid Angiopathy: New Data and Quantitative Meta-Analysis. Front. Aging Neurosci. 2022, 14, 783996. [Google Scholar] [CrossRef] [PubMed]
- Sembill, J.A.; Lusse, C.; Linnerbauer, M.; Sprügel, M.I.; Mrochen, A.; Knott, M.; Engelhorn, T.; Schmidt, M.A.; Doerfler, A.; Oberstein, T.J.; et al. Cerebrospinal fluid biomarkers for cerebral amyloid angiopathy. Brain Commun. 2023, 5, fcad159. [Google Scholar] [CrossRef] [PubMed]
- Hampel, H.; Elhage, A.; Cho, M.; Nicoll, J.A.; Atri, A. Amyloid-related imaging abnormalities (ARIA) and their radiological, biological and clinical characteristics: A plain language summary. Neurodegener. Dis. Manag. 2024, 14, 51–62. [Google Scholar] [CrossRef]
- Ashton, N.J.; Brum, W.S.; Di Molfetta, G.; Benedet, A.L.; Arslan, B.; Jonaitis, E.; Langhough, R.E.; Cody, K.; Wilson, R.; Carlsson, C.M.; et al. Diagnostic Accuracy of a Plasma Phosphorylated Tau 217 Immunoassay for Alzheimer Disease Pathology. JAMA Neurol. 2024, 81, 255–263. [Google Scholar] [CrossRef]
- Gonzalez-Ortiz, F.; Ferreira, P.C.L.; González-Escalante, A.; Montoliu-Gaya, L.; Ortiz-Romero, P.; Kac, P.R.; Turton, M.; Kvartsberg, H.; Ashton, N.J.; Zetterberg, H.; et al. A novel ultrasensitive assay for plasma p-tau217: Performance in individuals with subjective cognitive decline and early Alzheimer’s disease. Alzheimers Dement. 2024, 20, 1239–1249. [Google Scholar] [CrossRef]
- FDA. FDA Authorizes Marketing of First Blood Test to Aid in the Evaluation of Concussion in Adults. Available online: https://www.fda.gov/news-events/press-announcements/fda-authorizes-marketing-first-blood-test-aid-evaluation-concussion-adults (accessed on 28 January 2025).
- Abdelhak, A.; Foschi, M.; Abu-Rumeileh, S.; Yue, J.K.; D’Anna, L.; Huss, A.; Oeckl, P.; Ludolph, A.C.; Kuhle, J.; Petzold, A.; et al. Blood GFAP as an emerging biomarker in brain and spinal cord disorders. Nat. Rev. Neurol. 2022, 18, 158–172. [Google Scholar] [CrossRef]
- Beyer, L.; Stocker, H.; Rujescu, D.; Holleczek, B.; Stockmann, J.; Nabers, A.; Brenner, H.; Gerwert, K. Amyloid-beta misfolding and GFAP predict risk of clinical Alzheimer’s disease diagnosis within 17 years. Alzheimers Dement. 2023, 19, 1020–1028. [Google Scholar] [CrossRef]
- Oeckl, P.; Anderl-Straub, S.; Von Arnim, C.A.F.; Baldeiras, I.; Diehl-Schmid, J.; Grimmer, T.; Halbgebauer, S.; Kort, A.M.; Lima, M.; Marques, T.M.; et al. Serum GFAP differentiates Alzheimer’s disease from frontotemporal dementia and predicts MCI-to-dementia conversion. J. Neurol. Neurosurg. Psychiatry 2022, 93, 659–667. [Google Scholar] [CrossRef]
- Kim, K.Y.; Shin, K.Y.; Chang, K.-A. GFAP as a Potential Biomarker for Alzheimer’s Disease: A Systematic Review and Meta-Analysis. Cells 2023, 12, 1309. [Google Scholar] [CrossRef]
- Baiardi, S.; Quadalti, C.; Mammana, A.; Dellavalle, S.; Zenesini, C.; Sambati, L.; Pantieri, R.; Polischi, B.; Romano, L.; Suffritti, M.; et al. Diagnostic value of plasma p-tau181, NfL, and GFAP in a clinical setting cohort of prevalent neurodegenerative dementias. Alzheimers Res. Ther. 2022, 14, 153. [Google Scholar] [CrossRef] [PubMed]
