
Landscape of Global Gene Expression Reveals Distinctive Tissue Characteristics in Bactrian Camels (Camelus bactrianus)

 ,
,  , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Ethics Statement
2.2. Tissue Collection and RNA Extraction
2.3. cDNA Library Construction and RNA Sequencing
2.4. Sequence Analysis and Transcriptome Mapping
2.5. Classification of Protein-Coding Genes
2.6. Weighted Gene Co-Expressed Network Analysis
2.7. Functional Enrichment Analysis
2.8. Quantitative Real-Time PCR
3. Results
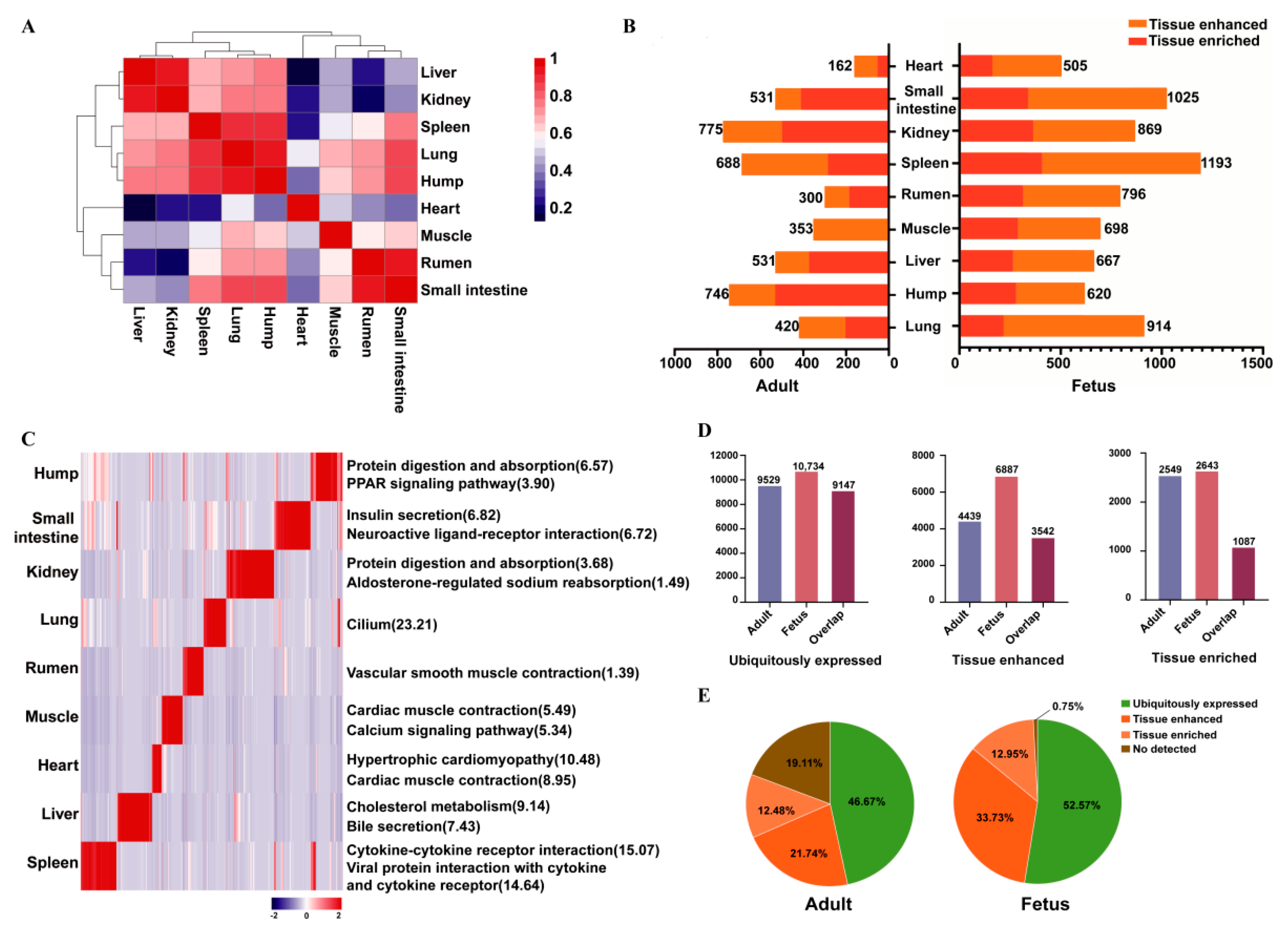
3.1. Identification and Characterizations of Coding RNAs
3.2. Classification of Protein-Coding Genes Based on Tissue Levels
3.3. Weighted Gene Co-Expression Network Analysis
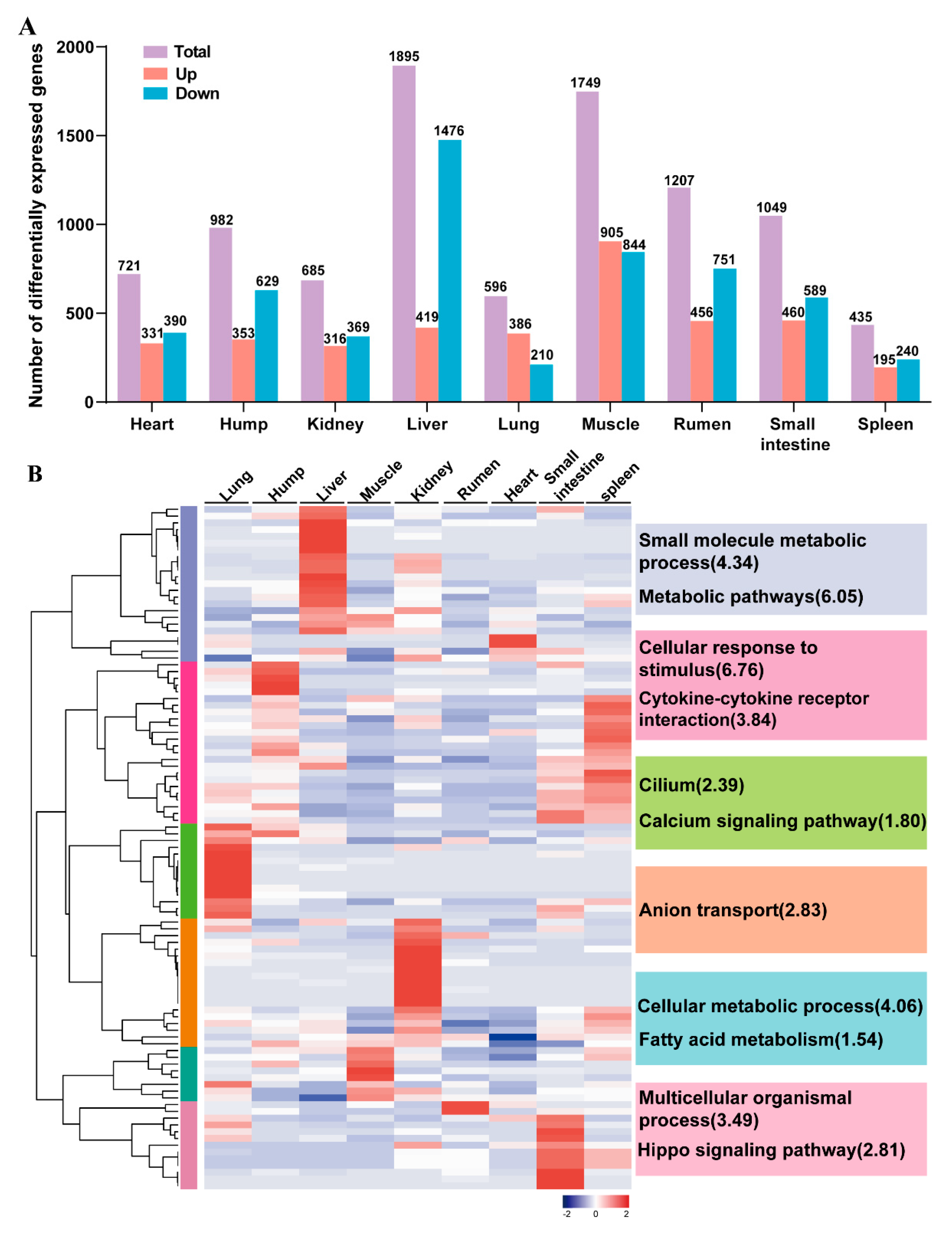
3.4. Development-Dependent Genes in Nine tissues
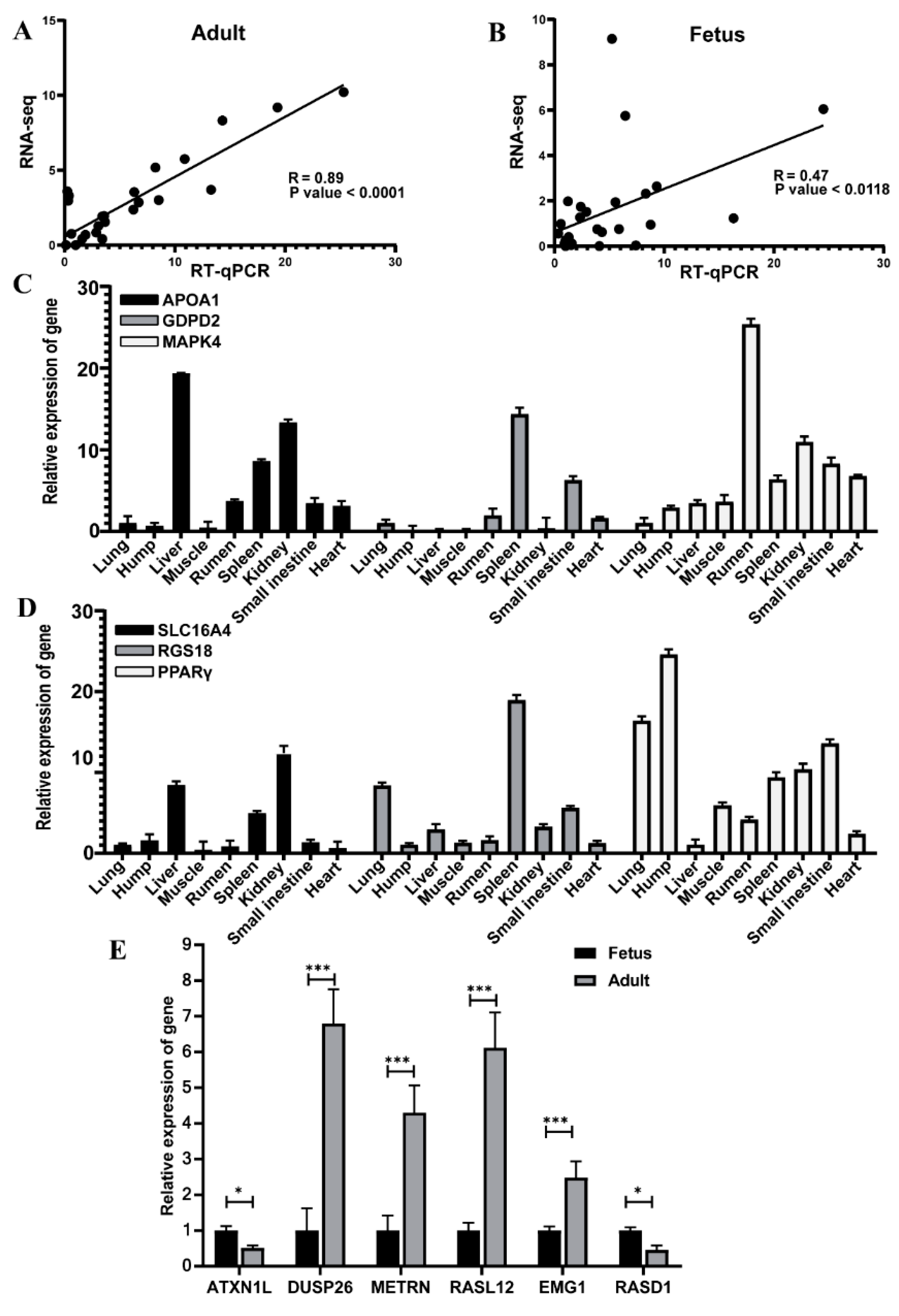
3.5. Validation of RNA-Seq Data by Quantitative Real-Time Polymerase chain Reaction (qRT-PCR)
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Stark, R.; Grzelak, M.; Hadfield, J. RNA sequencing: The teenage years. Nat. Rev. Genet. 2019, 20, 631–656. [Google Scholar] [CrossRef] [PubMed]
- Freeman, T.C.; Ivens, A.; Baillie, J.K.; Beraldi, D.; Barnett, M.W.; Dorward, D.; Downing, A.; Fairbairn, L.; Kapetanovic, R.; Raza, S.; et al. A gene expression atlas of the domestic pig. BMC Biol. 2012, 10, 90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harhay, G.P.; Smith, T.P.; Alexander, L.J.; Haudenschild, C.D.; Keele, J.W.; Matukumalli, L.K.; Schroeder, S.G.; Van Tassell, C.P.; Gresham, C.R.; Bridges, S.M.; et al. An atlas of bovine gene expression reveals novel distinctive tissue characteristics and evidence for improving genome annotation. Genome Biol. 2010, 11, R102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, A.I.; Wiltshire, T.; Batalov, S.; Lapp, H.; Ching, K.A.; Block, D.; Zhang, J.; Soden, R.; Hayakawa, M.; Kreiman, G.; et al. A gene atlas of the mouse and human protein-encoding transcriptomes. Proc. Natl. Acad. Sci. USA 2004, 101, 6062–6067. [Google Scholar] [CrossRef] [Green Version]
- Yu, Y.; Fuscoe, J.C.; Zhao, C.; Guo, C.; Jia, M.; Qing, T.; Bannon, D.I.; Lancashire, L.; Bao, W.; Du, T.; et al. A rat RNA-Seq transcriptomic BodyMap across 11 organs and 4 developmental stages. Nat. Commun. 2014, 5, 3230. [Google Scholar] [CrossRef]
- Andersson, R.; Gebhard, C.; Miguel-Escalada, I.; Hoof, I.; Bornholdt, J.; Boyd, M.; Chen, Y.; Zhao, X.; Schmidl, C.; Suzuki, T.; et al. An atlas of active enhancers across human cell types and tissues. Nature 2014, 507, 455–461. [Google Scholar] [CrossRef]
