Neuroinflammation in Primary Open-Angle Glaucoma

 ,
, {kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Methods
3. Oxidative Stress and Mitochondria Dysfunction
3.1. Oxidative Stress-Related Trabecular Meshwork Damage
3.2. Oxidative Stress and Neural Damage
3.3. AH Composition in POAG Patients
4. The NF-κB Pathway
4.1. Cross-Talk among NF-κB and NRF2
4.2. NF-κB and TM
5. Trabecular Meshwork (TM) and Location of Outflow Resistance
5.1. Local Mediators in Conventional Outflow Pathway
5.2. The Main Changes in POAG TM
5.3. LncRNA-miRNA-mRNA: Role inTM and in POAG
6. Neurodegeneration in POAG
6.1. Axonal Transport Deficit
6.2. Microglial Activation
6.3. Astrocytes Activation
6.4. Glaucoma Neuroinflammation Signaling
6.5. Glutamate Excitotoxicity
7. Concluding Remarks
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AC | Anterior Chamber |
| AH | Aqueous Humor |
| CB | Ciliar Body |
| ECM | Extra Cellular Matrix |
| ER | Endoplasmic Reticulum |
| HSPs | Heat Shock Proteins |
| HTG | High Tension Glaucoma |
| IOP | Introcular Pressure |
| JCT | Juxtacanalicular Connective Tissue |
| LC | Lamina Cribrosa |
| MMPs | Matrix Metalloproteinases |
| mtDNA | Mitochondria DNA |
| NO | Nitric Oxide |
| NTG | Normal Tension Glaucoma |
| ONH | Optic Nerve Head |
| OS | Oxidative Stress |
| POAG | Primary Open-Angle Glaucoma |
| RGCs | Retinal Ganglion Cells |
| ROS | Reactive Oxygen Species |
| SC | Schlemm’s Canal |
| SEM | Schlemm Endothelial Cell |
| Th 1/2 | T-helper 1/2 |
| TLRs | Toll Like Recepeptors |
| TM | Trabecular Meshwork |
| TME | Trabecular Endothelial Cell |
References
- McMonnies, C.W. Glaucoma history and risk factors. J. Optom. 2017, 10, 71–78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, D.; Chen, P.P. Glaucoma. Am. Fam. Physician 2016, 93, 668–674. [Google Scholar] [PubMed]
- Napoli, C.; Ignarro, L.J. Nitric oxide and pathogenic mechanisms involved in the development of vascular diseases. Arch. Pharmacal Res. 2009, 32, 1103–1108. [Google Scholar] [CrossRef]
- Bell, K.; Gramlich, O.W.; Von Thun Und Hohenstein-Blaul, N.; Beck, S.; Funke, S.; Wilding, C.; Pfeiffer, N.; Grus, F.H. Does autoimmunity play a part in the pathogenesis of glaucoma? Prog. Retin. Eye Res. 2013, 36, 199–216. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, S.K.; Lee, R.K.; Grus, F.H. Molecular Biomarkers in Glaucoma. Investig. Ophthalmol. Vis. Sci. 2013, 54, 121. [Google Scholar] [CrossRef] [Green Version]
- Salinas-Navarro, M.; Alarcón-Martínez, L.; Valiente-Soriano, F.J.; Jiménez-López, M.; Mayor-Torroglosa, S.; Avilés-Trigueros, M.; Villegas-Pérez, M.P.; Vidal-Sanz, M. Ocular hypertension impairs optic nerve axonal transport leading to progressive retinal ganglion cell degeneration. Exp. Eye Res. 2010, 90, 168–183. [Google Scholar] [CrossRef]
- Ju, W.-K.; Kim, K.-Y.; Lindsey, J.D.; Angert, M.; Patel, A.; Scott, R.T.; Liu, Q.; Crowston, J.G.; Ellisman, M.H.; Perkins, G.A.; et al. Elevated hydrostatic pressure triggers release of OPA1 and cytochrome C, and induces apoptotic cell death in differentiated RGC-5 cells. Mol. Vis. 2009, 15, 120–134. [Google Scholar]
- Salinas-Navarro, M.; Alarcón-Martínez, L.; Valiente-Soriano, F.J.; Ortín-Martínez, A.; Jiménez-López, M.; Avilés-Trigueros, M.; Villegas-Pérez, M.P.; de la Villa, P.; Vidal-Sanz, M. Functional and morphological effects of laser-induced ocular hypertension in retinas of adult albino Swiss mice. Mol. Vis. 2009, 15, 2578–2598. [Google Scholar]
- Flammer, J.; Mozaffarieh, M. What Is the Present Pathogenetic Concept of Glaucomatous Optic Neuropathy? Surv. Ophthalmol. 2007, 52, S162–S173. [Google Scholar] [CrossRef]
- Grieshaber, M.C.; Mozaffarieh, M.; Flammer, J. What Is the Link Between Vascular Dysregulation and Glaucoma? Surv. Ophthalmol. 2007, 52, S144–S154. [Google Scholar] [CrossRef]
- Grzybowski, A.; Och, M.; Kanclerz, P.; Leffler, C.; De Moraes, C.G. Primary Open Angle Glaucoma and Vascular Risk Factors: A Review of Population Based Studies from 1990 to 2019. J. Clin. Med. 2020, 9, 761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saccà, S.C.; Gandolfi, S.; Bagnis, A.; Manni, G.; Damonte, G.; Traverso, C.E.; Izzotti, A. From DNA damage to functional changes of the trabecular meshwork in aging and glaucoma. Ageing Res. Rev. 2016, 29, 26–41. [Google Scholar] [CrossRef] [PubMed]
- Izzotti, A.; Longobardi, M.; Cartiglia, C.; Saccà, S.C. Proteome Alterations in Primary Open Angle Glaucoma Aqueous Humor. J. Proteome Res. 2010, 9, 4831–4838. [Google Scholar] [CrossRef] [PubMed]
- Saccà, S.C.; Gandolfi, S.; Bagnis, A.; Manni, G.; Damonte, G.; Traverso, C.E.; Izzotti, A. The outflow pathway: A tissue with morphological and functional unity. J. Cell. Physiol. 2016, 231, 1876–1893. [Google Scholar] [CrossRef]
- Drance, S.M.; Sweeney, V.P.; Morgan, R.W.; Feldman, F. Studies of Factors Involved in the Production of Low Tension Glaucoma. Arch. Ophthalmol. 1973, 89, 457–465. [Google Scholar] [CrossRef]
- Tamm, E.R. The trabecular meshwork outflow pathways: Structural and functional aspects. Exp. Eye Res. 2009, 88, 648–655. [Google Scholar] [CrossRef]
- Nga, A.D.; Yap, S.-L.; Samsudin, A.; Abdul-Rahman, P.S.; Hashim, O.H.; Mimiwati, Z. Matrix metalloproteinases and tissue inhibitors of metalloproteinases in the aqueous humour of patients with primary angle closure glaucoma—A quantitative study. BMC Ophthalmol. 2014, 14, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Tezel, G. Oxidative stress in glaucomatous neurodegeneration: Mechanisms and consequences. Prog. Retin. Eye Res. 2006, 25, 490–513. [Google Scholar] [CrossRef] [Green Version]
- Mozaffarieh, M.; Grieshaber, M.C.; Flammer, J. Oxygen and blood flow: Players in the pathogenesis of glaucoma. Mol. Vis. 2008, 14, 224–233. [Google Scholar]
- Acott, T.S.; Kelley, M.J.; Keller, K.E.; Vranka, J.A.; Abu-Hassan, D.W.; Li, X.; Aga, M.; Bradley, J.M. Intraocular Pressure Homeostasis: Maintaining Balance in a High-Pressure Environment. J. Ocul. Pharmacol. Ther. 2014, 30, 94–101. [Google Scholar] [CrossRef]
- Kwon, Y.H.; Fingert, J.H.; Kuehn, M.H.; Alward, W.L.M. Primary Open-Angle Glaucoma. N. Engl. J. Med. 2009, 360, 1113–1124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calkins, D.J. Critical pathogenic events underlying progression of neurodegeneration in glaucoma. Prog. Retin. Eye Res. 2012, 31, 702–719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dreyer, E.B. Elevated Glutamate Levels in the Vitreous Body of Humans and Monkeys with Glaucoma. Arch. Ophthalmol. 1996, 114, 299. [Google Scholar] [CrossRef] [PubMed]
- Izzotti, A.; Saccà, S.C.; Cartiglia, C.; De Flora, S. Oxidative deoxyribonucleic acid damage in the eyes of glaucoma patients. Am. J. Med. 2003, 114, 638–646. [Google Scholar] [CrossRef]
- Saccà, S.C. Oxidative DNA Damage in the Human Trabecular Meshwork: Clinical Correlation in Patients with Primary Open-Angle Glaucoma. Arch. Ophthalmol. 2005, 123, 458. [Google Scholar] [CrossRef] [Green Version]
- Tezel, G.; Yang, X.; Cai, J. Proteomic Identification of Oxidatively Modified Retinal Proteins in a Chronic Pressure-Induced Rat Model of Glaucoma. Investig. Ophthalmol. Vis. Sci. 2005, 46, 3177. [Google Scholar] [CrossRef]
- Junqueira, V.B.C.; Barros, S.B.M.; Chan, S.S.; Rodrigues, L.; Giavarotti, L.; Abud, R.L.; Deucher, G.P. Aging and oxidative stress. Mol. Asp. Med. 2004, 25, 5–16. [Google Scholar] [CrossRef]
- Lin, M.T.; Beal, M.F. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 2006, 443, 787–795. [Google Scholar] [CrossRef]
- Izzotti, A.; Longobardi, M.; Cartiglia, C.; Saccà, S.C. Mitochondrial Damage in the Trabecular Meshwork Occurs Only in Primary Open-Angle Glaucoma and in Pseudoexfoliative Glaucoma. PLoS ONE 2011, 6, e14567. [Google Scholar] [CrossRef] [Green Version]
- Zündorf, G.; Kahlert, S.; Bunik, V.I.; Reiser, G. α-Ketoglutarate dehydrogenase contributes to production of reactive oxygen species in glutamate-stimulated hippocampal neurons in situ. Neuroscience 2009, 158, 610–616. [Google Scholar] [CrossRef]
- Crish, S.D.; Calkins, D.J. Neurodegeneration in glaucoma: Progression and calcium-dependent intracellular mechanisms. Neuroscience 2011, 176, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Batandier, C.; Leverve, X.; Fontaine, E. Opening of the Mitochondrial Permeability Transition Pore Induces Reactive Oxygen Species Production at the Level of the Respiratory Chain Complex I. J. Biol. Chem. 2004, 279, 17197–17204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feissner, R.F.; Skalska, J.; Gaum, W.E.; Sheu, S.-S. Crosstalk signaling between mitochondrial Ca2+ and ROS. Front. Biosci. 2009, 14, 1197–1218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, L.; Sha, X.-Y.; Wu, Y.-N.; Chen, M.-T.; Zhong, J.-X. Lycium barbarum polysaccharides protects retinal ganglion cells against oxidative stress injury. Neural Regen. Res. 2020, 15, 1526. [Google Scholar] [CrossRef]
- Nita, M.; Grzybowski, A. The Role of the Reactive Oxygen Species and Oxidative Stress in the Pathomechanism of the Age-Related Ocular Diseases and Other Pathologies of the Anterior and Posterior Eye Segments in Adults. Oxid. Med. Cell. Longev. 2016. [Google Scholar] [CrossRef] [Green Version]
- Green, K. Free Radicals and Aging of Anterior Segment Tissues of the Eye: A Hypothesis. ORE 1995, 27, 143–149. [Google Scholar] [CrossRef]
- Flammer, J.; Haefliger, I.O.; Orgül, S.; Resink, T. Vascular dysregulation: A principal risk factor for glaucomatous damage? J. Glaucoma 1999, 8, 212–219. [Google Scholar] [CrossRef]
- Yeh, L.-H.; Park, Y.J.; Hansalia, R.J.; Ahmed, I.S.; Deshpande, S.S.; Goldschmidt-Clermont, P.J.; Irani, K.; Alevriadou, B.R. Shear-Induced Tyrosine Phosphorylation in Endothelial Cells Requires Rac1-Dependent Production of ROS. Am. J. Physiol. Cell Physiol. 1999, 276, C838–C847. [Google Scholar] [CrossRef]
- Izzotti, A.; Saccà, S.C.; Longobardi, M.; Cartiglia, C. Sensitivity of Ocular Anterior Chamber Tissues to Oxidative Damage and Its Relevance to the Pathogenesis of Glaucoma. Investig. Ophthalmol. Vis. Sci. 2009, 50, 5251. [Google Scholar] [CrossRef] [Green Version]
- Babizhayev, M.A. Biomarkers and special features of oxidative stress in the anterior segment of the eye linked to lens cataract and the trabecular meshwork injury in primary open-angle glaucoma: Challenges of dual combination therapy with N-acetylcarnosine lubricant eye d: Enhanced oxidative stress in cataract and glaucoma. Fundam. Clin. Pharmacol. 2012, 26, 86–117. [Google Scholar] [CrossRef]
- Liu, T.; Xie, L.; Ye, J.; Liu, Y.; He, X. Screening of candidate genes for primary open angle glaucoma. Mol. Vis. 2012, 18, 2119–2126. [Google Scholar] [PubMed]
- Alvarado, J.A. A new insight into the cellular regulation of aqueous outflow: How trabecular meshwork endothelial cells drive a mechanism that regulates the permeability of Schlemm’s canal endothelial cells. Br. J. Ophthalmol. 2005, 89, 1500–1505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saccà, S.C.; Cutolo, C.A.; Rossi, T. Glaucoma: An Overview. In Handbook of Nutrition, Diet, and the Eye; Elsevier: Amsterdam, The Netherlands, 2019; pp. 167–187. ISBN 978-0-12-815245-4. [Google Scholar]
- Sorkhabi, R.; Ghorbanihaghjo, A.; Javadzadeh, A.; Rashtchizadeh, N.; Moharrery, M. Oxidative DNA damage and total antioxidant status in glaucoma patients. Mol. Vis. 2011, 17, 41–46. [Google Scholar] [PubMed]
- Bagnis, A.; Izzotti, A.; Centofanti, M.; Saccà, S.C. Aqueous humor oxidative stress proteomic levels in primary open angle glaucoma. Exp. Eye Res. 2012, 103, 55–62. [Google Scholar] [CrossRef]