- Benedet, A.L.; Milà-Alomà, M.; Vrillon, A.; Ashton, N.J.; Pascoal, T.A.; Lussier, F.; Karikari, T.K.; Hourregue, C.; Cognat, E.; Dumurgier, J.; et al. Differences Between Plasma and Cerebrospinal Fluid Glial Fibrillary Acidic Protein Levels Across the Alzheimer Disease Continuum. JAMA Neurol. 2021, 78, 1471–1483. [Google Scholar] [CrossRef] [PubMed]
- Rasing, I.; Voigt, S.; Koemans, E.A.; de Kort, A.M.; van Harten, T.W.; van Etten, E.S.; van Zwet, E.W.; Stoops, E.; Francois, C.; Kuiperij, H.B.; et al. Serum and cerebrospinal fluid neurofilament light chain and glial fibrillary acid protein levels in early and advanced stages of cerebral amyloid Angiopathy. Alzheimers Res. Ther. 2024, 16, 86. [Google Scholar] [CrossRef] [PubMed]
- Coppens, S.; Lehmann, S.; Hopley, C.; Hirtz, C. Neurofilament-Light, a Promising Biomarker: Analytical, Metrological and Clinical Challenges. Int. J. Mol. Sci. 2023, 24, 11624. [Google Scholar] [CrossRef]
- Mielke, M.M.; Syrjanen, J.A.; Blennow, K.; Zetterberg, H.; Vemuri, P.; Skoog, I.; Machulda, M.M.; Kremers, W.K.; Knopman, D.S.; Jack, C.; et al. Plasma and CSF neurofilament light: Relation to longitudinal neuroimaging and cognitive measures. Neurology 2019, 93, e252–e260. [Google Scholar] [CrossRef]
- Wang, X.; Shi, Z.; Qiu, Y.; Sun, D.; Zhou, H. Peripheral GFAP and NfL as early biomarkers for dementia: Longitudinal insights from the UK Biobank. BMC Med. 2024, 22, 192. [Google Scholar] [CrossRef]
- TauRx. Neurofilament Light Chain (NfL). Available online: https://taurx.com/medical-professionals/neurofilament-light-chain-nfl (accessed on 28 January 2025).
- Lund, S.A.; Giachelli, C.M.; Scatena, M. The role of osteopontin in inflammatory processes. J. Cell Commun. Signal. 2009, 3, 311–322. [Google Scholar] [CrossRef]
- Jakovac, H.; Grubić Kezele, T.; Šućurović, S.; Mulac-Jeričević, B.; Radošević-Stašić, B. Osteopontin-metallothionein I/II interactions in experimental autoimmune encephalomyelitis. Neuroscience 2017, 350, 133–145. [Google Scholar] [CrossRef]
- Ghare, S.; Gardener, H.; Ariko, T.; Gutierrez, J.; Wright, C.B.; Goldberg, R.B.; Elkind, M.S.V.; Cooper, G.E.; Shields, C.B.; Barve, S.; et al. Osteopontin Is Associated with Dementia in the Presence of Cerebral Small Vessel Disease. Cerebrovasc. Dis. 2024, 53, 495–500. [Google Scholar] [CrossRef]
- Rosmus, D.-D.; Lange, C.; Ludwig, F.; Ajami, B.; Wieghofer, P. The Role of Osteopontin in Microglia Biology: Current Concepts and Future Perspectives. Biomedicines 2022, 10, 840. [Google Scholar] [CrossRef]
- Lopes, K.D.P.; Yu, L.; Shen, X.; Qiu, Y.; Tasaki, S.; Iatrou, A.; Beeri, M.S.; Seyfried, N.T.; Menon, V.; Wang, Y.; et al. Associations of cortical SPP1 and ITGAX with cognition and common neuropathologies in older adults. Alzheimers Dement. 2024, 20, 525–537. [Google Scholar] [CrossRef]
- Hainsworth, A.H.; Elahi, F.M.; Corriveau, R.A. An introduction to therapeutic approaches to vascular cognitive impairment. Cereb. Circ.-Cogn. Behav. 2021, 2, 100033. [Google Scholar] [CrossRef] [PubMed]
- Harris, E. Blood Biomarker Helps Distinguish Vascular Dementia. JAMA 2023, 329, 969. [Google Scholar] [CrossRef] [PubMed]
- Newell, L.F.; Holtan, S.G. Placental growth factor: What hematologists need to know. Blood Rev. 2017, 31, 57–62. [Google Scholar] [CrossRef] [PubMed]