- Brennan, A.; Leech, J.T.; Kad, N.M.; Mason, J.M. An Approach to Derive Functional Peptide Inhibitors of Transcription Factor Activity. JACS Au 2022, 2, 996–1006. [Google Scholar] [CrossRef]
- Alhaddad, H.; Maraqa, T.; Alabdulghafour, S.; Alaskar, H.; Alaqeely, R.; Almathen, F.; Alhajeri, B.H. Quality and quantity of dromedary camel DNA sampled from whole-blood, saliva, and tail-hair. PLoS ONE 2019, 14, e0211743. [Google Scholar] [CrossRef] [Green Version]
- Emmanuel, B.; Nahapetian, A. Fatty-Acid Composition of Depot Fats, and Rumen Wall of the Camel (Camelus-Dromedarius). Comp. Biochem. Phys. B 1980, 67, 701–704. [Google Scholar] [CrossRef]
- Schmidt-Nielsen, K. The Physiology of the Camel. Sci. Am. 1959, 201, 140–151. [Google Scholar] [CrossRef]
- Bactrian Camels Genome, S.; Analysis, C.; Wang, Z.; Ding, G.; Chen, G.; Sun, Y.; Sun, Z.; Zhang, H.; Wang, L.; Hasi, S.; et al. Genome sequences of wild and domestic bactrian camels. Nat. Commun. 2012, 3, 1202. [Google Scholar] [CrossRef]
- Ming, L.; Yuan, L.; Yi, L.; Ding, G.; Hasi, S.; Chen, G.; Jambl, T.; Hedayat-Evright, N.; Batmunkh, M.; Badmaevna, G.K.; et al. Whole-genome sequencing of 128 camels across Asia reveals origin and migration of domestic Bactrian camels. Commun. Biol. 2020, 3, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ali, M.A.; Abu Damir, H.; Ali, O.M.; Amir, N.; Tariq, S.; Greenwood, M.P.; Lin, P.; Gillard, B.; Murphy, D.; Adem, A. The effect of long-term dehydration and subsequent rehydration on markers of inflammation, oxidative stress and apoptosis in the camel kidney. BMC Veter Res. 2020, 16, 458. [Google Scholar] [CrossRef]
- Zhang, D.; Pan, J.; Cao, J.; Cao, Y.; Zhou, H. Screening of drought-resistance related genes and analysis of promising regulatory pathway in camel renal medulla. Genomics 2020, 112, 2633–2639. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Zhang, D.; Zhou, H. Key genes differential expressions and pathway involved in salt and water-deprivation stresses for renal cortex in camel. BMC Mol. Biol. 2019, 20, 11. [Google Scholar] [CrossRef] [PubMed]
- Guo, F.; Si, R.; He, J.; Yuan, L.; Hai, L.; Ming, L.; Yi, L.; Ji, R. Comprehensive transcriptome analysis of adipose tissue in the Bactrian camel reveals fore hump has more specific physiological functions in immune and endocrine systems. Livest. Sci. 2019, 228, 195–200. [Google Scholar] [CrossRef]