- Knepper, P.A.; Goossens, W.; Palmberg, P.F. Glycosaminoglycan stratification of the juxtacanalicular tissue in normal and primary open-angle glaucoma. Investig. Ophthalmol. Vis. Sci. 1996, 37, 2414–2425. [Google Scholar]
- Knepper, P.A.; Goossens, W.; Hvizd, M.; Palmberg, P.F. Glycosaminoglycans of the human trabecular meshwork in primary open-angle glaucoma. Investig. Ophthalmol. Vis. Sci. 1996, 37, 1360–1367. [Google Scholar]
- Zhao, J.; Wang, S.; Zhong, W.; Yang, B.; Sun, L.; Zheng, Y. Oxidative stress in the trabecular meshwork (Review). Int. J. Mol. Med. 2016, 38, 995–1002. [Google Scholar] [CrossRef] [Green Version]
- Saccà, S.C.; Tirendi, S.; Scarfì, S.; Passalacqua, M.; Oddone, F.; Traverso, C.E.; Vernazza, S.; Bassi, A.M. An advanced in vitro model to assess glaucoma onset. ALTEX Altern. Anim. Exp. 2020. [Google Scholar] [CrossRef]
- Vernazza, S.; Tirendi, S.; Scarfì, S.; Passalacqua, M.; Oddone, F.; Traverso, C.E.; Rizzato, I.; Bassi, A.M.; Saccà, S.C. 2D- and 3D-cultures of human trabecular meshwork cells: A preliminary assessment of an in vitro model for glaucoma study. PLoS ONE 2019, 14, e0221942. [Google Scholar] [CrossRef]
- Babizhayev, M.A.; Bunin, A.Y. Lipid peroxidation in open-angle glaucoma. Acta Ophthalmol. 2009, 67, 371–377. [Google Scholar] [CrossRef]
- Welge-Lüßen, U.; Birke, K. Oxidativer Stress im Trabekelwerk beim POWG. Klin. Mon. Augenheilkd. 2010, 227, 99–107. [Google Scholar] [CrossRef] [PubMed]
- Saccà, S.C.; Izzotti, A.; Rossi, P.; Traverso, C. Glaucomatous outflow pathway and oxidative stress. Exp. Eye Res. 2007, 84, 389–399. [Google Scholar] [CrossRef] [PubMed]
- Abu-Amero, K.K.; Morales, J.; Bosley, T.M. Mitochondrial Abnormalities in Patients with Primary Open-Angle Glaucoma. Investig. Ophthalmol. Vis. Sci. 2006, 47, 2533. [Google Scholar] [CrossRef] [PubMed]
- Arruda, A.P.; Pers, B.M.; Parlakgül, G.; Güney, E.; Inouye, K.; Hotamisligil, G.S. Chronic enrichment of hepatic endoplasmic reticulum–mitochondria contact leads to mitochondrial dysfunction in obesity. Nat. Med. 2014, 20, 1427–1435. [Google Scholar] [CrossRef] [Green Version]
- Liton, P.B.; Lin, Y.; Luna, C.; Li, G.; Gonzalez, P.; Epstein, D.L. Cultured Porcine Trabecular Meshwork Cells Display Altered Lysosomal Function When Subjected to Chronic Oxidative Stress. Investig. Ophthalmol. Vis. Sci. 2008, 49, 3961. [Google Scholar] [CrossRef]
- Liton, P.B.; Challa, P.; Stinnett, S.; Luna, C.; Epstein, D.L.; Gonzalez, P. Cellular senescence in the glaucomatous outflow pathway. Exp. Gerontol. 2005, 40, 745–748. [Google Scholar] [CrossRef] [Green Version]
- Porter, K.; Nallathambi, J.; Lin, Y.; Liton, P.B. Lysosomal basification and decreased autophagic flux in oxidatively stressed trabecular meshwork cells: Implications for glaucoma pathogenesis. Autophagy 2013, 9, 581–594. [Google Scholar] [CrossRef] [Green Version]
- Fossel, M. Cell Senescence in Human Aging and Disease. Ann. N. Y. Acad. Sci. 2002, 959, 14–23. [Google Scholar] [CrossRef]
- Li, G.; Luna, C.; Qiu, J.; Epstein, D.L.; Gonzalez, P. Modulation of Inflammatory Markers by miR-146a during Replicative Senescence in Trabecular Meshwork Cells. Investig. Ophthalmol. Vis. Sci. 2010, 51, 2976. [Google Scholar] [CrossRef]
- Fumagalli, M.; d’Adda di Fagagna, F. SASPense and DDRama in cancer and ageing. Nat. Cell Biol. 2009, 11, 921–923. [Google Scholar] [CrossRef]
- Wang, N.; Chintala, S.K.; Fini, M.E.; Schuman, J.S. Activation of a tissue-specific stress response in the aqueous outflow pathway of the eye defines the glaucoma disease phenotype. Nat. Med. 2001, 7, 304–309. [Google Scholar] [CrossRef] [PubMed]
- Anholt, R.R.H.; Carbone, M.A. A molecular mechanism for glaucoma: Endoplasmic reticulum stress and the unfolded protein response. Trends Mol. Med. 2013, 19, 586–593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cullinan, S.B.; Diehl, J.A. Coordination of ER and oxidative stress signaling: The PERK/Nrf2 signaling pathway. Int. J. Biochem. Cell Biol. 2006, 38, 317–332. [Google Scholar] [CrossRef] [PubMed]
- Muriach, M.; Flores-Bellver, M.; Romero, F.J.; Barcia, J.M. Diabetes and the Brain: Oxidative Stress, Inflammation, and Autophagy. Oxidative Med. Cell. Longev. 2014, 2014, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rao, R.V.; Ellerby, H.M.; Bredesen, D.E. Coupling endoplasmic reticulum stress to the cell death program. Cell Death Differ. 2004, 11, 372–380. [Google Scholar] [CrossRef] [Green Version]
- Halliwell, B. Oxidative stress and neurodegeneration: Where are we now? J. Neurochem. 2006, 97, 1634–1658. [Google Scholar] [CrossRef]
- Broman, A.T.; Quigley, H.A.; West, S.K.; Katz, J.; Munoz, B.; Bandeen-Roche, K.; Tielsch, J.M.; Friedman, D.S.; Crowston, J.; Taylor, H.R.; et al. Estimating the Rate of Progressive Visual Field Damage in Those with Open-Angle Glaucoma, from Cross-Sectional Data. Investig. Ophthalmol. Vis. Sci. 2008, 49, 66. [Google Scholar] [CrossRef]
- Feilchenfeld, Z.; Yücel, Y.H.; Gupta, N. Oxidative injury to blood vessels and glia of the pre-laminar optic nerve head in human glaucoma. Exp. Eye Res. 2008, 87, 409–414. [Google Scholar] [CrossRef]
- Barnham, K.J.; Masters, C.L.; Bush, A.I. Neurodegenerative diseases and oxidative stress. Nat. Rev. Drug Discov. 2004, 3, 205–214. [Google Scholar] [CrossRef]
- Andersen, J.K. Oxidative stress in neurodegeneration: Cause or consequence? Nat. Med. 2004, 10, S18–S25. [Google Scholar] [CrossRef]
- Tezel, G.; Luo, C.; Yang, X. Accelerated Aging in Glaucoma: Immunohistochemical Assessment of Advanced Glycation End Products in the Human Retina and Optic Nerve Head. Investig. Ophthalmol. Vis. Sci. 2007, 48, 1201. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, M.R.; Luo, X.X.; Andrzejewska, W.; Neufeld, A.H. Age-Related Changes in the Extracellular Matrix of the Human Optic Nerve Head. Am. J. Ophthalmol. 1989, 107, 476–484. [Google Scholar] [CrossRef]
- Streit, W.J. Microglial senescence: Does the brain’s immune system have an expiration date? Trends Neurosci. 2006, 29, 506–510. [Google Scholar] [CrossRef]
- Ramírez, A.I.; Fernández-Albarral, J.A.; de Hoz, R.; López-Cuenca, I.; Salobrar-García, E.; Rojas, P.; Valiente-Soriano, F.J.; Avilés-Trigueros, M.; Villegas-Pérez, M.P.; Vidal-Sanz, M.; et al. Microglial changes in the early aging stage in a healthy retina and an experimental glaucoma model. In Progress in Brain Research; Elsevier: Amsterdam, The Netherlands, 2020. [Google Scholar]
- Xu, H.; Chen, M.; Forrester, J.V. Para-inflammation in the aging retina. Prog. Retin. Eye Res. 2009, 28, 348–368. [Google Scholar] [CrossRef] [PubMed]
- Tezel, G.; Li, L.Y.; Patil, R.V.; Wax, M.B. TNF-alpha and TNF-alpha receptor-1 in the retina of normal and glaucomatous eyes. Investig. Ophthalmol. Vis. Sci. 2001, 42, 1787–1794. [Google Scholar]
- Neufeld, A.H.; Hernandez, M.R.; Gonzalez, M.; Geller, A. Cyclooxygenase-1 and Cyclooxygenase-2 in the Human Optic Nerve Head. Exp. Eye Res. 1997, 65, 739–745. [Google Scholar] [CrossRef]
- Liu, B.; Neufeld, A.H. Expression of nitric oxide synthase-2 (NOS-2) in reactive astrocytes of the human glaucomatous optic nerve head. Glia 2000, 30, 178–186. [Google Scholar] [CrossRef]
- Tezel, G.; Yang, X.; Luo, C.; Kain, A.D.; Powell, D.W.; Kuehn, M.H.; Kaplan, H.J. Oxidative Stress and the Regulation of Complement Activation in Human Glaucoma. Investig. Ophthalmol. Vis. Sci. 2010, 51, 5071. [Google Scholar] [CrossRef]
- Pun, P.B.L.; Lu, J.; Moochhala, S. Involvement of ROS in BBB dysfunction. Free Radic. Res. 2009, 43, 348–364. [Google Scholar] [CrossRef]
- Tezel, G. Hypoxia-Inducible Factor 1α in the Glaucomatous Retina and OpticNerve Head. Arch. Ophthalmol. 2004, 122, 1348. [Google Scholar] [CrossRef] [Green Version]
- Ischiropoulos, H.; Zhu, L.; Chen, J.; Tsai, M.; Martin, J.C.; Smith, C.D.; Beckman, J.S. Peroxynitrite-mediated tyrosine nitration catalyzed by superoxide dismutase. Arch. Biochem. Biophys. 1992, 298, 431–437. [Google Scholar] [CrossRef]
- Luthra, A.; Gupta, N.; Kaufman, P.L.; Weinreb, R.N.; Yücel, Y.H. Oxidative injury by peroxynitrite in neural and vascular tissue of the lateral geniculate nucleus in experimental glaucoma. Exp. Eye Res. 2005, 80, 43–49. [Google Scholar] [CrossRef] [PubMed]
- Kuo, W.N.; Kanadia, R.N.; Shanbhag, V.P.; Morgan, R. Modification of proteins and polynucleotides by peroxynitrite. Cytobios 1999, 99, 47–55. [Google Scholar] [PubMed]
- Crow, J.P.; Ye, Y.Z.; Strong, M.; Kirk, M.; Barnes, S.; Beckman, J.S. Superoxide Dismutase Catalyzes Nitration of Tyrosines by Peroxynitrite in the Rod and Head Domains of Neurofilament-L. J. Neurochem. 2002, 69, 1945–1953. [Google Scholar] [CrossRef]
- Jang, B.; Han, S. Biochemical properties of cytochrome c nitrated by peroxynitrite. Biochimie 2006, 88, 53–58. [Google Scholar] [CrossRef] [PubMed]
- Tezel, G. Immunostaining of Heat Shock Proteins in the Retina and Optic Nerve Head of Normal and Glaucomatous Eyes. Arch. Ophthalmol. 2000, 118, 511. [Google Scholar] [CrossRef] [Green Version]
- Myer, C.; Perez, J.; Abdelrahman, L.; Mendez, R.; Khattri, R.B.; Junk, A.K.; Bhattacharya, S.K. Differentiation of soluble aqueous humor metabolites in primary open angle glaucoma and controls. Exp. Eye Res. 2020, 194, 108024. [Google Scholar] [CrossRef]
- Ferreira, S.M.; Lerner, S.F.; Brunzini, R.; Evelson, P.A.; Llesuy, S.F. Oxidative stress markers in aqueous humor of glaucoma patients. Am. J. Ophthalmol. 2004, 137, 62–69. [Google Scholar] [CrossRef]
- Ghanem, A.A.; Elewa, A.M.; Arafa, L.F. Endothelin-1 and Nitric Oxide Levels in Patients with Glaucoma. Ophthalmic Res. 2011, 46, 98–102. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Zhu, H.; Yang, Y.; Ye, Y.; Yao, Y.; Huang, X.; Zhang, Y.; Shu, X.; Chen, X.; Yang, Y.; et al. AQP1 suppression by ATF4 triggers trabecular meshwork tissue remodelling in ET-1-induced POAG. J. Cell. Mol. Med. 2020, 24, 3469–3480. [Google Scholar] [CrossRef] [PubMed]
- Buisset, A.; Gohier, P.; Leruez, S.; Muller, J.; Amati-Bonneau, P.; Lenaers, G.; Bonneau, D.; Simard, G.; Procaccio, V.; Annweiler, C.; et al. Metabolomic Profiling of Aqueous Humor in Glaucoma Points to Taurine and Spermine Deficiency: Findings from the Eye-D Study. J. Proteome Res. 2019, 18, 1307–1315. [Google Scholar] [CrossRef] [PubMed]
- Hannappel, E.; Pankow, G.; Grassl, F.; Brand, K.; Naumann, G.O.H. Amino Acid Pattern in Human Aqueous Humor of Patients with Senile Cataract and Primary Open-Angle Glaucoma. Ophthalmic Res. 1985, 17, 341–343. [Google Scholar] [CrossRef] [PubMed]
- Ozcan, A.A.; Ozdemir, N.; Canataroglu, A. The Aqueous Levels of TGF-2 in Patients with Glaucoma. Int. Ophthalmol. 2004, 25, 19–22. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, N.; Itonaga, K.; Marunouchi, T.; Majima, K. Concentration of Transforming Growth Factor β2 in Aqueous Humor. Ophthalmic Res. 2005, 37, 29–33. [Google Scholar] [CrossRef]
- Dan, J. Plasminogen Activator Inhibitor-1 in the Aqueous Humor of Patients with and Without Glaucoma. Arch. Ophthalmol. 2005, 123, 220. [Google Scholar] [CrossRef] [Green Version]
- Duan, X.; Xue, P.; Wang, N.; Dong, Z.; Lu, Q.; Yang, F. Proteomic analysis of aqueous humor from patients with primary open angle glaucoma. Mol. Vis. 2010, 16, 2839–2846. [Google Scholar]
- Alvarado, J.; Murphy, C.; Juster, R. Trabecular Meshwork Cellularity in Primary Open-angle Glaucoma and Nonglaucomatous Normals. Ophthalmology 1984, 91, 564–579. [Google Scholar] [CrossRef]
- Chen, Y.-S.; Green, C.R.; Danesh-Meyer, H.V.; Rupenthal, I.D. Neuroprotection in the treatment of glaucoma—A focus on connexin43 gap junction channel blockers. Eur. J. Pharm. Biopharm. 2015, 95, 182–193. [Google Scholar] [CrossRef]
- Advances in Immunology; Academic Press: Salt Lake, UT, USA, 1995; ISBN 978-0-08-057834-7.
