Glucocorticoid Receptor Modulates EGFR Feedback upon Acquisition of Resistance to Monoclonal Antibodies

 , , and
, , and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Material and Methods
2.1. Cell Culture
2.2. Colony Forming Assay
2.3. RNA Isolation and qPCR
2.4. Clustering and Heat-Map
2.5. Patients Data
2.6. Statistical Analysis
3. Results
3.1. Generation and Validation of Caco-2 Cetuximab-Resistant Cells
3.2. EGFR Positive Feedback Loops are Increased in Cetuximab Resistant Cells
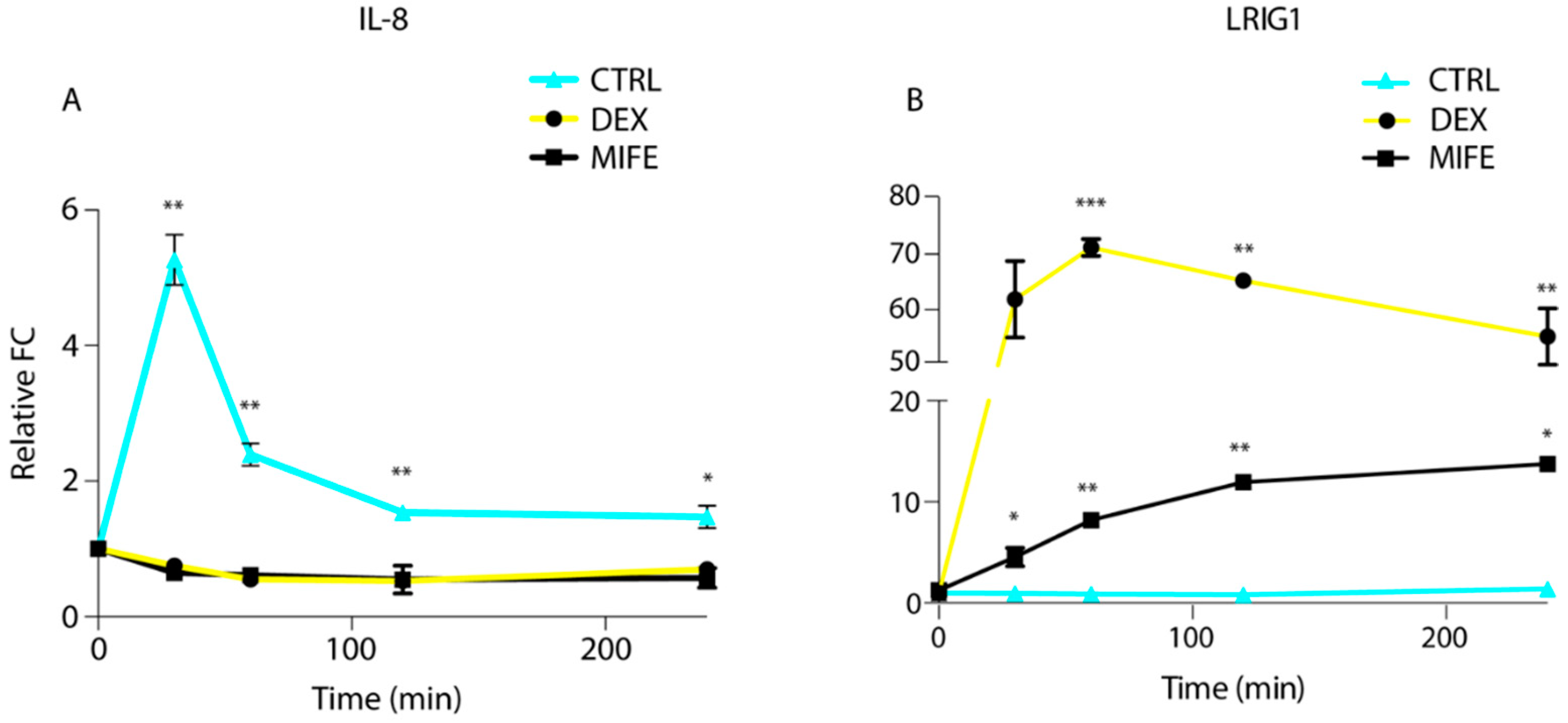
3.3. Glucocorticoids Administration Downregulates EGFR Positive Feedback Loops and Boosts EGFR Negative Feedback Loops in Cetuximab-Resistant Cells
3.4. GR Modulation Re-Activates EGFR Negative Feedback Loops in Cetuximab-Resistant Cells
3.5. LRIG1 Expression Predicts Resistance to Cetuximab in Colorectal Cancer Patients
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- D’Uva, G.; Lauriola, M. Towards the emerging crosstalk: ERBB family and steroid hormones. Semin. Cell Dev. Biol. 2016. [Google Scholar] [CrossRef] [PubMed]
- Troyer, K.L.; Lee, D.C. Regulation of mouse mammary gland development and tumorigenesis by the ERBB signaling network. J. Mammary Gland Biol. Neoplasia 2001. [Google Scholar] [CrossRef]
- Wintermantel, T.M.; Bock, D.; Fleig, V.; Greiner, E.F.; Schütz, G. The epithelial glucocorticoid receptor is required for the normal timing of cell proliferation during mammary lobuloalveolar development but is dispensable for milk production. Mol. Endocrinol. 2005. [Google Scholar] [CrossRef]
- Obradović, M.M.S.; Hamelin, B.; Manevski, N.; Couto, J.P.; Sethi, A.; Coissieux, M.-M.; Münst, S.; Okamoto, R.; Kohler, H.; Schmidt, A.; et al. Glucocorticoids promote breast cancer metastasis. Nature 2019. [Google Scholar] [CrossRef]
- Enuka, Y.; Feldman, M.E.; Chowdhury, A.; Srivastava, S.; Lindzen, M.; Sas-Chen, A.; Massart, R.; Cheishvili, D.; Suderman, M.J.; Zaltsman, Y.; et al. Epigenetic mechanisms underlie the crosstalk between growth factors and a steroid hormone. Nucleic Acids Res. 2017. [Google Scholar] [CrossRef] [PubMed]
- Cai, C.; Portnoy, D.C.; Wang, H.; Jiang, X.; Chen, S.; Balk, S.P. Androgen receptor expression in prostate cancer cells is suppressed by activation of epidermal growth factor receptor and ErbB2. Cancer Res. 2009. [Google Scholar] [CrossRef]
- Pradeep, C.R.; Köstler, W.J.; Lauriola, M.; Granit, R.Z.; Zhang, F.; Jacob-Hirsch, J.; Rechavi, G.; Nair, H.B.; Hennessy, B.T.; Gonzalez-Angulo, A.M.; et al. Modeling ductal carcinoma in situ: A HER2-Notch3 collaboration enables luminal filling. Oncogene 2012. [Google Scholar] [CrossRef]
- Real, P.J.; Tosello, V.; Palomero, T.; Castillo, M.; Hernando, E.; De Stanchina, E.; Sulis, M.L.; Barnes, K.; Sawai, C.; Homminga, I.; et al. γ-secretase inhibitors reverse glucocorticoid resistance in T cell acute lymphoblastic leukemia. Nat. Med. 2009. [Google Scholar] [CrossRef]