- Zhang, D.; Pan, J.; Zhou, H.; Cao, Y. Evidence from ileum and liver transcriptomes of resistance to high-salt and water-deprivation conditions in camel. Zool. Lett. 2020, 6, 8. [Google Scholar] [CrossRef]
- Hedayat-Evrigh, N.; Khalkhali-Evrigh, R.; Bakhtiarizadeh, M.R. Genome-Wide Identification and Analysis of Variants in Domestic and Wild Bactrian Camels Using Whole-Genome Sequencing Data. J. Genom. 2020, 2020, 2430846. [Google Scholar] [CrossRef]
- Wu, H.; Guang, X.; Al-Fageeh, M.B.; Cao, J.; Pan, S.; Zhou, H.; Zhang, L.; AbuTarboush, M.H.; Xing, Y.; Xie, Z.; et al. Camelid genomes reveal evolution and adaptation to desert environments. Nat. Commun. 2014, 5, 5188. [Google Scholar] [CrossRef] [Green Version]
- Trapnell, C.; Roberts, A.; Goff, L.; Pertea, G.; Kim, D.; Kelley, D.R.; Pimentel, H.; Salzberg, S.L.; Rinn, J.L.; Pachter, L. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 2012, 7, 562–578. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. 1000 Genome Project Data Processing Subgroup. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uhlén, M.; Fagerberg, L.; Hallström, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, Å.; Kampf, C.; Sjöstedt, E.; Asplund, A.; et al. Tissue-Based Map of the Human Proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef] [PubMed]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef] [Green Version]
- Langfelder, P.; Zhang, B.; Horvath, S. Defining clusters from a hierarchical cluster tree: The Dynamic Tree Cut package for R. Bioinformatics 2008, 24, 719–720. [Google Scholar] [CrossRef]
- Min, Y.; Kramer, E.M. Transcriptome profiling and weighted gene co-expression network analysis of early floral development in Aquilegia coerulea. Sci. Rep. 2020, 10, 19637. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of Biomolecular Interaction Networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Reimand, J.; Arak, T.; Adler, P.; Kolberg, L.; Reisberg, S.; Peterson, H.; Vilo, J. g:Profiler—A web server for functional interpretation of gene lists (2016 update). Nucleic Acids Res. 2016, 44, W83–W89. [Google Scholar] [CrossRef]
- Hebenstreit, D.; Fang, M.; Gu, M.; Charoensawan, V.; Van Oudenaarden, A.; Teichmann, S. RNA sequencing reveals two major classes of gene expression levels in metazoan cells. Mol. Syst. Biol. 2011, 7, 497. [Google Scholar] [CrossRef] [Green Version]