- Brigelius-Flohé, R.; Flohé, L. Basic principles and emerging concepts in the redox control of transcription factors. Antioxid. Redox Signal. 2011, 15, 2335–2381. [Google Scholar] [CrossRef] [Green Version]
- Barnes, P.J.; Karin, M. Nuclear Factor-κB—A Pivotal Transcription Factor in Chronic Inflammatory Diseases. New Engl. J. Med. 1997, 336, 1066–1071. [Google Scholar] [CrossRef]
- Willems, M.; Dubois, N.; Musumeci, L.; Bours, V.; Robe, P.A. IκBζ: An emerging player in cancer. Oncotarget 2016, 7. [Google Scholar] [CrossRef]
- Siebenlist, U.; Franzoso, G.; Brown, K. Structure, Regulation and Function of NF-kappaB. Annu. Rev. Cell Biol. 1994, 10, 405–455. [Google Scholar] [CrossRef] [PubMed]
- Baeuerle, P.A.; Baltimore, D. NF-κB: Ten Years after. Cell 1996, 87, 13–20. [Google Scholar] [CrossRef] [Green Version]
- Baldwin, A.S. THE NF-κB AND IκB PROTEINS: New Discoveries and Insights. Annu. Rev. Immunol. 1996, 14, 649–681. [Google Scholar] [CrossRef] [Green Version]
- Dejardin, E. The alternative NF-κB pathway from biochemistry to biology: Pitfalls and promises for future drug development. Biochem. Pharmacol. 2006, 72, 1161–1179. [Google Scholar] [CrossRef]
- Pahl, H.L. Activators and target genes of Rel/NF-κB transcription factors. Oncogene 1999, 18, 6853–6866. [Google Scholar] [CrossRef] [Green Version]
- Chung, K. Cytokines as Targets in Chronic Obstructive Pulmonary Disease. CDT 2006, 7, 675–681. [Google Scholar] [CrossRef]
- Williams, R.O.; Paleolog, E.; Feldmann, M. Cytokine inhibitors in rheumatoid arthritis and other autoimmune diseases. Curr. Opin. Pharmacol. 2007, 7, 412–417. [Google Scholar] [CrossRef]
- Holgate, S.T. Pathogenesis of Asthma. Clin. Exp. Allergy 2008, 38, 872–897. [Google Scholar] [CrossRef]
- Ghosh, S.; Karin, M. Missing Pieces in the NF-κB Puzzle. Cell 2002, 109, S81–S96. [Google Scholar] [CrossRef] [Green Version]
- Sigala, J.L.D.; Bottero, V.; Young, D.B.; Shevchenko, A.; Mercurio, F.; Verma, I.M. Activation of Transcription Factor NF-κB Requires ELKS, an IκB Kinase Regulatory Subunit. Science 2004, 304, 1963–1967. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, G.; Cao, P.; Goeddel, D.V. TNF-Induced Recruitment and Activation of the IKK Complex Require Cdc37 and Hsp90. Mol. Cell 2002, 9, 401–410. [Google Scholar] [CrossRef]
- Malhotra, D.; Portales-Casamar, E.; Singh, A.; Srivastava, S.; Arenillas, D.; Happel, C.; Shyr, C.; Wakabayashi, N.; Kensler, T.W.; Wasserman, W.W.; et al. Global mapping of binding sites for Nrf2 identifies novel targets in cell survival response through ChIP-Seq profiling and network analysis. Nucleic Acids Res. 2010, 38, 5718–5734. [Google Scholar] [CrossRef] [PubMed]
- Leonard, M.O.; Kieran, N.E.; Howell, K.; Burne, M.J.; Varadarajan, R.; Dhakshinamoorthy, S.; Porter, A.G.; O’Farrelly, C.; Rabb, H.; Taylor, C.T.; et al. Reoxygenation-specific activation of the antioxidant transcription factor Nrf2 mediates cytoprotective gene expression in ischemia-reperfusion injury. FASEB J. 2006, 20, 2624–2626. [Google Scholar] [CrossRef]
- Chorley, B.N.; Campbell, M.R.; Wang, X.; Karaca, M.; Sambandan, D.; Bangura, F.; Xue, P.; Pi, J.; Kleeberger, S.R.; Bell, D.A. Identification of novel NRF2-regulated genes by ChIP-Seq: Influence on retinoid X receptor alpha. Nucleic Acids Res. 2012, 40, 7416–7429. [Google Scholar] [CrossRef] [Green Version]
- Ishii, T.; Itoh, K.; Takahashi, S.; Sato, H.; Yanagawa, T.; Katoh, Y.; Bannai, S.; Yamamoto, M. Transcription Factor Nrf2 Coordinately Regulates a Group of Oxidative Stress-inducible Genes in Macrophages. J. Biol. Chem. 2000, 275, 16023–16029. [Google Scholar] [CrossRef] [Green Version]
- Park, E.Y.; Rho, H.M. The trascriptional activation of the human copper/zinc superoxide dismutase gene by 2,3,7,8-tetrachlorodibenzo-p-dioxin through two different regulator sites, the anitoxidant responsive element and xenobiotic responsive element. Mol. Cell Biochem. 2002, 240, 47–55. [Google Scholar] [CrossRef]
- Thimmulappa, R.K.; Mai, K.H.; Srisuma, S.; Kensler, T.W.; Yamamoto, M.; Biswal, S. Identification of Nrf2-regulated Genes Induced by the Chemopreventive Agent Sulforaphane by Oligonucleotide Microarray. Cancer Res. 2002, 62. Available online: https://cancerres.aacrjournals.org/content/62/18/5196.short (accessed on 24 April 2020).
- Motohashi, H.; Yamamoto, M. Nrf2–Keap1 defines a physiologically important stress response mechanism. Trends Mol. Med. 2004, 10, 549–557. [Google Scholar] [CrossRef]
- Eggler, A.L.; Small, E.; Hannink, M.; Mesecar, A.D. Cul3-mediated Nrf2 ubiquitination and antioxidant response element (ARE) activation are dependent on the partial molar volume at position 151 of Keap1. Biochem. J. 2009, 422, 171–180. [Google Scholar] [CrossRef] [Green Version]
- Takaya, K.; Suzuki, T.; Motohashi, H.; Onodera, K.; Satomi, S.; Kensler, T.W.; Yamamoto, M. Validation of the multiple sensor mechanism of the Keap1-Nrf2 system. Free Radic. Biol. Med. 2012, 53, 817–827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jain, A.K.; Jaiswal, A.K. GSK-3β Acts Upstream of Fyn Kinase in Regulation of Nuclear Export and Degradation of NF-E2 Related Factor 2. J. Biol. Chem. 2007, 282, 16502–16510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buelna-Chontal, M.; Zazueta, C. Redox activation of Nrf2 & NF-κB: A double end sword? Cell. Signal. 2013, 25, 2548–2557. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.-Y.; Jang, J.-H.; Li, M.-H.; Surh, Y.-J. Resveratrol upregulates heme oxygenase-1 expression via activation of NF-E2-related factor 2 in PC12 cells. Biochem. Biophys. Res. Commun. 2005, 331, 993–1000. [Google Scholar] [CrossRef] [PubMed]
- Surh, Y.-J. Cancer chemoprevention with dietary phytochemicals. Nat. Rev. Cancer 2003, 3, 768–780. [Google Scholar] [CrossRef]
- Balogun, E.; Hoque, M.; Gong, P.; Killeen, E.; Green, C.J.; Foresti, R.; Alam, J.; Motterlini, R. Curcumin activates the haem oxygenase-1 gene via regulation of Nrf2 and the antioxidant-responsive element. Biochem. J. 2003, 371, 887–895. [Google Scholar] [CrossRef] [Green Version]
- Bellezza, I.; Mierla, A.L.; Minelli, A. Nrf2 and NF-κB and Their Concerted Modulation in Cancer Pathogenesis and Progression. Cancers 2010, 2, 483–497. [Google Scholar] [CrossRef]
- Gloire, G.; Legrand-Poels, S.; Piette, J. NF-κB activation by reactive oxygen species: Fifteen years later. Biochem. Pharmacol. 2006, 72, 1493–1505. [Google Scholar] [CrossRef]
- Minelli, A.; Grottelli, S.; Mierla, A.; Pinnen, F.; Cacciatore, I.; Bellezza, I. Cyclo (His-Pro) exerts anti-inflammatory effects by modulating NF-κB and Nrf2 signalling. Int. J. Biochem. Cell Biol. 2012, 44, 525–535. [Google Scholar] [CrossRef]
- Ross, R. Cell Biology of Atherosclerosis. Annu. Rev. Physiol. 1995, 57, 791–804. [Google Scholar] [CrossRef]
- Saccà, S.C.; Izzotti, A. Focus on molecular events in the anterior chamber leading to glaucoma. Cell. Mol. Life Sci. 2014, 71, 2197–2218. [Google Scholar] [CrossRef] [PubMed]
- Schmetterer, L.; Polak, K. Role of Nitric Oxide in the Control of Ocular Blood Flow. Prog. Retin. Eye Res. 2001, 20, 823–847. [Google Scholar] [CrossRef]
- Saccà, S.C.; Pulliero, A.; Izzotti, A. The Dysfunction of the Trabecular Meshwork during Glaucoma Course. J. Cell. Physiol. 2015, 230, 510–525. [Google Scholar] [CrossRef]
- Xie, Q.W.; Kashiwabara, Y.; Nathan, C. Role of transcription factor NF-kappa B/Rel in induction of nitric oxide synthase. J. Biol. Chem. 1994, 269, 4705–4708. [Google Scholar] [PubMed]
- Bredt, D.S.; Snyder, S.H. NITRIC OXIDE: A Physiologic Messenger Molecule. Annu. Rev. Biochem. 1994, 63, 175–195. [Google Scholar] [CrossRef] [PubMed]
- Esterbauer, H.; Schaur, R.J.; Zollner, H. Chemistry and biochemistry of 4-hydroxynonenal, malonaldehyde and related aldehydes. Free Radic. Biol. Med. 1991, 11, 81–128. [Google Scholar] [CrossRef]
- Mattson, M.P. Modification of ion homeostasis by lipid peroxidation: Roles in neuronal degeneration and adaptive plasticity. Trends Neurosci. 1998, 21, 53–57. [Google Scholar] [CrossRef]
- Fernández-Durango, R.; Fernández-Martínez, A.; García-Feijoo, J.; Castillo, A.; de la Casa, J.M.; García-Bueno, B.; Pérez-Nievas, B.G.; Fernández-Cruz, A.; Leza, J.C. Expression of Nitrotyrosine and Oxidative Consequences in the Trabecular Meshwork of Patients with Primary Open-Angle Glaucoma. Investigative Ophthalmol. Vis. Sci. 2008, 49, 2506–2511. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.; Luo, C.; Cai, J.; Powell, D.W.; Yu, D.; Kuehn, M.H.; Tezel, G. Neurodegenerative and Inflammatory Pathway Components Linked to TNF-α/TNFR1 Signaling in the Glaucomatous Human Retina. Investig. Ophthalmol. Vis. Sci. 2011, 52, 8442–8454. [Google Scholar] [CrossRef]
- Luo, C.; Yang, X.; Kain, A.D.; Powell, D.W.; Kuehn, M.H.; Tezel, G. Glaucomatous Tissue Stress and the Regulation of Immune Response through Glial Toll-like Receptor Signaling. Investig. Ophthalmol. Vis. Sci. 2010, 51, 5697. [Google Scholar] [CrossRef]
- Tezel, G. TNF-α signaling in glaucomatous neurodegeneration. Prog. Brain Res. 2008, 173, 409–421. [Google Scholar] [PubMed] [Green Version]
- Harari, O.A.; Liao, J.K. NF-κB and innate immunity in ischemic stroke. Ann. N. Y. Acad. Sci. 2010, 1207, 32–40. [Google Scholar] [CrossRef] [PubMed]
- Genestra, M. Oxyl radicals, redox-sensitive signalling cascades and antioxidants. Cell. Signal. 2007, 19, 1807–1819. [Google Scholar] [CrossRef] [PubMed]
- Zambrano, S.; Toma, I.D.; Piffer, A.; Bianchi, M.E.; Agresti, A. NF-κB oscillations translate into functionally related patterns of gene expression. Elife 2016, 5, e09100. [Google Scholar] [CrossRef]
- Dinarello, C.A. Biology of interleukin 1. FASEB J. 1988, 2, 108–115. [Google Scholar] [CrossRef] [Green Version]
- Schreck, R.; Rieber, P.; Baeuerle, P.A. Reactive oxygen intermediates as apparently widely used messengers in the activation of the NF-kappa B transcription factor and HIV-1. EMBO J. 1991, 10, 2247–2258. [Google Scholar] [CrossRef]
- Cook, J.R.; Mody, M.K.; Fini, M.E. Failure to Activate Transcription Factor NF-κB in Corneal Stromal Cells (Keratocytes). Investig. Ophthalmol. Vis. Sci. 1999, 40, 3122–3131. [Google Scholar]
- Beg, A.A.; Baltimore, D. An Essential Role for NF-κB in Preventing TNF-α-Induced Cell Death. Science 1996, 274, 782–784. [Google Scholar] [CrossRef]
- Kee, C.; Seo, K. The effect of interleukin-1alpha on outflow facility in rat eyes. J. Glaucoma 1997, 6, 246–249. [Google Scholar] [CrossRef]
- Alexander, J.P.; Samples, J.R.; Buskirk, E.M.V.; Acott, T.S. Expression of matrix metalloproteinases and inhibitor by human trabecular meshwork. Investig. Ophthalmol. Vis. Sci. 1991, 32, 172–180. [Google Scholar]
- Civan, M.M.; Macknight, A.D.C. The ins and outs of aqueous humour secretion. Exp. Eye Res. 2004, 78, 625–631. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Tun, T.A.; Baskaran, M.; Atalay, E.; Thakku, S.G.; Liang, Z.; Milea, D.; Strouthidis, N.G.; Aung, T.; Girard, M.J. Effect of acute intraocular pressure elevation on the minimum rim width in normal, ocular hypertensive and glaucoma eyes. Br. J. Ophthalmol. 2018, 102, 131–135. [Google Scholar] [CrossRef] [PubMed]