- Varricchio, L.; Tirelli, V.; Masselli, E.; Ghinassi, B.; Saha, N.; Besmer, P.; Migliaccio, A.R. The expression of the glucocorticoid receptor in human erythroblasts is uniquely regulated by KIT ligand: Implications for stress erythropoiesis. Stem Cells Dev. 2012. [Google Scholar] [CrossRef]
- Treviño, L.S.; Weigel, N.L. Phosphorylation: A fundamental regulator of steroid receptor action. Trends Endocrinol. Metab. 2013. [Google Scholar] [CrossRef]
- Michalides, R.; Griekspoor, A.; Balkenende, A.; Verwoerd, D.; Janssen, L.; Jalink, K.; Floore, A.; Velds, A.; Van’t Veer, L.; Neefjes, J. Tamoxifen resistance by a conformational arrest of the estrogen receptor α after PKA activation in breast cancer. Cancer Cell 2004. [Google Scholar] [CrossRef] [PubMed]
- Lauriola, M.; Enuka, Y.; Zeisel, A.; D’Uva, G.; Roth, L.; Sharon-Sevilla, M.; Lindzen, M.; Sharma, K.; Nevo, N.; Feldman, M.; et al. Diurnal suppression of EGFR signalling by glucocorticoids and implications for tumour progression and treatment. Nat. Commun. 2014, 5, 5073. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, S.V.; Settleman, J. Oncogene addiction: Setting the stage for molecularly targeted cancer therapy. Genes Dev. 2007, 21, 3214–3231. [Google Scholar] [CrossRef] [PubMed]
- Chong, C.R.; Jänne, P.A. The quest to overcome resistance to EGFR-targeted therapies in cancer. Nat. Med. 2013. [Google Scholar] [CrossRef] [PubMed]
- Bardelli, A.; Siena, S. Molecular mechanisms of resistance to cetuximab and panitumumab in colorectal cancer. J. Clin. Oncol. 2010. [Google Scholar] [CrossRef] [PubMed]
- Misale, S.; Di Nicolantonio, F.; Sartore-Bianchi, A.; Siena, S.; Bardelli, A. Resistance to anti-EGFR therapy in colorectal cancer: From heterogeneity to convergent evolution. Cancer Discov. 2014. [Google Scholar] [CrossRef]
- Trusolino, L.; Bertotti, A. Compensatory pathways in oncogenic kinase signaling and resistance to targeted therapies: Six degrees of separation. Cancer Discov. 2012, 2, 876–880. [Google Scholar] [CrossRef] [PubMed]
- Hrustanovic, G.; Lee, B.J.; Bivona, T.G. Mechanisms of resistance to EGFR targeted therapies. Cancer Biol. Ther. 2013, 14, 304–314. [Google Scholar] [CrossRef] [Green Version]
- Bivona, T.G.; Doebele, R.C. A framework for understanding and targeting residual disease in oncogene-driven solid cancers. Nat. Med. 2016. [Google Scholar] [CrossRef]
- Lee, H.J.; Zhuang, G.; Cao, Y.; Du, P.; Kim, H.J.; Settleman, J. Drug resistance via feedback activation of stat3 in oncogene-addicted cancer cells. Cancer Cell 2014. [Google Scholar] [CrossRef]
- Gelfo, V.; Rodia, M.T.; Pucci, M.; Dall’Ora, M.; Santi, S.; Solmi, R.; Roth, L.; Lindzen, M.; Bonafè, M.; Bertotti, A.; et al. A module of inflammatory cytokines defines resistance of colorectal cancer to EGFR inhibitors. Oncotarget 2016. [Google Scholar] [CrossRef] [PubMed]
- Gelfo, V.; Mazzeschi, M.; Grilli, G.; Lindzen, M.; Santi, S.; D’Uva, G.; Győrffy, B.; Ardizzoni, A.; Yarden, Y.; Lauriola, M. A novel role for the interleukin-1 receptor axis in resistance to anti-EGFR therapy. Cancers (Basel) 2018, 10, 355. [Google Scholar] [CrossRef] [PubMed]