- Uhlén, M.; Hallström, B.M.; Lindskog, C.; Mardinoglu, A.; Pontén, F.; Nielsen, J. Transcriptomics resources of human tissues and organs. Mol. Syst. Biol. 2016, 12, 862. [Google Scholar] [CrossRef]
- Hawrylycz, M.J.; Lein, E.S.; Guillozet-Bongaarts, A.L.; Shen, E.H.; Ng, L.; Miller, J.A.; Van De Lagemaat, L.N.; Smith, K.A.; Ebbert, A.; Riley, Z.L.; et al. An anatomically comprehensive atlas of the adult human brain transcriptome. Nature 2012, 489, 391–399. [Google Scholar] [CrossRef]
- Keen, J.C.; Moore, H.M. The Genotype-Tissue Expression (GTEx) Project: Linking Clinical Data with Molecular Analysis to Advance Personalized Medicine. J. Pers. Med. 2015, 5, 22–29. [Google Scholar] [CrossRef] [PubMed]
- Yu, N.Y.-L.; Hallström, B.M.; Fagerberg, L.; Ponten, F.; Kawaji, H.; Carninci, P.; Forrest, A.; The FANTOM consortium; Hayashizaki, Y.; Uhlen, M.; et al. Complementing tissue characterization by integrating transcriptome profiling from the Human Protein Atlas and from the FANTOM5 consortium. Nucleic Acids Res. 2015, 43, 6787–6798. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diez-Roux, G.; Banfi, S.; Sultan, M.; Geffers, L.; Anand, S.; Rozado, D.; Magen, A.; Canidio, E.; Pagani, M.; Peluso, I.; et al. A High-Resolution Anatomical Atlas of the Transcriptome in the Mouse Embryo. PLOS Biol. 2011, 9, e1000582. [Google Scholar] [CrossRef] [PubMed]
- Carson, J.; Thaller, C.; Eichele, G. A transcriptome atlas of the mouse brain at cellular resolution. Curr. Opin. Neurobiol. 2002, 12, 562–565. [Google Scholar] [CrossRef]
- Sun, H.-Z.; Zhao, K.; Zhou, M.; Chen, Y.; Guan, L.L. Landscape of multi-tissue global gene expression reveals the regulatory signatures of feed efficiency in beef cattle. Bioinformatics 2018, 35, 1712–1719. [Google Scholar] [CrossRef]
- Clark, E.L.; Bush, S.J.; McCulloch, M.E.B.; Farquhar, I.L.; Young, R.; Lefevre, L.; Pridans, C.; Tsang, H.G.; Wu, C.; Afrasiabi, C.; et al. A high resolution atlas of gene expression in the domestic sheep (Ovis aries). PLoS Genet. 2017, 13, e1006997. [Google Scholar] [CrossRef] [Green Version]
- Qi, X.; Zhang, Q.; He, Y.; Yang, L.; Zhang, X.; Shi, P.; Yang, L.; Liu, Z.; Zhang, F.; Liu, F.; et al. The Transcriptomic Landscape of Yaks Reveals Molecular Pathways for High Altitude Adaptation. Genome Biol. Evol. 2018, 11, 72–85. [Google Scholar] [CrossRef]