- Alvarado, J.A.; Yeh, R.-F.; Franse-Carman, L.; Marcellino, G.; Brownstein, M.J. Interactions between endothelia of the trabecular meshwork and of Schlemm’s canal: A new insight into the regulation of aqueous outflow in the eye. Trans. Am. Ophthalmol. Soc. 2005, 103, 148–162; discussion 162–163. [Google Scholar]
- Abu-Hassan, D.; Acott, T.; Kelley, M. The Trabecular Meshwork: A Basic Review of Form and Function. J. Ocul. Biol. 2014, 2. [Google Scholar] [CrossRef]
- Costagliola, C.; dell’Omo, R.; Agnifili, L.; Bartollino, S.; Fea, A.M.; Uva, M.G.; Zeppa, L.; Mastropasqua, L. How many aqueous humor outflow pathways are there? Surv. Ophthalmol. 2020, 65, 144–170. [Google Scholar] [CrossRef]
- Rohen, J.W.; Futa, R.; Lütjen-Drecoll, E. The fine structure of the cribriform meshwork in normal and glaucomatous eyes as seen in tangential sections. Investig. Ophthalmol. Vis. Sci. 1981, 21, 574–585. [Google Scholar]
- Epstein, D.L.; Rohen, J.W. Morphology of the trabecular meshwork and inner-wall endothelium after cationized ferritin perfusion in the monkey eye. Investig. Ophthalmol. Vis. Sci. 1991, 32, 160–171. [Google Scholar]
- Ye, W.; Gong, H.; Sit, A.; Johnson, M.; Freddo, T.F. Interendothelial junctions in normal human Schlemm’s canal respond to changes in pressure. Investig. Ophthalmol. Vis. Sci. 1997, 38, 2460–2468. [Google Scholar]
- Raviola, G. Schwalbe line’s cells: A new cell type in the trabecular meshwork of Macaca mulatta. Investig. Ophthalmol. Vis. Sci. 1982, 22, 45–56. [Google Scholar]
- Keller, K.E.; Aga, M.; Bradley, J.M.; Kelley, M.J.; Acott, T.S. Extracellular matrix turnover and outflow resistance. Exp. Eye Res. 2009, 88, 676–682. [Google Scholar] [CrossRef] [Green Version]
- Tian, B.; Gabelt, B.T.; Geiger, B.; Kaufman, P.L. The role of the actomyosin system in regulating trabecular fluid outflow. Exp. Eye Res. 2009, 88, 713–717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alm, A.; Nilsson, S.F.E. Uveoscleral outflow—A review. Exp. Eye Res. 2009, 88, 760–768. [Google Scholar] [CrossRef] [PubMed]
- Flügel-Koch, C.; Ohlmann, A.; Fuchshofer, R.; Welge-Lüssen, U.; Tamm, E.R. Thrombospondin-1 in the trabecular meshwork: Localization in normal and glaucomatous eyes, and induction by TGF-β1 and dexamethasone in vitro. Exp. Eye Res. 2004, 79, 649–663. [Google Scholar] [CrossRef] [PubMed]
- Acott, T.S.; Kelley, M.J. Extracellular matrix in the trabecular meshwork. Exp. Eye Res. 2008, 86, 543–561. [Google Scholar] [CrossRef] [Green Version]
- Rhee, D.J.; Fariss, R.N.; Brekken, R.; Helene Sage, E.; Russell, P. The matricellular protein SPARC is expressed in human trabecular meshwork. Exp. Eye Res. 2003, 77, 601–607. [Google Scholar] [CrossRef]
- Chatterjee, A.; Villarreal, G.; Rhee, D.J. Matricellular Proteins in the Trabecular Meshwork: Review and Update. J. Ocul. Pharmacol. Ther. 2014, 30, 447–463. [Google Scholar] [CrossRef] [Green Version]
- Stamer, W.D.; Acott, T.S. Current understanding of conventional outflow dysfunction in glaucoma. Curr. Opin. Ophthalmol. 2012, 23, 135–143. [Google Scholar] [CrossRef] [Green Version]
- Gong, H.; Ruberti, J.; Overby, D.; Johnson, M.; Freddo, T.F. A new view of the human trabecular meshwork using quick-freeze, deep-etch electron microscopy. Exp. Eye Res. 2002, 75, 347–358. [Google Scholar] [CrossRef]
- Karl, M.O.; Fleischhauer, J.C.; Stamer, W.D.; Peterson-Yantorno, K.; Mitchell, C.H.; Stone, R.A.; Civan, M.M. Differential P1-purinergic modulation of human Schlemm’s canal inner-wall cells. Am. J. Physiol. Cell Physiol. 2005, 288, C784–C794. [Google Scholar] [CrossRef]
- Overby, D.R.; Stamer, W.D.; Johnson, M. The changing paradigm of outflow resistance generation: Towards synergistic models of the JCT and inner wall endothelium. Exp. Eye Res. 2009, 88, 656–670. [Google Scholar] [CrossRef] [Green Version]
- Pedrigi, R.M.; Simon, D.; Reed, A.; Stamer, W.D.; Overby, D.R. A model of giant vacuole dynamics in human Schlemm’s canal endothelial cells. Exp. Eye Res. 2011, 92, 57–66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ethier, C.R.; Coloma, F.M.; Sit, A.J.; Johnson, M. Two pore types in the inner-wall endothelium of Schlemm’s canal. Investig. Ophthalmol. Vis. Sci. 1998, 39, 2041–2048. [Google Scholar]
- Johnstone, M.A.; Grant, W.M. Pressure-Dependent Changes in Structures of the Aqueous Outflow System of Human and Monkey Eyes. Am. J. Ophthalmol. 1973, 75, 365–383. [Google Scholar] [CrossRef]
- Allingham, R.R.; de KATER, A.W.; Ethier, R.C. Schlemm’s Canal and Primary Open Angle Glaucoma: Correlation between Schlemm’s Canal Dimensions and Outflow Facility. Exp. Eye Res. 1996, 62, 101–110. [Google Scholar] [CrossRef]
- Johnson, M. What controls aqueous humour outflow resistance? Exp. Eye Res. 2006, 82, 545–557. [Google Scholar] [CrossRef] [Green Version]
- Overby, D.R.; Bertrand, J.; Tektas, O.-Y.; Boussommier-Calleja, A.; Schicht, M.; Ethier, C.R.; Woodward, D.F.; Stamer, W.D.; Lütjen-Drecoll, E. Ultrastructural Changes Associated with Dexamethasone-Induced Ocular Hypertension in Mice. Investig. Ophthalmol. Vis. Sci. 2014, 55, 4922. [Google Scholar] [CrossRef] [Green Version]
- Hann, C.R.; Vercnocke, A.J.; Bentley, M.D.; Jorgensen, S.M.; Fautsch, M.P. Anatomic Changes in Schlemm’s Canal and Collector Channels in Normal and Primary Open-Angle Glaucoma Eyes Using Low and High Perfusion Pressures. Investig. Ophthalmol. Vis. Sci. 2014, 55, 5834. [Google Scholar] [CrossRef]
- Stamer, W.D.; Clark, A.F. The many faces of the trabecular meshwork cell. Exp. Eye Res. 2017, 158, 112–123. [Google Scholar] [CrossRef] [Green Version]
- Teng, C.C.; Paton, R.T.; Katzin, H.M. Primary Degeneration in the Vicinity of the Chamber Angle. Am. J. Ophthalmol. 1955, 40, 619–631. [Google Scholar] [CrossRef]
- Dvorak-Theobald Further Studies on the Canal of Schlemm. Am. J. Ophthalmol. 1955, 39, 65–89. [CrossRef]
- Bahler, C.K.; Howell, K.G.; Hann, C.R.; Fautsch, M.P.; Johnson, D.H. Prostaglandins Increase Trabecular Meshwork Outflow Facility in Cultured Human Anterior Segments. Am. J. Ophthalmol. 2008, 145, 114–119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Millard, L.H.; Woodward, D.F.; Stamer, W.D. The Role of the Prostaglandin EP 4 Receptor in the Regulation of Human Outflow Facility. Investig. Ophthalmol. Vis. Sci. 2011, 52, 3506. [Google Scholar] [CrossRef] [Green Version]
- Wan, Z.; Woodward, D.F.; Cornell, C.L.; Fliri, H.G.; Martos, J.L.; Pettit, S.N.; Wang, J.W.; Kharlamb, A.B.; Wheeler, L.A.; Garst, M.E.; et al. Bimatoprost, Prostamide Activity, and Conventional Drainage. Investig. Ophthalmol. Vis. Sci. 2007, 48, 4107. [Google Scholar] [CrossRef] [PubMed]
- Woodward, D.F.; Nilsson, S.F.E.; Toris, C.B.; Kharlamb, A.B.; Nieves, A.L.; Krauss, A.H.-P. Prostanoid EP4 Receptor Stimulation Produces Ocular Hypotension by a Mechanism That Does Not Appear to Involve Uveoscleral Outflow. Investig. Ophthalmol. Vis. Sci. 2009, 50, 3320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toris, C.B.; Gabelt, B.T.; Kaufman, P.L. Update on the Mechanism of Action of Topical Prostaglandins for Intraocular Pressure Reduction. Surv. Ophthalmol. 2008, 53, S107–S120. [Google Scholar] [CrossRef] [Green Version]
- Ziai, N. The Effects on Aqueous Dynamics of PhXA41, a New Prostaglandin F2α Analogue, After Topical Application in Normal and Ocular Hypertensive Human Eyes. Arch. Ophthalmol. 1993, 111, 1351. [Google Scholar] [CrossRef]
- Mettu, P.S.; Deng, P.-F.; Misra, U.K.; Gawdi, G.; Epstein, D.L.; Rao, P.V. Role of Lysophospholipid Growth Factors in the Modulation of Aqueous Humor Outflow Facility. Investig. Ophthalmol. Vis. Sci. 2004, 45, 2263. [Google Scholar] [CrossRef]
- Stamer, W.D.; Read, A.T.; Sumida, G.M.; Ethier, C.R. Sphingosine-1-phosphate effects on the inner wall of Schlemm’s canal and outflow facility in perfused human eyes. Exp. Eye Res. 2009, 89, 980–988. [Google Scholar] [CrossRef] [Green Version]
- Kuchtey, J.; Rezaei, K.A.; Jaru-Ampornpan, P.; Sternberg, P.; Kuchtey, R.W. Multiplex Cytokine Analysis Reveals Elevated Concentration of Interleukin-8 in Glaucomatous Aqueous Humor. Investig. Ophthalmol. Vis. Sci. 2010, 51, 6441. [Google Scholar] [CrossRef] [Green Version]
- Tripathi, R.C.; Li, J.; Chan, W.A.; Tripathi, B.J. Aqueous Humor in Glaucomatous Eyes Contains an Increased Level of TGF-β2. Exp. Eye Res. 1994, 59, 723–728. [Google Scholar] [CrossRef]
- Fuchshofer, R.; Stephan, D.A.; Russell, P.; Tamm, E.R. Gene expression profiling of TGFβ2- and/or BMP7-treated trabecular meshwork cells: Identification of Smad7 as a critical inhibitor of TGF-β2 signaling. Exp. Eye Res. 2009, 88, 1020–1032. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shin, D.-M.; Jeon, J.-H.; Kim, C.-W.; Cho, S.-Y.; Lee, H.-J.; Jang, G.-Y.; Jeong, E.M.; Lee, D.-S.; Kang, J.-H.; Melino, G.; et al. TGFβ mediates activation of transglutaminase 2 in response to oxidative stress that leads to protein aggregation. FASEB J. 2008, 22, 2498–2507. [Google Scholar] [CrossRef] [PubMed]
- Huang, P.; Qi, Y.; Xu, Y.-S.; Liu, J.; Liao, D.; Zhang, S.S.-M.; Zhang, C. Serum Cytokine Alteration is Associated with Optic Neuropathy in Human Primary Open Angle Glaucoma. J. Glaucoma 2009, 19, 324–330. [Google Scholar] [CrossRef] [PubMed]
- Huang, P.; Zhang, S.S.-M.; Zhang, C. The two sides of cytokine signaling and glaucomatous optic neuropathy. J. Ocul. Biol. Dis. Inform. 2009, 2, 78–83. [Google Scholar] [CrossRef] [Green Version]
- Avotri, S.; Eatman, D.; Russell-Randall, K. Effects of Resveratrol on Inflammatory Biomarkers in Glaucomatous Human Trabecular Meshwork Cells. Nutrients 2019, 11, 984. [Google Scholar] [CrossRef] [Green Version]
- De Groef, L.; Van Hove, I.; Dekeyster, E.; Stalmans, I.; Moons, L. MMPs in the Trabecular Meshwork: Promising Targets for Future Glaucoma Therapies? Investig. Ophthalmol. Vis. Sci. 2013, 54, 7756. [Google Scholar] [CrossRef] [Green Version]
- Chua, J.; Vania, M.; Cheung, C.M.G.; Ang, M.; Chee, S.P.; Yang, H.; Li, J.; Wong, T.T. Expression profile of inflammatory cytokines in aqueous from glaucomatous eyes. Mol. Vis. 2012, 18, 431–438. [Google Scholar]
- Li, A.; Leung, C.T.; Peterson-Yantorno, K.; Stamer, W.D.; Civan, M.M. Cytoskeletal Dependence of Adenosine Triphosphate Release by Human Trabecular Meshwork Cells. Investig. Ophthalmol. Vis. Sci. 2011, 52, 7996. [Google Scholar] [CrossRef]
- Li, A.; Leung, C.T.; Peterson-Yantorno, K.; Stamer, W.D.; Mitchell, C.H.; Civan, M.M. Mechanisms of ATP release by human trabecular meshwork cells, the enabling step in purinergic regulation of aqueous humor outflow. J. Cell. Physiol. 2012, 227, 172–182. [Google Scholar] [CrossRef] [Green Version]
- Chowdhury, U.R.; Bahler, C.K.; Hann, C.R.; Chang, M.; Resch, Z.T.; Romero, M.F.; Fautsch, M.P. ATP-Sensitive Potassium (KATP) Channel Activation Decreases Intraocular Pressure in the Anterior Chamber of the Eye. Investig. Ophthalmol. Vis. Sci. 2011, 52, 6435. [Google Scholar] [CrossRef] [Green Version]