- Troiani, T.; Martinelli, E.; Napolitano, S.; Vitagliano, D.; Ciuffreda, L.P.; Costantino, S.; Morgillo, F.; Capasso, A.; Sforza, V.; Nappi, A.; et al. Increased TGF-α as a mechanism of acquired resistance to the anti-EGFR inhibitor cetuximab through EGFR-MET interaction and activation of MET signaling in colon cancer cells. Clin. Cancer Res. 2013. [Google Scholar] [CrossRef] [PubMed]
- Hobor, S.; Van Emburgh, B.O.; Crowley, E.; Misale, S.; Di Nicolantonio, F.; Bardelli, A. TGFα and amphiregulin paracrine network promotes resistance to EGFR blockade in colorectal cancer cells. Clin. Cancer Res. 2014. [Google Scholar] [CrossRef]
- Khambata-Ford, S.; Garrett, C.R.; Meropol, N.J.; Basik, M.; Harbison, C.T.; Wu, S.; Wong, T.W.; Huang, X.; Takimoto, C.H.; Godwin, A.K.; et al. Expression of epiregulin and amphiregulin and K-ras mutation status predict disease control in metastatic colorectal cancer patients treated with cetuximab. J. Clin. Oncol. 2007. [Google Scholar] [CrossRef] [PubMed]
- Segatto, O.; Anastasi, S.; Alema, S. Regulation of epidermal growth factor receptor signalling by inducible feedback inhibitors. J. Cell Sci. 2011. [Google Scholar] [CrossRef] [PubMed]
- Saldanha, A.J. Java Treeview—Extensible visualization of microarray data. Bioinformatics 2004, 20, 3246–3248. [Google Scholar] [CrossRef]
- Bertotti, A.; Bertotti, A.; Migliardi, G.; Galimi, F.; Sassi, F.; Torti, D.; Isella, C.; Corà, D.; Di Nicolantonio, F.; Buscarino, M.; Petti, C.; et al. A molecularly annotated platform of patient-derived xenografts (“xenopatients”) identifies HER2 as an effective therapeutic target in cetuximab-resistant colorectal cancer. Cancer Discov. 2011, 1, 508–523. [Google Scholar] [CrossRef]
- Isella, C.; Brundu, F.; Bellomo, S.E.; Galimi, F.; Zanella, E.; Porporato, R.; Petti, C.; Fiori, A.; Orzan, F.; Senetta, R.; et al. Selective analysis of cancer-cell intrinsic transcriptional traits defines novel clinically relevant subtypes of colorectal cancer. Nat. Commun. 2017. [Google Scholar] [CrossRef] [PubMed]
- Teng, F.; Xu, Z.; Chen, J.; Zheng, G.; Zheng, G.; Lv, H.; Wang, Y.; Wang, L.; Cheng, X. DUSP1 induces apatinib resistance by activating the MAPK pathway in gastric cancer. Oncol. Rep. 2018. [Google Scholar] [CrossRef] [PubMed]
- Zeisel, A.; Köstler, W.J.; Molotski, N.; Tsai, J.M.; Krauthgamer, R.; Jacob-Hirsch, J.; Rechavi, G.; Soen, Y.; Jung, S.; Yarden, Y.; et al. Coupled pre-mRNA and mRNA dynamics unveil operational strategies underlying transcriptional responses to stimuli. Mol. Syst. Biol. 2011. [Google Scholar] [CrossRef]
- Xu, D.; Makkinje, A.; Kyriakis, J.M. Gene 33 is an endogenous inhibitor of Epidermal Growth Factor (EGF) receptor signaling and mediates dexamethasone-induced suppression of EGF function. J. Biol. Chem. 2005. [Google Scholar] [CrossRef] [PubMed]
- Auphan, N.; DiDonato, J.A.; Rosette, C.; Helmberg, A.; Karin, M. Immunosuppression by glucocorticoids: Inhibition of NF-κB activity through induction of IκB synthesis. Science 1995. [Google Scholar] [CrossRef]