- Tang, Q.; Guosong, W.; Zhou, X.; Jin, L.; Guan, J.; Liu, R.; Li, J.; Long, K.; Tiandong, C.; Che, T.; et al. Comparative transcriptomics of 5 high-altitude vertebrates and their low-altitude relatives. GigaScience 2017, 6, gix105. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, W.; Wu, X.; Wu, C.; Qian, L.; Wang, L.; Zhang, X.; Yang, M.; Li, D.; Ding, J.; et al. Transcriptomic comparison of liver tissue between Anqing six-end-white pigs and Yorkshire pigs based on RNA sequencing. Genome 2020, 63, 203–214. [Google Scholar] [CrossRef]
- Li, H.; Wang, T.; Xu, C.; Wang, D.; Ren, J.; Li, Y.; Tian, Y.; Wang, Y.; Jiao, Y.; Kang, X.; et al. Transcriptome profile of liver at different physiological stages reveals potential mode for lipid metabolism in laying hens. BMC Genom. 2015, 16, 763. [Google Scholar] [CrossRef] [Green Version]
- Li, L.Y.; Wu, J.X. Analysis of hub genes and molecular mechanisms in non-alcoholic steatohepatitis based on the gene expression omnibus database. Zhonghua Yi Xue Za Zhi 2021, 101, 3317–3322. [Google Scholar] [CrossRef] [PubMed]
- Song, X.; Jiang, H.; Qi, Z.; Shen, X.; Xue, M.; Hu, J.; Liu, H.; Zhou, X.; Tu, J.; Qi, K. APEC infection affects cytokine–cytokine receptor interaction and cell cycle pathways in chicken trachea. Res. Veter Sci. 2020, 130, 144–152. [Google Scholar] [CrossRef] [PubMed]
- Vu, T.H.; Hong, Y.; Truong, A.D.; Lee, J.; Lee, S.; Song, K.-D.; Cha, J.; Dang, H.V.; Tran, H.T.T.; Lillehoj, H.S.; et al. Cytokine-cytokine receptor interactions in the highly pathogenic avian influenza H5N1 virus-infected lungs of genetically disparate Ri chicken lines. Anim. Biosci. 2022, 35, 367–376. [Google Scholar] [CrossRef] [PubMed]
- Ye, M.; Bao, H.; Meng, Y.; Guan, L.; Stothard, P.; Plastow, G. Comparative transcriptomic analysis of porcine peripheral blood reveals differentially expressed genes from the cytokine–cytokine receptor interaction pathway related to health status. Genome 2017, 60, 1021–1028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olech, M.; Ropka-Molik, K.; Szmatoła, T.; Piórkowska, K.; Kuźmak, J. Transcriptome Analysis for Genes Associated with Small Ruminant Lentiviruses Infection in Goats of Carpathian Breed. Viruses 2021, 13, 2054. [Google Scholar] [CrossRef] [PubMed]
- Kershaw, E.E.; Flier, J.S. Adipose Tissue as an Endocrine Organ. J. Clin. Endocrinol. Metab. 2004, 89, 2548–2556. [Google Scholar] [CrossRef]
- Chung, K.-J.; Nati, M.; Chavakis, T.; Chatzigeorgiou, A. Innate immune cells in the adipose tissue. Rev. Endocr. Metab. Disord. 2018, 19, 283–292. [Google Scholar] [CrossRef]