- Zhou, L.; Fukuchi, T.; Kawa, J.E.; Higginbotham, E.J.; Yue, B.Y. Loss of cell-matrix cohesiveness after phagocytosis by trabecular meshwork cells. Investig. Ophthalmol. Vis. Sci. 1995, 36, 787–795. [Google Scholar]
- Calthorpe, C.M.; Grierson, I. Fibronectin induces migration of bovine trabecular meshwork cells in vitro. Exp. Eye Res. 1990, 51, 39–48. [Google Scholar] [CrossRef]
- Stumpff, F.; Wiederholt, M. Regulation of trabecular meshwork contractility. Ophthalmologica 2000, 214, 33–53. [Google Scholar] [CrossRef] [PubMed]
- Weinreb, R.; Cotlier, E.; Yue, B.Y. The extracellular matrix and its modulation in the trabecular meshwork. Surv. Ophthalmol. 1996, 40, 379–390. [Google Scholar] [CrossRef]
- Nathanson, J.A.; McKee, M. Alterations of ocular nitric oxide synthase in human glaucoma. Investig. Ophthalmol. Vis. Sci. 1995, 36, 1774–1784. [Google Scholar]
- Wiederholt, M.; Sturm, A.; Lepple-Wienhues, A. Relaxation of trabecular meshwork and ciliary muscle by release of nitric oxide. Investig. Ophthalmol. Vis. Sci. 1994, 35, 2515–2520. [Google Scholar]
- Haynes, W.G.; Webb, D.J. Endothelin as a regulator of cardiovascular function in health and disease. J. Hypertens. 1998, 16, 1081–1098. [Google Scholar] [CrossRef]
- Saccà, S.C.; Centofanti, M.; Izzotti, A. New Proteins as Vascular Biomarkers in Primary Open Angle Glaucomatous Aqueous Humor. Investig. Ophthalmol. Vis. Sci. 2012, 53, 4242. [Google Scholar] [CrossRef]
- Blondin, C.; Hamard, P.; Brignole, F.; Baudouin, C. Human Trabecular Meshwork Cells Produce the Pro-inflammatory Chemokines Interleukin-8 (IL-8) and Monocyte Chemoattractant Protein-1 (MCP-1) in vitro. Investig. Ophthalmol. Vis. Sci. 2003, 44, 679. [Google Scholar]
- Micera, A.; Quaranta, L.; Esposito, G.; Floriani, I.; Pocobelli, A.; Saccà, S.C.; Riva, I.; Manni, G.; Oddone, F. Differential Protein Expression Profiles in Glaucomatous Trabecular Meshwork: An Evaluation Study on a Small Primary Open Angle Glaucoma Population. Adv. Ther. 2016, 33, 252–267. [Google Scholar] [CrossRef] [Green Version]
- Li, G.; Luna, C.; Liton, P.B.; Navarro, I.; Epstein, D.L.; Gonzalez, P. Sustained stress response after oxidative stress in trabecular meshwork cells. Mol. Vis. 2007, 13, 2282–2288. [Google Scholar] [PubMed]
- Luna, C.; Li, G.; Liton, P.B.; Epstein, D.L.; Gonzalez, P. Alterations in gene expression induced by cyclic mechanical stress in trabecular meshwork cells. Mol. Vis. 2009, 15, 534–544. [Google Scholar]
- Fuchshofer, R.; Tamm, E.R. Modulation of extracellular matrix turnover in the trabecular meshwork. Exp. Eye Res. 2009, 88, 683–688. [Google Scholar] [CrossRef] [PubMed]
- Liesenborghs, I.; Eijssen, L.M.T.; Kutmon, M.; Gorgels, T.G.M.F.; Evelo, C.T.; Beckers, H.J.M.; Webers, C.A.B.; Schouten, J.S.A.G. Comprehensive bioinformatics analysis of trabecular meshwork gene expression data to unravel the molecular pathogenesis of primary open-angle glaucoma. Acta Ophthalmol. 2020, 98, 48–57. [Google Scholar] [CrossRef] [PubMed]
- Tamm, E.R.; Siegner, A.; Baur, A.; Lütjen-Drecoll, E. Transforming Growth Factor-β1 Induces α-Smooth Muscle-Actin Expression in Cultured Human and Monkey Trabecular Meshwork. Exp. Eye Res. 1996, 62, 389–398. [Google Scholar] [CrossRef]
- Sanka, K.; Maddala, R.; Epstein, D.L.; Rao, P.V. Influence of Actin Cytoskeletal Integrity on Matrix Metalloproteinase-2 Activation in Cultured Human Trabecular Meshwork Cells. Investig. Ophthalmol. Vis. Sci. 2007, 48, 2105. [Google Scholar] [CrossRef] [Green Version]
- Sabanay, I.; Gabelt, B.T.; Tian, B.; Kaufman, P.L.; Geiger, B. H-7 Effects on the Structure and Fluid Conductance of Monkey Trabecular Meshwork. Arch. Ophthalmol. 2000, 118, 955–962. [Google Scholar]
- Vittitow, J.L.; Garg, R.; Rowlette, L.-L.S.; Epstein, D.L.; O’Brien, E.T.; Borrás, T. Gene transfer of dominant-negative RhoA increases outflow facility in perfused human anterior segment cultures. Mol. Vis. 2002, 8, 32–44. [Google Scholar]
- Lütjen-Drecoll, E. Functional morphology of the trabecular meshwork in primate eyes. Prog. Retin. Eye Res. 1999, 18, 91–119. [Google Scholar] [CrossRef]
- Epstein, D.L.; Rowlette, L.L.; Roberts, B.C. Acto-myosin drug effects and aqueous outflow function. Investig. Ophthalmol. Vis. Sci. 1999, 40, 74–81. [Google Scholar]
- Rönkkö, S.; Rekonen, P.; Kaarniranta, K.; Puustjärvi, T.; Teräsvirta, M.; Uusitalo, H. Matrix metalloproteinases and their inhibitors in the chamber angle of normal eyes and patients with primary open-angle glaucoma and exfoliation glaucoma. Graefe’s Arch. Clin. Exp. Ophthalmol. 2007, 245, 697–704. [Google Scholar] [CrossRef]
- Bradley, J.M.; Vranka, J.; Colvis, C.M.; Conger, D.M.; Alexander, J.P.; Fisk, A.S.; Samples, J.R.; Acott, T.S. Effect of matrix metalloproteinases activity on outflow in perfused human organ culture. Investig. Ophthalmol. Vis. Sci. 1998, 39, 2649–2658. [Google Scholar]
- Rhee, D.J.; Haddadin, R.I.; Kang, M.H.; Oh, D.-J. Matricellular proteins in the trabecular meshwork. Exp. Eye Res. 2009, 88, 694–703. [Google Scholar] [CrossRef]
- Downs, J.C.; Roberts, M.D.; Sigal, I.A. Glaucomatous Cupping of the Lamina Cribrosa: A Review of the Evidence for Active Progressive Remodeling as a Mechanism. Exp. Eye Res 2011, 93, 133–140. [Google Scholar] [CrossRef] [Green Version]
- Tomarev, S.I.; Wistow, G.; Raymond, V.; Dubois, S.; Malyukova, I. Gene Expression Profile of the Human Trabecular Meshwork: NEIBank Sequence Tag Analysis. Investig. Ophthalmol. Vis. Sci. 2003, 44, 2588–2596. [Google Scholar] [CrossRef] [Green Version]
- Kimura, S.; Suzuki, K.; Sagara, T.; Nishida, T.; Yamamoto, T.; Kitazawa, Y. Regulation of Connexin Phosphorylation and Cell–Cell Coupling in Trabecular Meshwork Cells. Investig. Ophthalmol. Vis. Sci. 2000, 41, 2222–2228. [Google Scholar]
- Goodenough, D.A.; Goliger, J.A.; Paul, D.L. Connexins, Connexons, and Intercellular Communication. Annu. Rev. Biochem. 1996, 65, 475–502. [Google Scholar] [CrossRef]
- Tellios, N.; Feng, M.; Chen, N.; Liu, H.; Tellios, V.; Wang, M.; Li, X.; Chang, C.A.; Hutnik, C. Mechanical stretch upregulates connexin43 in human trabecular meshwork cells. Clin. Exp. Ophthalmol. 2019, 47, 787–794. [Google Scholar] [CrossRef]
- Zhou, M.; Lu, B.; Tan, W.; Fu, M. Identification of lncRNA–miRNA–mRNA regulatory network associated with primary open angle glaucoma. BMC Ophthalmol. 2020, 20, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Yoon, J.-H.; Abdelmohsen, K.; Gorospe, M. Functional interactions among microRNAs and long noncoding RNAs. Semin. Cell Dev. Biol. 2014, 34, 9–14. [Google Scholar] [CrossRef] [Green Version]
- Pasquale, L.R.; Loomis, S.J.; Kang, J.H.; Yaspan, B.L.; Abdrabou, W.; Budenz, D.L.; Chen, T.C.; DelBono, E.; Friedman, D.S.; Gaasterland, D.; et al. CDKN2B-AS1 Genotype–Glaucoma Feature Correlations in Primary Open-Angle Glaucoma Patients from the United States. Am. J. Ophthalmol. 2013, 155, 342–353. [Google Scholar] [CrossRef] [Green Version]
- Wiggs, J.L.; Yaspan, B.L.; Hauser, M.A.; Kang, J.H.; Allingham, R.R.; Olson, L.M.; Abdrabou, W.; Fan, B.J.; Wang, D.Y.; Brodeur, W.; et al. Common Variants at 9p21 and 8q22 Are Associated with Increased Susceptibility to Optic Nerve Degeneration in Glaucoma. PLoS Genet. 2012, 8, e1002654. [Google Scholar] [CrossRef] [Green Version]
- Li, F.; Wen, X.; Zhang, H.; Fan, X. Novel Insights into the Role of Long Noncoding RNA in Ocular Diseases. Int. J. Mol. Sci. 2016, 17, 478. [Google Scholar] [CrossRef] [Green Version]
- Xie, L.; Mao, M.; Wang, C.; Zhang, L.; Pan, Z.; Shi, J.; Duan, X.; Jia, S.; Jiang, B. Potential Biomarkers for Primary Open-Angle Glaucoma Identified by Long Noncoding RNA Profiling in the Aqueous Humor. Am. J. Pathol. 2019, 189, 739–752. [Google Scholar] [CrossRef] [Green Version]
- Zhao, J.; Sun, H.; Zhang, J.-M.; Wang, M.; Du, X.-J.; Zhang, J.-L. Long non-coding RNA ANRIL down-regulates microRNA-7 to protect human trabecular meshwork cells in an experimental model for glaucoma. Eur. Rev. Med. Pharmacol. Sci. 2019, 23, 3173–3182. [Google Scholar] [CrossRef]
- Fan, C.-N.; Ma, L.; Liu, N. Systematic analysis of lncRNA–miRNA–mRNA competing endogenous RNA network identifies four-lncRNA signature as a prognostic biomarker for breast cancer. J. Transl. Med. 2018, 16. [Google Scholar] [CrossRef]
- Guo, X.; Yang, J.; Liang, B.; Shen, T.; Yan, Y.; Huang, S.; Zhou, J.; Huang, J.; Gu, L.; Su, L. Identification of Novel LncRNA Biomarkers and Construction of LncRNA-Related Networks in Han Chinese Patients with Ischemic Stroke. Cell. Physiol. Biochem. 2018, 50, 2157–2175. [Google Scholar] [CrossRef]
- Jiang, S.; Kametani, M.; Chen, D.F. Adaptive Immunity: New Aspects of Pathogenesis Underlying Neurodegeneration in Glaucoma and Optic Neuropathy. Front. Immunol. 2020, 11. [Google Scholar] [CrossRef] [Green Version]
- Meng, B.; Li, H.; Sun, X.; Qu, W.; Yang, B.; Cheng, F.; Shi, L.; Yuan, H. σ-1 receptor stimulation protects against pressure-induced damage through InsR-MAPK signaling in human trabecular meshwork cells. Mol. Med. Rep. 2017, 16, 617–624. [Google Scholar] [CrossRef] [Green Version]
- Beit-Yannai, E.; Shmulevich, A. Does the aqueous humor have a role in mitogen-activated protein kinase (MAPK) intracellular signaling in Glaucoma? Med. Hypotheses 2007, 68, 299–302. [Google Scholar] [CrossRef]
- Webber, H.C.; Bermudez, J.Y.; Millar, J.C.; Mao, W.; Clark, A.F. The Role of Wnt/β-Catenin Signaling and K-Cadherin in the Regulation of Intraocular Pressure. Investig. Ophthalmol. Vis. Sci. 2018, 59, 1454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahadome, S.D.; Zhang, C.; Tannous, E.; Shen, J.; Zheng, J.J. Small-molecule inhibition of Wnt signaling abrogates dexamethasone-induced phenotype of primary human trabecular meshwork cells. Exp. Cell Res. 2017, 357, 116–123. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Du, X.; Wang, M.; Yang, P.; Zhang, J. Salidroside mitigates hydrogen peroxide-induced injury by enhancement of microRNA-27a in human trabecular meshwork cells. Artif. Cells Nanomed. Biotechnol. 2019, 47, 1758–1765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tay, Y.; Rinn, J.; Pandolfi, P.P. The multilayered complexity of ceRNA crosstalk and competition. Nature 2014, 505, 344–352. [Google Scholar] [CrossRef] [Green Version]
- McKinnon, S.J.; Goldberg, L.D.; Peeples, P.; Walt, J.G.; Bramley, T.J. Current management of glaucoma and the need for complete therapy. Am. J. Manag. Care 2008, 14, S20–S27. [Google Scholar]
- Saccà, S.C.; Vernazza, S.; Iorio, E.L.; Tirendi, S.; Bassi, A.M.; Gandolfi, S.; Izzotti, A. Molecular changes in glaucomatous trabecular meshwork. Correlations with retinal ganglion cell death and novel strategies for neuroprotection. Prog. Brain Res. 2020, 256, 151–188. [Google Scholar]
- Bouhenni, R.; Dunmire, J.; Sewell, A.; Edward, D.P. Animal Models of Glaucoma. J. Biomed. Biotechnol. 2012, 2012. [Google Scholar] [CrossRef] [Green Version]