- Hinman, J.D.; Elahi, F.; Chong, D.; Radabaugh, H.; Ferguson, A.; Maillard, P.; Thompson, J.F.; Rosenberg, G.A.; Sagare, A.; Moghekar, A.; et al. Placental growth factor as a sensitive biomarker for vascular cognitive impairment. Alzheimers Dement. 2023, 19, 3519–3527. [Google Scholar] [CrossRef]
- Wu, L.-Y.; Chong, J.R.; Chong, J.P.C.; Hilal, S.; Venketasubramanian, N.; Tan, B.Y.; Richards, A.M.; Chen, C.P.; Lai, M.K.P. Serum Placental Growth Factor as a Marker of Cerebrovascular Disease Burden in Alzheimer’s Disease. J. Alzheimers Dis. 2024, 97, 1289–1298. [Google Scholar] [CrossRef]
- Mammana, S.; Fagone, P.; Cavalli, E.; Basile, M.S.; Petralia, M.C.; Nicoletti, F.; Bramanti, P.; Mazzon, E. The Role of Macrophages in Neuroinflammatory and Neurodegenerative Pathways of Alzheimer’s Disease, Amyotrophic Lateral Sclerosis, and Multiple Sclerosis: Pathogenetic Cellular Effectors and Potential Therapeutic Targets. Int. J. Mol. Sci. 2018, 19, 831. [Google Scholar] [CrossRef]
- Frautschy, S.A.; Yang, F.; Irrizarry, M.; Hyman, B.; Saido, T.C.; Hsiao, K.; Cole, G.M. Microglial response to amyloid plaques in APPsw transgenic mice. Am. J. Pathol. 1998, 152, 307–317. [Google Scholar]
- Prinz, M.; Masuda, T.; Wheeler, M.A.; Quintana, F.J. Microglia and Central Nervous System-Associated Macrophages-From Origin to Disease Modulation. Annu. Rev. Immunol. 2021, 39, 251–277. [Google Scholar] [CrossRef]
- Hickman, S.; Izzy, S.; Sen, P.; Morsett, L.; El Khoury, J. Microglia in neurodegeneration. Nat. Neurosci. 2018, 21, 1359–1369. [Google Scholar] [CrossRef]
- Feng, W.; Zhang, Y.; Wang, Z.; Xu, H.; Wu, T.; Marshall, C.; Gao, J.; Xiao, M. Microglia prevent beta-amyloid plaque formation in the early stage of an Alzheimer’s disease mouse model with suppression of glymphatic clearance. Alzheimers Res. Ther. 2020, 12, 125. [Google Scholar] [CrossRef]
- Nabizadeh, F.; Seyedmirzaei, H.; Karami, S. Neuroimaging biomarkers and CSF sTREM2 levels in Alzheimer’s disease: A longitudinal study. Sci. Rep. 2024, 14, 15318. [Google Scholar] [CrossRef] [PubMed]
- Cogswell, P.M.; Barakos, J.A.; Barkhof, F.; Benzinger, T.S.; Jack, C.R.; Poussaint, T.Y.; Raji, C.A.; Ramanan, V.K.; Whitlow, C.T. Amyloid-Related Imaging Abnormalities with Emerging Alzheimer Disease Therapeutics: Detection and Reporting Recommendations for Clinical Practice. AJNR Am. J. Neuroradiol. 2022, 43, E19–E35. [Google Scholar] [CrossRef] [PubMed]
- Hampel, H.; Elhage, A.; Cho, M.; Apostolova, L.G.; Nicoll, J.A.R.; Atri, A. Amyloid-related imaging abnormalities (ARIA): Radiological, biological and clinical characteristics. Brain J. Neurol. 2023, 146, 4414–4424. [Google Scholar] [CrossRef] [PubMed]
- Šimončičová, E.; Gonçalves de Andrade, E.; Vecchiarelli, H.A.; Awogbindin, I.O.; Delage, C.I.; Tremblay, M.-È. Present and future of microglial pharmacology. Trends Pharmacol. Sci. 2022, 43, 669–685. [Google Scholar] [CrossRef]
- d’Errico, P.; Ziegler-Waldkirch, S.; Aires, V.; Hoffmann, P.; Mezö, C.; Erny, D.; Monasor, L.S.; Liebscher, S.; Ravi, V.M.; Joseph, K.; et al. Microglia contribute to the propagation of Aβ Into unaffected brain tissue. Nat. Neurosci. 2022, 25, 20–25. [Google Scholar] [CrossRef]
- Zgorzynska, E. TREM2 in Alzheimer’s disease: Structure, function, therapeutic prospects, and activation challenges. Mol. Cell. Neurosci. 2024, 128, 103917. [Google Scholar] [CrossRef]