- Kane, H.; Lynch, L. Innate Immune Control of Adipose Tissue Homeostasis. Trends Immunol. 2019, 40, 857–872. [Google Scholar] [CrossRef]
- Jimenez, M.T.; Michieletto, M.F.; Henao-Mejia, J. A new perspective on mesenchymal-immune interactions in adipose tissue. Trends Immunol. 2021, 42, 375–388. [Google Scholar] [CrossRef]
- Kadegowda, A.K.; Bionaz, M.; Piperova, L.S.; Erdman, R.A.; Loor, J.J. Peroxisome proliferator-activated receptor-gamma activation and long-chain fatty acids alter lipogenic gene networks in bovine mammary epithelial cells to various extents. J. Dairy Sci. 2009, 92, 4276–4289. [Google Scholar] [CrossRef] [Green Version]
- Shi, H.B.; Luo, J.; Yao, D.W.; Zhu, J.J.; Xu, H.F.; Shi, H.P.; Loor, J.J. Peroxisome proliferator-activated receptor-gamma stimulates the synthesis of monounsaturated fatty acids in dairy goat mammary epithelial cells via the control of stearoyl-coenzyme A desaturase. J. Dairy Sci. 2013, 96, 7844–7853. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, H.; Luo, J.; Zhu, J.; Li, J.; Sun, Y.; Lin, X.; Zhang, L.; Yao, D.; Shi, H. PPAR gamma Regulates Genes Involved in Triacylglycerol Synthesis and Secretion in Mammary Gland Epithelial Cells of Dairy Goats. PPAR Res. 2013, 2013, 310948. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, H.B.; Zhao, W.S.; Luo, J.; Yao, D.W.; Sun, Y.T.; Li, J.; Shi, H.P.; Loor, J.J. Peroxisome proliferator-activated receptor gamma1 and gamma2 isoforms alter lipogenic gene networks in goat mammary epithelial cells to different extents. J. Dairy Sci. 2014, 97, 5437–5447. [Google Scholar] [CrossRef] [PubMed]
- Lorenzi, I.; Eckardstein, A.V.; Cavelier, C.; Radosavljevic, S.; Rohrer, L. Apolipoprotein A-I but not high-density lipoproteins are internalised by RAW macrophages: Roles of ATP-binding cassette transporter A1 and scavenger receptor BI. J. Mol. Med. 2008, 86, 171–183. [Google Scholar] [CrossRef] [Green Version]
- Izaola, O.; Primo, D.; Hoyos, E.G.; Gomez, J.J.L.; Ortola, A.; de Luis, D. Association of rs670 variant of APOA1 gene with lipid profile and insulin resistance after 9 months of a high protein/low carbohydrate vs a standard hypocaloric diet. Clin. Nutr. 2019, 39, 988–993. [Google Scholar] [CrossRef]
- Reichl, D.; Miller, N.E. Pathophysiology of reverse cholesterol transport. Insights from inherited disorders of lipoprotein metabolism. Arter. Off. J. Am. Heart Assoc. Inc. 1989, 9, 785–797. [Google Scholar] [CrossRef] [Green Version]