- Sappington, R.M.; Sidorova, T.; Long, D.J.; Calkins, D.J. TRPV1: Contribution to Retinal Ganglion Cell Apoptosis and Increased Intracellular Ca2+ with Exposure to Hydrostatic Pressure. Investig. Ophthalmol. Vis. Sci. 2009, 50, 717. [Google Scholar] [CrossRef] [Green Version]
- Moreno, M.C.; Campanelli, J.; Sande, P.; Sáenz, D.A.; Keller Sarmiento, M.I.; Rosenstein, R.E. Retinal Oxidative Stress Induced by High Intraocular Pressure. Free Radic. Biol. Med. 2004, 37, 803–812. [Google Scholar] [CrossRef]
- Ferreira, S.M.; Lerner, S.F.; Brunzini, R.; Reides, C.G.; Evelson, P.A.; Llesuy, S.F. Time Course Changes of Oxidative Stress Markers in a Rat Experimental Glaucoma Model. Investig. Ophthalmol. Vis. Sci. 2010, 51, 4635. [Google Scholar] [CrossRef] [Green Version]
- Henein, C.; Khaw, P.T. The interplay between inflammation, immunity and commensal microflora in glaucomatous neurodegeneration. Ann. Eye Sci. 2019, 4, 10. [Google Scholar] [CrossRef]
- Wostyn, P.; Audenaert, K.; De Deyn, P.P. Alzheimer’s disease and glaucoma: Is there a causal relationship? Br. J. Ophthalmol. 2009, 93, 1557–1559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cesareo, M.; Martucci, A.; Ciuffoletti, E.; Mancino, R.; Cerulli, A.; Sorge, R.P.; Martorana, A.; Sancesario, G.; Nucci, C. Association Between Alzheimer’s Disease and Glaucoma: A Study Based on Heidelberg Retinal Tomography and Frequency Doubling Technology Perimetry. Front. Neurosci. 2015, 9, 479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yücel, Y. Effects of retinal ganglion cell loss on magno-, parvo-, koniocellular pathways in the lateral geniculate nucleus and visual cortex in glaucoma. Prog. Retin. Eye Res. 2003, 22, 465–481. [Google Scholar] [CrossRef]
- Gupta, N. Human glaucoma and neural degeneration in intracranial optic nerve, lateral geniculate nucleus, and visual cortex. Br. J. Ophthalmol. 2006, 90, 674–678. [Google Scholar] [CrossRef]
- Weber, A.J.; Chen, H.; Hubbard, W.C.; Kaufman, P.L. Experimental Glaucoma and Cell Size, Density, and Number in the Primate Lateral Geniculate Nucleus. Investig. Ophthalmol. Vis. Sci. 2000, 41, 1370–1379. [Google Scholar]
- Quigley, H.A. Blockade of Rapid Axonal Transport: Effect of Intraocular Pressure Elevation in Primate Optic Nerve. Arch. Ophthalmol. 1979, 97, 525. [Google Scholar] [CrossRef]
- Takihara, Y.; Inatani, M.; Eto, K.; Inoue, T.; Kreymerman, A.; Miyake, S.; Ueno, S.; Nagaya, M.; Nakanishi, A.; Iwao, K.; et al. In vivo imaging of axonal transport of mitochondria in the diseased and aged mammalian CNS. Proc. Natl. Acad. Sci. USA 2015, 112, 10515–10520. [Google Scholar] [CrossRef] [Green Version]
- Spear, P.D.; Kim, C.B.Y.; Ahmad, A.; Tom, B.W. Relationship between numbers of retinal ganglion cells and lateral geniculate neurons in the rhesus monkey. Vis. Neurosci. 1996, 13, 199–203. [Google Scholar] [CrossRef]
- Gupta, N.; Yücel, Y.H. Glaucoma as a neurodegenerative disease. Curr. Opin. Ophthalmol. 2007, 18, 110–114. [Google Scholar] [CrossRef]
- Quigley, H.A.; Hohman, R.M.; Addicks, E.M.; Massof, R.W.; Green, W.R. Morphologic Changes in the Lamina Cribrosa Correlated with Neural Loss in Open-Angle Glaucoma. Am. J. Ophthalmol. 1983, 95, 673–691. [Google Scholar] [CrossRef]
- Perlson, E.; Maday, S.; Fu, M.; Moughamian, A.J.; Holzbaur, E.L.F. Retrograde axonal transport: Pathways to cell death? Trends Neurosci. 2010, 33, 335–344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fahy, E.T.; Chrysostomou, V.; Crowston, J.G. Impaired Axonal Transport and Glaucoma. Curr. Eye Res. 2015, 1–11. [Google Scholar] [CrossRef]
- Harada, C.; Harada, T.; Quah, H.-M.A.; Namekata, K.; Yoshida, K.; Ohno, S.; Tanaka, K.; Parada, L.F. Role of Neurotrophin-4/5 in Neural Cell Death during Retinal Development and Ischemic Retinal Injury In Vivo. Investig. Ophthalmol. Vis. Sci. 2005, 46, 669–673. [Google Scholar] [CrossRef]
- Johnson, E.C.; Guo, Y.; Cepurna, W.O.; Morrison, J.C. Neurotrophin roles in retinal ganglion cell survival: Lessons from rat glaucoma models. Exp. Eye Res. 2009, 88, 808–815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ren, R.; Jonas, J.B.; Tian, G.; Zhen, Y.; Ma, K.; Li, S.; Wang, H.; Li, B.; Zhang, X.; Wang, N. Cerebrospinal Fluid Pressure in Glaucoma. Ophthalmology 2010, 117, 259–266. [Google Scholar] [CrossRef]
- Hernandez, M.R. The optic nerve head in glaucoma: Role of astrocytes in tissue remodeling. Prog. Retin. Eye Res. 2000, 19, 297–321. [Google Scholar] [CrossRef]
- Tezel, G.; Wax, M.B. Increased Production of Tumor Necrosis Factor-α by Glial Cells Exposed to Simulated Ischemia or Elevated Hydrostatic Pressure Induces Apoptosis in Cocultured Retinal Ganglion Cells. J. Neurosci. 2000, 20, 8693–8700. [Google Scholar] [CrossRef]
- Yuan, L.; Neufeld, A.H. Tumor necrosis factor-α: A potentially neurodestructive cytokine produced by glia in the human glaucomatous optic nerve head. Glia 2000, 32, 42–50. [Google Scholar] [CrossRef]
- Chevalier-Larsen, E.; Holzbaur, E.L.F. Axonal transport and neurodegenerative disease. Biochim. Et Biophys. Acta Mol. Basis Dis. 2006, 1762, 1094–1108. [Google Scholar] [CrossRef] [Green Version]
- Crish, S.D.; Sappington, R.M.; Inman, D.M.; Horner, P.J.; Calkins, D.J. Distal axonopathy with structural persistence in glaucomatous neurodegeneration. Proc. Natl. Acad. Sci. USA 2010, 107, 5196–5201. [Google Scholar] [CrossRef] [Green Version]
- Ashhurst, T.M.; van Vreden, C.; Niewold, P.; King, N.J.C. The plasticity of inflammatory monocyte responses to the inflamed central nervous system. Cell. Immunol. 2014, 291, 49–57. [Google Scholar] [CrossRef]
- Glass, C.K.; Saijo, K.; Winner, B.; Marchetto, M.C.; Gage, F.H. Mechanisms Underlying Inflammation in Neurodegeneration. Cell 2010, 140, 918–934. [Google Scholar] [CrossRef] [Green Version]
- Magni, P.; Ruscica, M.; Dozio, E.; Rizzi, E.; Beretta, G.; Facino, R.M. Parthenolide Inhibits the LPS-induced Secretion of IL-6 and TNF-α and NF-κB Nuclear Translocation in BV-2 Microglia: PARTHENOLIDE INHIBITION OF BV-2 MICROGLIA. Phytother. Res. 2012, 26, 1405–1409. [Google Scholar] [CrossRef]
- Wei, X.; Yu, Z.; Cho, K.-S.; Chen, H.; Malik, M.T.A.; Chen, X.; Lo, E.H.; Wang, X.; Chen, D.F. Neuroglobin Is an Endogenous Neuroprotectant for Retinal Ganglion Cells against Glaucomatous Damage. Am. J. Pathol. 2011, 179, 2788–2797. [Google Scholar] [CrossRef]
- Bosco, A.; Crish, S.D.; Steele, M.R.; Romero, C.O.; Inman, D.M.; Horner, P.J.; Calkins, D.J.; Vetter, M.L. Early Reduction of Microglia Activation by Irradiation in a Model of Chronic Glaucoma. PLoS ONE 2012, 7, e43602. [Google Scholar] [CrossRef]
- Howell, G.R.; Macalinao, D.G.; Sousa, G.L.; Walden, M.; Soto, I.; Kneeland, S.C.; Barbay, J.M.; King, B.L.; Marchant, J.K.; Hibbs, M.; et al. Molecular clustering identifies complement and endothelin induction as early events in a mouse model of glaucoma. J. Clin. Investig. 2011, 121, 1429–1444. [Google Scholar] [CrossRef]
- Ebneter, A.; Casson, R.J.; Wood, J.P.M.; Chidlow, G. Microglial Activation in the Visual Pathway in Experimental Glaucoma: Spatiotemporal Characterization and Correlation with Axonal Injury. Investig. Ophthalmol. Vis. Sci. 2010, 51, 6448. [Google Scholar] [CrossRef] [Green Version]
- Kim, W.-K.; Hwang, S.-Y.; Oh, E.-S.; Piao, H.Z.; Kim, K.-W.; Han, I.-O. TGF-β1 Represses Activation and Resultant Death of Microglia via Inhibition of Phosphatidylinositol 3-Kinase Activity. J. Immunol. 2004, 172, 7015–7023. [Google Scholar] [CrossRef] [Green Version]
- Le, Y.; Iribarren, P.; Gong, W.; Cui, Y.; Zhang, X.; Wang, J.M. TGF-β1 Disrupts Endotoxin Signaling in Microglial Cells through Smad3 and MAPK Pathways. J. Immunol. 2004, 173, 962–968. [Google Scholar] [CrossRef] [Green Version]
- Zhou, X.; Zöller, T.; Krieglstein, K.; Spittau, B. TGFβ1 inhibits IFNγ-mediated microglia activation and protects mDA neurons from IFNγ-driven neurotoxicity. J. Neurochem. 2015, 134, 125–134. [Google Scholar] [CrossRef] [PubMed]
- Spittau, B.; Dokalis, N.; Prinz, M. The Role of TGFβ Signaling in Microglia Maturation and Activation. Trends Immunol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Shull, M.M.; Ormsby, I.; Kier, A.B.; Pawlowski, S.; Diebold, R.J.; Yin, M.; Allen, R.; Sidman, C.; Proetzel, G.; Calvin, D. Targeted disruption of the mouse transforming growth factor-β1 gene results in multifocal inflammatory disease. Nature 1992, 359, 693–699. [Google Scholar] [CrossRef]
- Spittau, B. Aging Microglia—Phenotypes, Functions and Implications for Age-Related Neurodegenerative Diseases. Front. Aging Neurosci. 2017, 9. [Google Scholar] [CrossRef]
- van Eden, W.; Jansen, M.A.A.; Ludwig, I.; van Kooten, P.; van der Zee, R.; Broere, F. The Enigma of Heat Shock Proteins in Immune Tolerance. Front. Immunol. 2017, 8. [Google Scholar] [CrossRef]
- Jin, C.; Cleveland, J.C.; Ao, L.; Li, J.; Zeng, Q.; Fullerton, D.A.; Meng, X. Human Myocardium Releases Heat Shock Protein 27 (HSP27) after Global Ischemia: The Proinflammatory Effect of Extracellular HSP27 through Toll-like Receptor (TLR)-2 and TLR4. Mol. Med. 2014, 20, 280–289. [Google Scholar] [CrossRef]
- Rosenberger, K.; Dembny, P.; Derkow, K.; Engel, O.; Krüger, C.; Wolf, S.A.; Kettenmann, H.; Schott, E.; Meisel, A.; Lehnardt, S. Intrathecal heat shock protein 60 mediates neurodegeneration and demyelination in the CNS through a TLR4- and MyD88-dependent pathway. Mol. Neurodegener. 2015, 10, 5. [Google Scholar] [CrossRef] [Green Version]
- Swaroop, S.; Sengupta, N.; Suryawanshi, A.R.; Adlakha, Y.K.; Basu, A. HSP60 plays a regulatory role in IL-1β-induced microglial inflammation via TLR4-p38 MAPK axis. J. Neuroinflamm. 2016, 13. [Google Scholar] [CrossRef] [Green Version]
- Murakami, Y.; Ishikawa, K.; Nakao, S.; Sonoda, K.-H. Innate immune response in retinal homeostasis and inflammatory disorders. Prog. Retin. Eye Res. 2020, 74, 100778. [Google Scholar] [CrossRef]
- Solà-Villà, D.; Camacho, M.; Solà, R.; Soler, M.; Diaz, J.-M.; Vila, L. IL-1β induces VEGF, independently of PGE2 induction, mainly through the PI3-K/mTOR pathway in renal mesangial cells. Kidney Int. 2006, 70, 1935–1941. [Google Scholar] [CrossRef] [Green Version]
- Pavilack, M.A.; Elner, V.M.; Elner, S.G.; Todd, R.F.; Huber, A.R. Differential expression of human corneal and perilimbal ICAM-1 by inflammatory cytokines. Investig. Ophthalmol. Vis. Sci. 1992, 33, 564–573. [Google Scholar]
- Khan, S.; Cole, N.; Hume, E.B.; Garthwaite, L.; Conibear, T.C.R.; Miles, D.H.; Aliwaga, Y.; Krockenberger, M.B.; Willcox, M.D.P. The role of CXC chemokine receptor 2 in Pseudomonas aeruginosa corneal infection. J. Leukoc. Biol. 2007, 81, 315–318. [Google Scholar] [CrossRef] [PubMed]
- Yerramothu, P.; Vijay, A.K.; Willcox, M.D.P. Inflammasomes, the eye and anti-inflammasome therapy. Eye 2018, 32, 491–505. [Google Scholar] [CrossRef] [PubMed]
- Nimmerjahn, A. Resting Microglial Cells Are Highly Dynamic Surveillants of Brain Parenchyma in Vivo. Science 2005, 308, 1314–1318. [Google Scholar] [CrossRef] [Green Version]