- Bonomi, C.G.; Assogna, M.; Di Donna, M.G.; Bernocchi, F.; De Lucia, V.; Nuccetelli, M.; Fiorelli, D.; Loizzo, S.; Mercuri, N.B.; Koch, G.; et al. Cerebrospinal Fluid sTREM-2, GFAP, and β-S100 in Symptomatic Sporadic Alzheimer’s Disease: Microglial, Astrocytic, and APOE Contributions Along the Alzheimer’s Disease Continuum. J. Alzheimers Dis. 2023, 92, 1385–1397. [Google Scholar] [CrossRef]
- Park, S.-H.; Lee, E.-H.; Kim, H.-J.; Jo, S.; Lee, S.; Seo, S.W.; Park, H.-H.; Koh, S.-H.; Lee, J.-H. The relationship of soluble TREM2 to other biomarkers of sporadic Alzheimer’s disease. Sci. Rep. 2021, 11, 13050. [Google Scholar] [CrossRef]
- Yaghmoor, F.; Noorsaeed, A.; Alsaggaf, S.; Aljohani, W.; Scholtzova, H.; Boutajangout, A.; Wisniewski, T. The Role of TREM2 in Alzheimer’s Disease and Other Neurological Disorders. J. Alzheimers Dis. Park. 2014, 4, 160. [Google Scholar] [CrossRef]
- Zhong, R.; Xu, Y.; Williams, J.W.; Li, L. Loss of TREM2 diminishes CAA despite an overall increase of amyloid load in Tg-SwDI mice. Alzheimers Dement. 2024, 20, 7595–7612. [Google Scholar] [CrossRef]
- ALZFORUM. Is ARIA an Inflammatory Reaction to Vascular Amyloid? Available online: https://www.alzforum.org/news/conference-coverage/aria-inflammatory-reaction-vascular-amyloid (accessed on 28 January 2025).
- Chen, Y.; Lu, P.; Wu, S.; Yang, J.; Liu, W.; Alzheimer’s Disease Neuroimaging Initiative; Zhang, Z.; Xu, Q. CD163-Mediated Small-Vessel Injury in Alzheimer’s Disease: An Exploration from Neuroimaging to Transcriptomics. Int. J. Mol. Sci. 2024, 25, 2293. [Google Scholar] [CrossRef]
- Hawkes, C.A.; McLaurin, J. Selective targeting of perivascular macrophages for clearance of beta-amyloid in cerebral amyloid angiopathy. Proc. Natl. Acad. Sci. USA 2009, 106, 1261–1266. [Google Scholar] [CrossRef] [PubMed]
- Murray, P.J.; Wynn, T.A. Protective and pathogenic functions of macrophage subsets. Nat. Rev. Immunol. 2011, 11, 723–737. [Google Scholar] [CrossRef] [PubMed]
- Taylor, X.; Cisternas, P.; You, Y.; You, Y.; Xiang, S.; Marambio, Y.; Zhang, J.; Vidal, R.; Lasagna-Reeves, C.A. A1 reactive astrocytes and a loss of TREM2 are associated with an early stage of pathology in a mouse model of cerebral amyloid angiopathy. J. Neuroinflamm. 2020, 17, 223–227. [Google Scholar] [CrossRef] [PubMed]
- Grand Moursel, L.; van der Graaf, L.M.; Bulk, M.; van Roon-Mom, W.M.C.; van der Weerd, L. Osteopontin and phospho-SMAD2/3 are associated with calcification of vessels in D-CAA, an hereditary cerebral amyloid angiopathy. Brain Pathol. 2019, 29, 793–802. [Google Scholar] [CrossRef]
- Handa, T.; Sasaki, H.; Takao, M.; Tano, M.; Uchida, Y. Proteomics-based investigation of cerebrovascular molecular mechanisms in cerebral amyloid angiopathy by the FFPE-LMD-PCT-SWATH method. Fluids Barriers CNS 2022, 19, 56. [Google Scholar] [CrossRef]
- Hondius, D.C.; Eigenhuis, K.N.; Morrema, T.H.J.; van der Schors, R.C.; van Nierop, P.; Bugiani, M.; Li, K.W.; Hoozemans, J.J.M.; Smit, A.B.; Rozemuller, A.J.M. Proteomics analysis identifies new markers associated with capillary cerebral amyloid angiopathy in Alzheimer’s disease. Acta Neuropathol. Commun. 2018, 6, 46. [Google Scholar] [CrossRef]