- Holleboom, A.G.; Vergeer, M.; Hovingh, G.K.; Kastelein, J.J.; Kuivenhoven, J.A. The value of HDL genetics. Curr. Opin. Lipidol. 2008, 19, 385–394. [Google Scholar] [CrossRef]
- Nikolaou, K.; Vatandaslar, H.; Meyer, C.; Schmid, M.W.; Tuschl, T.; Stoffel, M. The RNA-Binding Protein A1CF Regulates Hepatic Fructose and Glycerol Metabolism via Alternative RNA Splicing. Cell Rep. 2019, 29, 283–300. [Google Scholar] [CrossRef] [Green Version]
- Blanc, V.; Riordan, J.D.; Soleymanjahi, S.; Nadeau, J.H.; Nalbantoglu, I.; Xie, Y.; Molitor, E.A.; Madison, B.B.; Brunt, E.M.; Mills, J.C.; et al. Apobec1 complementation factor overexpression promotes hepatic steatosis, fibrosis, and hepatocellular cancer. J. Clin. Investig. 2021, 131, 138699. [Google Scholar] [CrossRef]
- Bartee, M.Y.; Lutsenko, S. Hepatic copper-transporting ATPase ATP7B: Function and inactivation at the molecular and cellular level. BioMetals 2007, 20, 627–637. [Google Scholar] [CrossRef]
- Pan, G.; Cavalli, M.; Carlsson, B.; Skrtic, S.; Kumar, C.; Wadelius, C. rs953413 Regulates Polyunsaturated Fatty Acid Metabolism by Modulating ELOVL2 Expression. iScience 2020, 23, 100808. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Ye, D.; Wang, H.; He, M.; Sun, Y. Elovl2 But Not Elovl5 Is Essential for the Biosynthesis of Docosahexaenoic Acid (DHA) in Zebrafish: Insight from a Comparative Gene Knockout Study. Mar. Biotechnol. 2020, 22, 613–619. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.Z.; Sun, B.; Hao, D.X.; Zhang, X.J.; Shi, T.P.; Ma, D.L. Human TMEM174 that is highly expressed in kidney tissue activates AP-1 and promotes cell proliferation. Biochem. Bioph. Res. Co. 2010, 394, 993–999. [Google Scholar] [CrossRef]
- Nagasawa, T.; Hirota, S.; Tachibana, K.; Takakura, N.; Nishikawa, S.-I.; Kitamura, Y.; Yoshida, N.; Kikutani, H.; Kishimoto, T. Defects of B-cell lymphopoiesis and bone-marrow myelopoiesis in mice lacking the CXC chemokine PBSF/SDF-1. Nature 1996, 382, 635–638. [Google Scholar] [CrossRef] [PubMed]
- Feig, C.; Jones, J.O.; Kraman, M.; Wells, R.J.; Deonarine, A.; Chan, D.S.; Connell, C.M.; Roberts, E.W.; Zhao, Q.; Caballero, O.L.; et al. Targeting CXCL12 from FAP-expressing carcinoma-associated fibroblasts synergizes with anti-PD-L1 immunotherapy in pancreatic cancer. Proc. Natl. Acad. Sci. USA 2013, 110, 20212–20217. [Google Scholar] [CrossRef] [Green Version]