- Kigerl, K.A.; de Rivero Vaccari, J.P.; Dietrich, W.D.; Popovich, P.G.; Keane, R.W. Pattern recognition receptors and central nervous system repair. Exp. Neurol. 2014, 258, 5–16. [Google Scholar] [CrossRef] [Green Version]
- Rojas, B.; Gallego, B.I.; Ramírez, A.I.; Salazar, J.J.; de Hoz, R.; Valiente-Soriano, F.J.; Avilés-Trigueros, M.; Villegas-Perez, M.P.; Vidal-Sanz, M.; Triviño, A.; et al. Microglia in mouse retina contralateral to experimental glaucoma exhibit multiple signs of activation in all retinal layers. J. Neuroinflamm. 2014, 11, 133. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Song, X.; Li, M.; Wang, X.; Tao, Y.; Xiya, X.; Liu, H.; Zhao, Y.; Chang, D.; Sha, Q. The role of TLR4/NF-κB signaling pathway in activated microglia of rats with chronic high intraocular pressure and vitro scratch injury-induced microglia. Int. Immunopharmacol. 2020, 83, 106395. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y. Editorial: Microglial Polarization in the Pathogenesis and Therapeutics of Neurodegenerative Diseases. Front. Aging Neurosci. 2018, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varnum, M.M.; Ikezu, T. The Classification of Microglial Activation Phenotypes on Neurodegeneration and Regeneration in Alzheimer’s Disease Brain. Arch. Immunol. Ther. Exp. 2012, 60, 251–266. [Google Scholar] [CrossRef] [PubMed]
- González, H.; Elgueta, D.; Montoya, A.; Pacheco, R. Neuroimmune regulation of microglial activity involved in neuroinflammation and neurodegenerative diseases. J. Neuroimmunol. 2014, 274, 1–13. [Google Scholar] [CrossRef]
- Jones, E.V.; Bouvier, D.S. Astrocyte-Secreted Matricellular Proteins in CNS Remodelling during Development and Disease. Neural Plast. 2014, 2014, 321209. [Google Scholar] [CrossRef] [Green Version]
- Ding, Y.M.; Jaumotte, J.D.; Signore, A.P.; Zigmond, M.J. Effects of 6-hydroxydopamine on primary cultures of substantia nigra: Specific damage to dopamine neurons and the impact of glial cell line-derived neurotrophic factor. J. Neurochem. 2004, 89, 776–787. [Google Scholar] [CrossRef]
- Simard, A.R.; Soulet, D.; Gowing, G.; Julien, J.-P.; Rivest, S. Bone Marrow-Derived Microglia Play a Critical Role in Restricting Senile Plaque Formation in Alzheimer’s Disease. Neuron 2006, 49, 489–502. [Google Scholar] [CrossRef] [Green Version]
- Glezer, I.; Simard, A.R.; Rivest, S. Neuroprotective role of the innate immune system by microglia. Neuroscience 2007, 147, 867–883. [Google Scholar] [CrossRef]
- Block, M.L.; Zecca, L.; Hong, J.-S. Microglia-mediated neurotoxicity: Uncovering the molecular mechanisms. Nat. Rev. Neurosci. 2007, 8, 57–69. [Google Scholar] [CrossRef]
- Lull, M.E.; Block, M.L. Microglial activation and chronic neurodegeneration. Neurotherapeutics 2010, 7, 354–365. [Google Scholar] [CrossRef] [Green Version]
- Burguillos, M.A.; Deierborg, T.; Kavanagh, E.; Persson, A.; Hajji, N.; Garcia-Quintanilla, A.; Cano, J.; Brundin, P.; Englund, E.; Venero, J.L.; et al. Caspase signalling controls microglia activation and neurotoxicity. Nature 2011, 472, 319–324. [Google Scholar] [CrossRef]
- Kettenmann, H.; Hanisch, U.-K.; Noda, M.; Verkhratsky, A. Physiology of Microglia. Physiol. Rev. 2011, 91, 461–553. [Google Scholar] [CrossRef]
- Suh, H.-S.; Zhao, M.-L.; Derico, L.; Choi, N.; Lee, S.C. Insulin-like growth factor 1 and 2 (IGF1, IGF2) expression in human microglia: Differential regulation by inflammatory mediators. J. Neuroinflamm. 2013, 10. [Google Scholar] [CrossRef] [Green Version]
- Tang, Y.; Le, W. Differential Roles of M1 and M2 Microglia in Neurodegenerative Diseases. Mol. Neurobiol. 2016, 53, 1181–1194. [Google Scholar] [CrossRef]
- Ramirez, A.I.; de Hoz, R.; Salobrar-Garcia, E.; Salazar, J.J.; Rojas, B.; Ajoy, D.; López-Cuenca, I.; Rojas, P.; Triviño, A.; Ramírez, J.M. The Role of Microglia in Retinal Neurodegeneration: Alzheimer’s Disease, Parkinson, and Glaucoma. Front. Aging Neurosci. 2017, 9. [Google Scholar] [CrossRef] [Green Version]
- Koizumi, S.; Shigemoto-Mogami, Y.; Nasu-Tada, K.; Shinozaki, Y.; Ohsawa, K.; Tsuda, M.; Joshi, B.V.; Jacobson, K.A.; Kohsaka, S.; Inoue, K. UDP acting at P2Y6 receptors is a mediator of microglial phagocytosis. Nature 2007, 446, 1091–1095. [Google Scholar] [CrossRef] [Green Version]
- Wu, L.-J.; Vadakkan, K.I.; Zhuo, M. ATP-induced chemotaxis of microglial processes requires P2Y receptor-activated initiation of outward potassium currents. Glia 2007, 55, 810–821. [Google Scholar] [CrossRef]
- Nakazawa, T.; Nakazawa, C.; Matsubara, A.; Noda, K.; Hisatomi, T.; She, H.; Michaud, N.; Hafezi-Moghadam, A.; Miller, J.W.; Benowitz, L.I. Tumor Necrosis Factor- Mediates Oligodendrocyte Death and Delayed Retinal Ganglion Cell Loss in a Mouse Model of Glaucoma. J. Neurosci. 2006, 26, 12633–12641. [Google Scholar] [CrossRef] [Green Version]
- Neufeld, A.H.; Liu, B. Glaucomatous Optic Neuropathy: When Glia Misbehave. Neurosci. 2003, 9, 485–495. [Google Scholar] [CrossRef]
- Soto, I.; Howell, G.R. The Complex Role of Neuroinflammation in Glaucoma. Cold Spring Harb. Perspect. Med. 2014, 4, a017269. [Google Scholar] [CrossRef]
- Bosco, A.; Inman, D.M.; Steele, M.R.; Wu, G.; Soto, I.; Marsh-Armstrong, N.; Hubbard, W.C.; Calkins, D.J.; Horner, P.J.; Vetter, M.L. Reduced Retina Microglial Activation and Improved Optic Nerve Integrity with Minocycline Treatment in the DBA/2J Mouse Model of Glaucoma. Investig. Ophthalmol. Vis. Sci. 2008, 49, 1437. [Google Scholar] [CrossRef]
- Bosco, A.; Romero, C.O.; Breen, K.T.; Chagovetz, A.A.; Steele, M.R.; Ambati, B.K.; Vetter, M.L. Neurodegeneration severity can be predicted from early microglia alterations monitored in vivo in a mouse model of chronic glaucoma. Dis. Models Mech. 2015, 8, 443–455. [Google Scholar] [CrossRef] [Green Version]
- Colombo, E.; Farina, C. Astrocytes: Key Regulators of Neuroinflammation. Trends Immunol. 2016, 37, 608–620. [Google Scholar] [CrossRef]
- Sofroniew, M.V. Molecular dissection of reactive astrogliosis and glial scar formation. Trends Neurosci. 2009, 32, 638–647. [Google Scholar] [CrossRef] [Green Version]
- Hatten, M.E.; Liem, R.K.H.; Shelanski, M.L.; Mason, C.A. Astroglia in CNS injury. Glia 1991, 4, 233–243. [Google Scholar] [CrossRef]
- Yan, X. Matrix Metalloproteinases and Tumor Necrosis Factor α in Glaucomatous Optic Nerve Head. Arch. Ophthalmol. 2000, 118, 666. [Google Scholar] [CrossRef] [Green Version]
- Davey, P. Glaucoma: Current Clinical and Research Aspects; BoD—Books on Demand: Norderstedt, Germany, 2011; ISBN 978-953-307-263-0. [Google Scholar]
- Mélik Parsadaniantz, S.; Réaux-le Goazigo, A.; Sapienza, A.; Habas, C.; Baudouin, C. Glaucoma: A Degenerative Optic Neuropathy Related to Neuroinflammation? Cells 2020, 9, 535. [Google Scholar] [CrossRef] [Green Version]
- Hu, B.; Yip, H.K.; So, K.-F. Localization of p75 neurotrophin receptor in the retina of the adult SD rat: An immunocytochemical study at light and electron microscopic levels. Glia 1998, 24, 187–197. [Google Scholar] [CrossRef]
- Lebrun-Julien, F.; Morquette, B.; Douillette, A.; Saragovi, H.U.; Di Polo, A. Inhibition of p75NTR in glia potentiates TrkA-mediated survival of injured retinal ganglion cells. Mol. Cell. Neurosci. 2009, 40, 410–420. [Google Scholar] [CrossRef]
- Bringmann, A.; Iandiev, I.; Pannicke, T.; Wurm, A.; Hollborn, M.; Wiedemann, P.; Osborne, N.N.; Reichenbach, A. Cellular signaling and factors involved in Müller cell gliosis: Neuroprotective and detrimental effects. Prog. Retin. Eye Res. 2009, 28, 423–451. [Google Scholar] [CrossRef]
- Bringmann, A.; Pannicke, T.; Grosche, J.; Francke, M.; Wiedemann, P.; Skatchkov, S.; Osborne, N.; Reichenbach, A. Müller cells in the healthy and diseased retina. Prog. Retin. Eye Res. 2006, 25, 397–424. [Google Scholar] [CrossRef]
- Garcia, T.B.; Pannicke, T.; Vogler, S.; Berk, B.-A.; Grosche, A.; Wiedemann, P.; Seeger, J.; Reichenbach, A.; Herculano, A.M.; Bringmann, A. Nerve growth factor inhibits osmotic swelling of rat retinal glial (Müller) and bipolar cells by inducing glial cytokine release. J. Neurochem. 2014, 131, 303–313. [Google Scholar] [CrossRef] [Green Version]
- Xu, F.; Wei, Y.; Lu, Q.; Zheng, D.; Zhang, F.; Gao, E.; Wang, N. Immunohistochemical localization of sortilin and p75NTR in normal and ischemic rat retina. Neurosci. Lett. 2009, 454, 81–85. [Google Scholar] [CrossRef]
- Bringmann, A.; Uckermann, O.; Pannicke, T.; Iandiev, I.; Reichenbach, A.; Wiedemann, P. Neuronal versus glial cell swelling in the ischaemic retina: Acta Ophthalmologica Scandinavica 2005. Acta Ophthalmol. Scand. 2005, 83, 528–538. [Google Scholar] [CrossRef]
- Hernandez, M.R.; Miao, H.; Lukas, T. Astrocytes in glaucomatous optic neuropathy. In Progress in Brain Research; Elsevier: Amsterdam, The Netherlands, 2008; Volume 173, pp. 353–373. ISBN 978-0-444-53256-5. [Google Scholar]
- Kerr, N.M.; Johnson, C.S.; Green, C.R.; Danesh-Meyer, H.V. Gap junction protein connexin43 (GJA1) in the human glaucomatous optic nerve head and retina. J. Clin. Neurosci. 2011, 18, 102–108. [Google Scholar] [CrossRef]
- Tomimoto, H.; Akiguchi, I.; Wakita, H.; Suenaga, T.; Nakamura, S.; Kimura, J. Regressive changes of astroglia in white matter lesions in cerebrovascular disease and Alzheimer’s disease patients. Acta Neuropathol. 1997, 94, 146–152. [Google Scholar] [CrossRef]
- Mac Nair, C.E.; Nickells, R.W. Neuroinflammation in Glaucoma and Optic Nerve Damage. In Progress in Molecular Biology and Translational Science; Elsevier: Amsterdam, The Netherlands, 2015; Volume 134, pp. 343–363. ISBN 978-0-12-801059-4. [Google Scholar]
- Arroba, A.I.; Campos-Caro, A.; Aguilar-Diosdado, M.; Valverde, Á.M. IGF-1, Inflammation and Retinal Degeneration: A Close Network. Front. Aging Neurosci. 2018, 10. [Google Scholar] [CrossRef]
- Nanetti, L.; Taffi, R.; Vignini, A.; Moroni, C.; Raffaelli, F.; Bacchetti, T.; Silvestrini, M.; Provinciali, L.; Mazzanti, L. Reactive oxygen species plasmatic levels in ischemic stroke. Mol. Cell. Biochem. 2007, 303, 19–25. [Google Scholar] [CrossRef]
- Williams, P.A.; Marsh-Armstrong, N.; Howell, G.R.; Bosco, A.; Danias, J.; Simon, J.; Di Polo, A.; Kuehn, M.H.; Przedborski, S.; Raff, M.; et al. Neuroinflammation in glaucoma: A new opportunity. Exp. Eye Res. 2017, 157, 20–27. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, F.; Brown, K.M.; Stephan, D.A.; Morrison, J.C.; Johnson, E.C.; Tomarev, S.I. Microarray Analysis of Changes in mRNA Levels in the Rat Retina after Experimental Elevation of Intraocular Pressure. Investig. Ophthalmol. Vis. Sci. 2004, 45, 1247. [Google Scholar] [CrossRef] [Green Version]