- Vervuurt, M.; Schrader, J.M.; de Kort, A.M.; Kersten, I.; Wessels, H.J.C.T.; Klijn, C.J.M.; Schreuder, F.H.B.M.; Kuiperij, H.B.; Gloerich, J.; Van Nostrand, W.E.; et al. Cerebrospinal fluid shotgun proteomics identifies distinct proteomic patterns in cerebral amyloid angiopathy rodent models and human patients. Acta Neuropathol. Commun. 2024, 12, 6. [Google Scholar] [CrossRef]
- Westwood, S.; Baird, A.L.; Anand, S.N.; Nevado-Holgado, A.J.; Kormilitzin, A.; Shi, L.; Hye, A.; Ashton, N.J.; Morgan, A.R.; Bos, I.; et al. Validation of Plasma Proteomic Biomarkers Relating to Brain Amyloid Burden in the EMIF-Alzheimer’s Disease Multimodal Biomarker Discovery Cohort. J. Alzheimers Dis. 2020, 74, 213–225. [Google Scholar] [CrossRef]
- Li, Y.-B.; Fu, Q.; Guo, M.; Du, Y.; Chen, Y.; Cheng, Y. MicroRNAs: Pioneering regulators in Alzheimer’s disease pathogenesis, diagnosis, and therapy. Transl. Psychiatry 2024, 14, 367. [Google Scholar] [CrossRef]
- Weldon Furr, J.; Morales-Scheihing, D.; Manwani, B.; Lee, J.; McCullough, L.D. Cerebral Amyloid Angiopathy, Alzheimer’s Disease and MicroRNA: MiRNA as Diagnostic Biomarkers and Potential Therapeutic Targets. NeuroMol. Med. 2019, 21, 369–390. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Rapp, J.; Rainone, S.; Goupil, C.; Dorval, V.; Smith, P.Y.; Saint-Pierre, M.; Vallée, M.; Planel, E.; Droit, A.; Calon, F.; et al. microRNA-132/212 deficiency enhances Aβ production and senile plaque deposition in Alzheimer’s disease triple transgenic mice. Sci. Rep. 2016, 6, 30953. [Google Scholar] [CrossRef] [PubMed]
- Hong, H.; Li, Y.; Su, B. Identification of Circulating miR-125b as a Potential Biomarker of Alzheimer’s Disease in APP/PS1 Transgenic Mouse. J. Alzheimers Dis. 2017, 59, 1449–1458. [Google Scholar] [CrossRef]
- Cheng, L.; Doecke, J.D.; Sharples, R.A.; Villemagne, V.L.; Fowler, C.J.; Rembach, A.; Martins, R.N.; Rowe, C.C.; Macaulay, S.L.; Masters, C.L.; et al. Prognostic serum miRNA biomarkers associated with Alzheimer’s disease shows concordance with neuropsychological and neuroimaging assessment. Mol. Psychiatry 2015, 20, 1188–1196. [Google Scholar] [CrossRef]
- Huang, X.; Yuan, T.; Tschannen, M.; Sun, Z.; Jacob, H.; Du, M.; Liang, M.; Dittmar, R.L.; Liu, Y.; Liang, M.; et al. Characterization of human plasma-derived exosomal RNAs by deep sequencing. BMC Genom. 2013, 14, 319. [Google Scholar] [CrossRef]
- Soreq, H.; Wolf, Y. NeurimmiRs: microRNAs in the neuroimmune interface. Trends Mol. Med. 2011, 17, 548–555. [Google Scholar] [CrossRef]
- Walgrave, H.; Zhou, L.; De Strooper, B.; Salta, E. The promise of microRNA-based therapies in Alzheimer’s disease: Challenges and perspectives. Mol. Neurodegener. 2021, 16, 76. [Google Scholar] [CrossRef]
- Abidin, S.Z.; Mat Pauzi, N.A.; Mansor, N.I.; Mohd Isa, N.I.; Hamid, A.A. A new perspective on Alzheimer’s disease: microRNAs and circular RNAs. Front. Genet. 2023, 14, 1231486. [Google Scholar] [CrossRef]
- Hospitalization–Health, United States. Available online: https://www.cdc.gov/nchs/hus/topics/hospitalization.htm (accessed on 28 January 2025).
- 2023 National Population Projections Tables: Main Series. Available online: https://www.census.gov/data/tables/2023/demo/popproj/2023-summary-tables.html (accessed on 28 January 2025).
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).