- Zhang, S.; Jonklaas, J.; Danielsen, M. The glucocorticoid agonist activities of mifepristone (RU486) and progesterone are dependent on glucocorticoid receptor levels but not on EC50 values. Steroids 2007. [Google Scholar] [CrossRef]
- Lemmon, M.A.; Schlessinger, J. Cell signaling by receptor tyrosine kinases. Cell 2010. [Google Scholar] [CrossRef] [PubMed]
- Avraham, R.; Yarden, Y. Feedback regulation of EGFR signalling: Decision making by early and delayed loops. Nat. Rev. Mol. Cell Biol. 2011, 12, 104–117. [Google Scholar] [CrossRef]
- Stelling, J.; Sauer, U.; Szallasi, Z.; Doyle, F.J.; Doyle, J. Robustness of cellular functions. Cell 2004. [Google Scholar] [CrossRef]
- Grieco, L.; Calzone, L.; Bernard-Pierrot, I.; Radvanyi, F.; Kahn-Perlès, B.; Thieffry, D. Integrative modelling of the influence of MAPK network on cancer cell fate decision. PLoS Comput. Biol. 2013, 9. [Google Scholar] [CrossRef]
- Amit, I.; Citri, A.; Shay, T.; Lu, Y.; Katz, M.; Zhang, F.; Tarcic, G.; Siwak, D.; Lahad, J.; Jacob-Hirsch, J.; et al. A module of negative feedback regulators defines growth factor signaling. Nat. Genet. 2007, 39, 503–512. [Google Scholar] [CrossRef]
- Barozzi, C.; Ravaioli, M.; D’Errico, A.; Grazi, G.L.; Poggioli, G.; Cavrini, G.; Mazziotti, A.; Grigioni, W.F. Relevance of biologic markers in colorectal carcinoma: A comparative study of a broad panel. Cancer 2002. [Google Scholar] [CrossRef]
- Prahallad, A.; Sun, C.; Huang, S.; Di Nicolantonio, F.; Salazar, R.; Zecchin, D.; Beijersbergen, R.L.; Bardelli, A.; Bernards, R. Unresponsiveness of colon cancer to BRAF(V600E) inhibition through feedback activation of EGFR. Nature 2012, 483, 100–103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Citri, A.; Yarden, Y. EGF–ERBB signalling: Towards the systems level. Nat. Rev. Mol. Cell Biol. 2006, 7, 505–516. [Google Scholar] [CrossRef] [PubMed]
- Kholodenko, B.N. Cell-signalling dynamics in time and space. Nat. Rev. Mol. Cell Biol. 2006. [Google Scholar] [CrossRef] [PubMed]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gelfo, V.; Pontis, F.; Mazzeschi, M.; Sgarzi, M.; Mazzarini, M.; Solmi, R.; D’Uva, G.; Lauriola, M. Glucocorticoid Receptor Modulates EGFR Feedback upon Acquisition of Resistance to Monoclonal Antibodies. J. Clin. Med. 2019, 8, 600. https://doi.org/10.3390/jcm8050600
Gelfo V, Pontis F, Mazzeschi M, Sgarzi M, Mazzarini M, Solmi R, D’Uva G, Lauriola M. Glucocorticoid Receptor Modulates EGFR Feedback upon Acquisition of Resistance to Monoclonal Antibodies. Journal of Clinical Medicine. 2019; 8(5):600. https://doi.org/10.3390/jcm8050600
Chicago/Turabian StyleGelfo, Valerio, Francesca Pontis, Martina Mazzeschi, Michela Sgarzi, Maria Mazzarini, Rossella Solmi, Gabriele D’Uva, and Mattia Lauriola. 2019. "Glucocorticoid Receptor Modulates EGFR Feedback upon Acquisition of Resistance to Monoclonal Antibodies" Journal of Clinical Medicine 8, no. 5: 600. https://doi.org/10.3390/jcm8050600