- Howell, G.R.; Soto, I.; Zhu, X.; Ryan, M.; Macalinao, D.G.; Sousa, G.L.; Caddle, L.B.; MacNicoll, K.H.; Barbay, J.M.; Porciatti, V.; et al. Radiation treatment inhibits monocyte entry into the optic nerve head and prevents neuronal damage in a mouse model of glaucoma. J. Clin. Investig. 2012, 122, 1246–1261. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z.; Quigley, H.A.; Pease, M.E.; Yang, Y.; Qian, J.; Valenta, D.; Zack, D.J. Changes in Gene Expression in Experimental Glaucoma and Optic Nerve Transection: The Equilibrium between Protective and Detrimental Mechanisms. Investig. Ophthalmol. Vis. Sci. 2007, 48, 5539. [Google Scholar] [CrossRef]
- Pinazo-Duran, M.D.; Muñoz-Negrete, F.J.; Sanz-Gonzalez, S.; Benitez-del-Castillo, J.; Gimenez-Gomez, R.; Valero-Vello, M.; Zanon-Moreno, V.; Garcia-Medina, J.J. The Role of Neuroinflammation in the Pathogenesis of Glaucoma Neurodegeneration. In Progress in Brain Research; Elsevier: Amsterdam, The Netherlands, 2020. [Google Scholar]
- Tezel, G.; Wax, M.B. Glaucoma. In Chemical Immunology and Allergy; Niederkorn, J.Y., Kaplan, H.J., Eds.; KARGER: Basel, Switzerland, 2007; pp. 221–227. ISBN 978-3-8055-8187-5. [Google Scholar]
- Grus, F.H.; Joachim, S.C.; Wuenschig, D.; Rieck, J.; Pfeiffer, N. Autoimmunity and Glaucoma. J. Glaucoma 2008, 17, 79–84. [Google Scholar] [CrossRef]
- Grus, F.H.; Joachim, S.C.; Hoffmann, E.M.; Pfeiffer, N. Complex autoantibody repertoires in patients with glaucoma. Mol. Vis. 2004, 10, 13–17. [Google Scholar]
- Grus, F.; Joachim, S.; Pfeiffer, N. Diagnosis of Glaucoma by Complex Autoantibody Repertoires in Body Fluids. PCT Patent WO2004036220A1, 29 April 2004. [Google Scholar]
- Joachim, S.C.; Grus, F.H.; Pfeiffer, N. Analysis of Autoantibody Repertoires in Sera of Patients with Glaucoma. Eur. J. Ophthalmol. 2003, 13, 752–758. [Google Scholar] [CrossRef]
- Yang, J.; Patil, R.V.; Yu, H.; Gordon, M.; Wax, M.B. T cell subsets and sIL-2R/IL-2 levels in patients with glaucoma. Am. J. Ophthalmol. 2001, 131, 421–426. [Google Scholar] [CrossRef]
- Marlottl, S.; Caturegll, P.; Barbesino, G.; Marinò, M.; Prete, G.F.; Chlovato, L.; Tonacchera, M.; Carll, M.; Pinchers, A. Thyroid function and thyroid autoimmunity independently modulate serum concentration of soluble interleukin 2 (IL-2) receptor (sIL-2R) in thyroid diseases. Clin. Endocrinol. 1992, 37, 415–422. [Google Scholar] [CrossRef]
- Yamaguchi, M.; Yamamoto, T.; Tate, G.; Matsumoto, T.; Matsumiya, A.; Kuzume, M.; Sanada, Y.; Kumada, K. Specific detection of Epstein-Barr virus in inflammatory pseudotumor of the spleen in a patient with a high serum level of soluble IL-2 receptor. J. Gastroenterol. 2000, 35, 563–566. [Google Scholar] [CrossRef]
- Ferrarini, A.M.; Sivieri, S.; Bulian, P.; Buttarello, M.; Biasi, G.; Tavolato, B.; Gallo, P. Time-course of interleukin-2 receptor expression in interferon beta-treated multiple sclerosis patients. J. Neuroimmunol. 1998, 84, 213–217. [Google Scholar] [CrossRef]
- Zorn, U.; Dallmann, I.; Groβe, J.; Kirchner, H.; Poliwoda, H.; Atzpodien, J. Soluble interleukin 2 receptors abrogate IL-2 induced activation of peripheral mononuclear cells. Cytokine 1994, 6, 358–364. [Google Scholar] [CrossRef]
- Joachim, S.C.; Bruns, K.; Lackner, K.J.; Pfeiffer, N.; Grus, F.H. Antibodies to α B-Crystallin, Vimentin, and Heat Shock Protein 70 in Aqueous Humor of Patients with Normal Tension Glaucoma and IgG Antibody Patterns Against Retinal Antigen in Aqueous Humor. Curr. Eye Res. 2007, 32, 501–509. [Google Scholar] [CrossRef]
- Wax, M.B.; Tezel, G.; Saito, I.; Gupta, R.S.; Harley, J.B.; Li, Z.; Romano, C. Anti-Ro/SS-a positivity and heat shock protein antibodies in patients with normal-pressure glaucoma. Am. J. Ophthalmol. 1998, 125, 145–157. [Google Scholar] [CrossRef]
- Chen, H.; Cho, K.-S.; Vu, T.H.K.; Shen, C.-H.; Kaur, M.; Chen, G.; Mathew, R.; McHam, M.L.; Fazelat, A.; Lashkari, K.; et al. Commensal microflora-induced T cell responses mediate progressive neurodegeneration in glaucoma. Nat. Commun. 2018, 9. [Google Scholar] [CrossRef]
- Howell, G.R.; Walton, D.O.; King, B.L.; Libby, R.T.; John, S.W. Datgan, a reusable software system for facile interrogation and visualization of complex transcription profiling data. BMC Genom. 2011, 12. [Google Scholar] [CrossRef] [Green Version]
- Gallego, B.I.; Salazar, J.J.; de Hoz, R.; Rojas, B.; Ramírez, A.I.; Salinas-Navarro, M.; Ortín-Martínez, A.; Valiente-Soriano, F.J.; Avilés-Trigueros, M.; Villegas-Perez, M.P.; et al. IOP induces upregulation of GFAP and MHC-II and microglia reactivity in mice retina contralateral to experimental glaucoma. J. Neuroinflamm. 2012, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nucci, C.; Piccirilli, S.; Nisticò, R.; Morrone, L.A.; Cerulli, L.; Bagetta, G. Apoptosis in the Mechanisms of Neuronal Plasticity in the Developing Visual System. Eur. J. Ophthalmol. 2003, 13, 36–43. [Google Scholar] [CrossRef] [PubMed]
- Danbolt, N.C. Glutamate uptake. Prog. Neurobiol. 2001, 65, 1–105. [Google Scholar] [CrossRef]
- Epstein, F.H.; Lipton, S.A.; Rosenberg, P.A. Excitatory Amino Acids as a Final Common Pathway for Neurologic Disorders. New Engl. J. Med. 1994, 330, 613–622. [Google Scholar] [CrossRef]
- Monaghan, D.T.; Bridges, R.J.; Cotman, C.W. The Excitatory Amino Acid Receptors: Their Classes, Pharmacology, and Distinct Properties in the Function of the Central Nervous System. Annu. Rev. Pharmacol. Toxicol. 1989, 29, 365–402. [Google Scholar] [CrossRef]
- Hollmann, M.; Heinemann, S. Cloned Glutamate Receptors. Annu. Rev. Neurosci. 1994, 17, 31–108. [Google Scholar] [CrossRef]
- Bannai, S.; Christensen, H.N.; Vadgama, J.V.; Ellory, J.C.; Englesberg, E.; Guidotti, G.G.; Gazzola, G.C.; Kilberg, M.S.; Lajtha, A.; Sacktor, B. Amino acid transport systems. Nature 1984, 311, 308. [Google Scholar] [CrossRef] [Green Version]
- Danbolt, N.C.; Storm-Mathisen, J.; Kanner, B.I. An [Na+ + K+] coupledl-glutamate transporter purified from rat brain is located in glial cell processes. Neuroscience 1992, 51, 295–310. [Google Scholar] [CrossRef]
- Balcar, V.J.; Shen, J.; Bao, S.; King, N.J.C. Na+ -dependent high affinity uptake of l -glutamate in primary cultures of human fibroblasts isolated from three different types of tissue. FEBS Lett. 1994, 339, 50–54. [Google Scholar] [CrossRef] [Green Version]
- Grünewald, R.A. Ascorbic acid in the brain. Brain Res. Rev. 1993, 18, 123–133. [Google Scholar] [CrossRef]
- Seki, M.; Lipton, S.A. Targeting excitotoxic/free radical signaling pathways for therapeutic intervention in glaucoma. In Progress in Brain Research; Elsevier: Amsterdam, The Netherlands, 2008; Volume 173, pp. 495–510. ISBN 978-0-444-53256-5. [Google Scholar]
- Lotery, A.J. Glutamate excitotoxicity in glaucoma: Truth or fiction? Eye 2005, 19, 369–370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conn, P.J.; Pin, J.-P. Pharmacology and functions of metabotropic glutamate receptors. Annu. Rev. Pharm. Toxicol. 1997, 37, 205–237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carter-Dawson, L.; Crawford, M.L.J.; Harwerth, R.S.; Smith, E.L.; Feldman, R.; Shen, F.F.; Mitchell, C.K.; Whitetree, A. Vitreal Glutamate Concentration in Monkeys with Experimental Glaucoma. Investig. Ophthalmol. Vis. Sci. 2002, 43, 2633–2637. [Google Scholar]
- Lee, D.; Shim, M.S.; Kim, K.-Y.; Noh, Y.H.; Kim, H.; Kim, S.Y.; Weinreb, R.N.; Ju, W.-K. Coenzyme Q10 Inhibits Glutamate Excitotoxicity and Oxidative Stress–Mediated Mitochondrial Alteration in a Mouse Model of Glaucoma. Investig. Ophthalmol. Vis. Sci. 2014, 55, 993. [Google Scholar] [CrossRef] [Green Version]
- Azuma, N.; Kawamura, M.; Kohsaka, S. Morphological and immunohistochemical studies on degenerative changes of the retina and the optic nerve in neonatal rats injected with monosodium-L-glutamate. Nippon Ganka Gakkai Zasshi 1989, 93, 72–79. [Google Scholar]
- Siliprandi, R.; Canella, R.; Carmignoto, G.; Schiavo, N.; Zanellato, A.; Zanoni, R.; Vantini, G. N-methyl-D-aspartate-induced neurotoxicity in the adult rat retina. Vis. Neurosci. 1992, 8, 567–573. [Google Scholar] [CrossRef]
- Ying, W. Deleterious Network: A Testable Pathogenetic Concept of Alzheimer’s Disease. Gerontology 1997, 43, 242–253. [Google Scholar] [CrossRef]
- Green, D.R.; Reed, J.C. Mitochondria and Apoptosis. Science 1998, 281, 1309–1312. [Google Scholar] [CrossRef]
- Savolainen, K.M.; Loikkanen, J.; Naarala, J. Amplification of glutamate-induced oxidative stress. Toxicol. Lett. 1995, 82–83, 399–405. [Google Scholar] [CrossRef]
- Mailly, F.; Marin, P.; Israël, M.; Glowinski, J.; Prémont, J. Increase in External Glutamate and NMDA Receptor Activation Contribute to H2O2-Induced Neuronal Apoptosis. J. Neurochem. 2001, 73, 1181–1188. [Google Scholar] [CrossRef] [Green Version]
- Guo, Z.H. Neurotrophic Factors Protect Cortical Synaptic Terminals Against Amyloid- and Oxidative Stress-induced Impairment of Glucose Transport, Glutamate Transport and Mitochondrial Function. Cereb. Cortex 2000, 10, 50–57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brines, M.L.; Dare, A.O.; de Lanerolle, N.C. The cardiac glycoside ouabain potentiates excitotoxic injury of adult neurons in rat hippocampus. Neurosci. Lett. 1995, 191, 145–148. [Google Scholar] [CrossRef]
- Kohmura, E.; Yamada, K.; Hayakawa, T.; Kinoshita, A.; Matsumoto, K.; Mogami, H. Hippocampal Neurons Become More Vulnerable to Glutamate after Subcritical Hypoxia: An in vitro Study. J. Cereb. Blood Flow Metab. 1990, 10, 877–884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schinder, A.F.; Olson, E.C.; Spitzer, N.C.; Montal, M. Mitochondrial Dysfunction Is a Primary Event in Glutamate Neurotoxicity. J. Neurosci. 1996, 16, 6125–6133. [Google Scholar] [CrossRef] [Green Version]
- Cheung, N.S.; Pascoe, C.J.; Giardina, S.F.; John, C.A.; Beart, P.M. Micromolar l-glutamate induces extensive apoptosis in an apoptotic-necrotic continuum of insult-dependent, excitotoxic injury in cultured cortical neurones. Neuropharmacology 1998, 37, 1419–1429. [Google Scholar] [CrossRef]
- Thompson, C. Apoptosis in the pathogenesis and treatment of disease. Science 1995, 267, 1456–1462. [Google Scholar] [CrossRef]
- Uren, A.G.; Vaux, D.L. Molecular and clinical aspects of apoptosis. Pharmacol. Ther. 1996, 72, 37–50. [Google Scholar] [CrossRef]

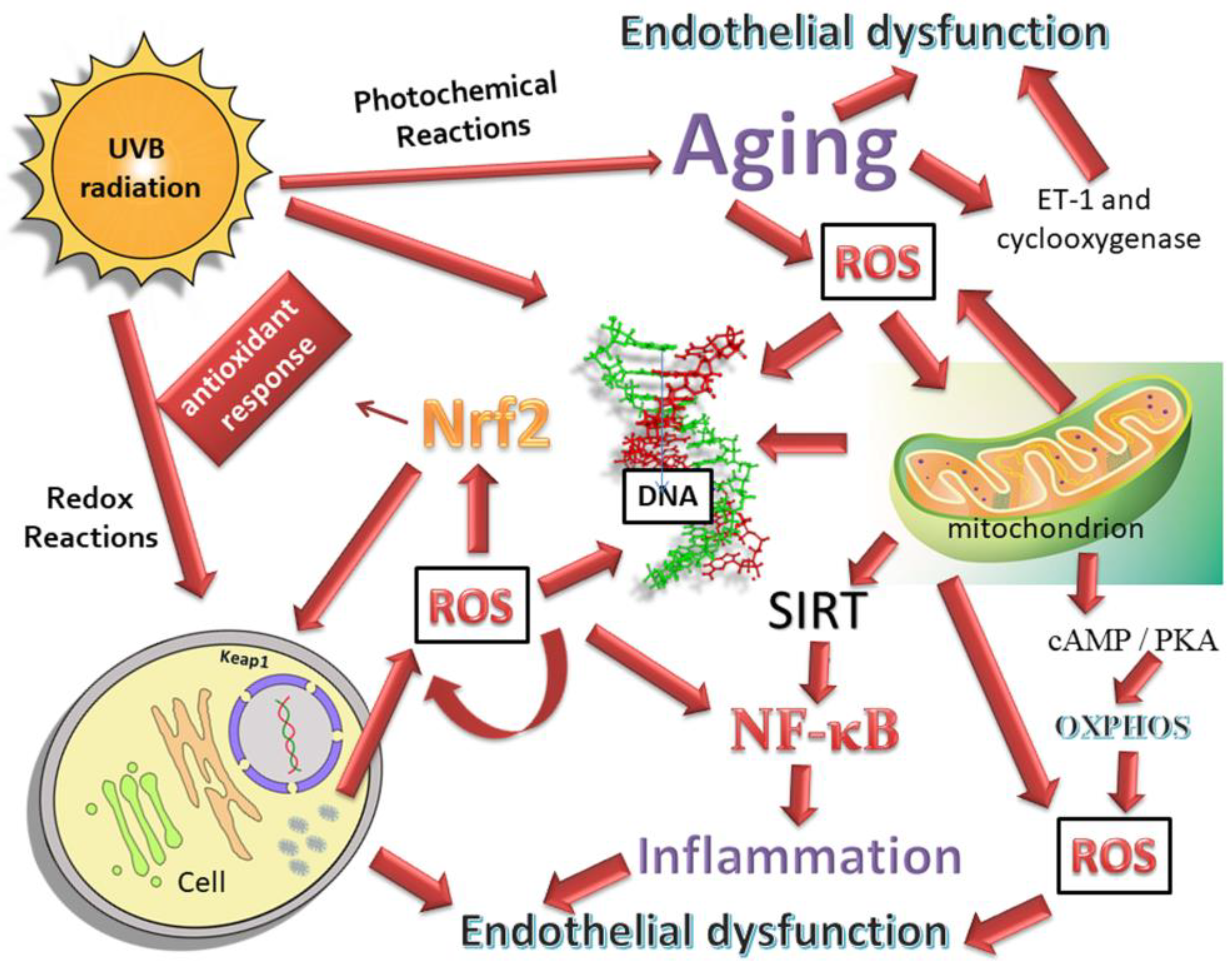
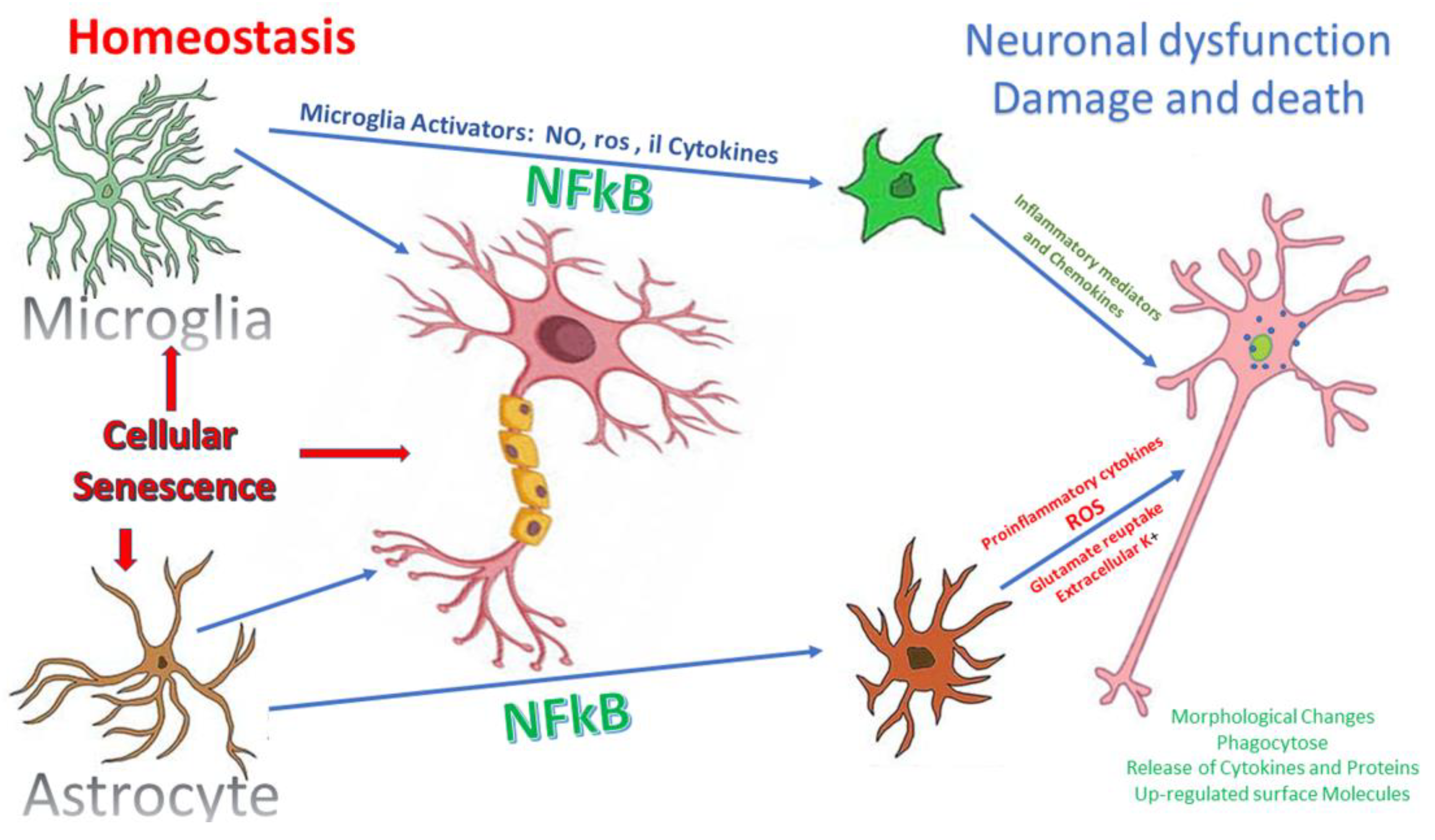
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vernazza, S.; Tirendi, S.; Bassi, A.M.; Traverso, C.E.; Saccà, S.C. Neuroinflammation in Primary Open-Angle Glaucoma. J. Clin. Med. 2020, 9, 3172. https://doi.org/10.3390/jcm9103172
Vernazza S, Tirendi S, Bassi AM, Traverso CE, Saccà SC. Neuroinflammation in Primary Open-Angle Glaucoma. Journal of Clinical Medicine. 2020; 9(10):3172. https://doi.org/10.3390/jcm9103172
Chicago/Turabian StyleVernazza, Stefania, Sara Tirendi, Anna Maria Bassi, Carlo Enrico Traverso, and Sergio Claudio Saccà. 2020. "Neuroinflammation in Primary Open-Angle Glaucoma" Journal of Clinical Medicine 9, no. 10: 3172. https://doi.org/10.3390/jcm9103172
APA StyleVernazza, S., Tirendi, S., Bassi, A. M., Traverso, C. E., & Saccà, S. C. (2020). Neuroinflammation in Primary Open-Angle Glaucoma. Journal of Clinical Medicine, 9(10), 3172. https://doi.org/10.3390/jcm9103172