- Schiraldi, M.; Raucci, A.; Muñoz, L.M.; Livoti, E.; Celona, B.; Venereau, E.; Apuzzo, T.; De Marchis, F.; Pedotti, M.; Bachi, A.; et al. HMGB1 promotes recruitment of inflammatory cells to damaged tissues by forming a complex with CXCL12 and signaling via CXCR4. J. Exp. Med. 2012, 209, 551–563. [Google Scholar] [CrossRef] [Green Version]
- Vinnitskaya, A.; Svitich, O.A.; Ammour, Y.; Zverev, V. Effect of CXCL12 and TLR ligands on tumor cell migration and expression of innate immunity factors. Allergy 2020, 75, 188. [Google Scholar]
- Adler, M.W.; Rogers, T.J. Are chemokines the third major system in the brain? J. Leukoc. Biol. 2005, 78, 1204–1209. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Xie, S.; Qian, L.; Cai, C.; Bi, H.; Cui, W. Identification of genes related to skeletal muscle growth and development by integrated analysis of transcriptome and proteome in myostatin-edited Meishan pigs. J. Proteom. 2020, 213, 103628. [Google Scholar] [CrossRef]
- Tan, I.; Ng, C.H.; Lim, L.; Leung, T. Phosphorylation of a Novel Myosin Binding Subunit of Protein Phosphatase 1 Reveals a Conserved Mechanism in the Regulation of Actin Cytoskeleton. J. Biol. Chem. 2001, 276, 21209–21216. [Google Scholar] [CrossRef] [Green Version]
- Meinild, A.-K.; Loo, D.D.F.; Pajor, A.M.; Zeuthen, T.; Wright, E.M. Water transport by the renal Na+-dicarboxylate cotransporter. Am. J. Physiol. Physiol. 2000, 278, F777–F783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takvam, M.; Wood, C.M.; Kryvi, H.; Nilsen, T.O. Ion Transporters and Osmoregulation in the Kidney of Teleost Fishes as a Function of Salinity. Front. Physiol. 2021, 12, 664588. [Google Scholar] [CrossRef] [PubMed]
- Kanazawa, N.; Nakamura, T.; Tashiro, K.; Muramatsu, M.; Morita, K.; Yoneda, K.; Inaba, K.; Imamura, S.; Honjo, T. Fractalkine and macrophage-derived chemokine: T cell-attracting chemokines expressed in T cell area dendritic cells. Eur. J. Immunol. 1999, 29, 1925–1932. [Google Scholar] [CrossRef]
- Verge, G.M.; Milligan, E.D.; Maier, S.F.; Watkins, L.R.; Naeve, G.S.; Foster, A.C. Fractalkine (CX3CL1) and fractalkine receptor (CX3CR1) distribution in spinal cord and dorsal root ganglia under basal and neuropathic pain conditions. Eur. J. Neurosci. 2004, 20, 1150–1160. [Google Scholar] [CrossRef]
- Deiva, K.; Geeraerts, T.; Salim, H.; Leclerc, P.; Hery, C.; Hugel, B.; Freyssinet, J.-M.; Tardieu, M. Fractalkine reduces N-methyl-d-aspartate-induced calcium flux and apoptosis in human neurons through extracellular signal-regulated kinase activation. Eur. J. Neurosci. 2004, 20, 3222–3232. [Google Scholar] [CrossRef]
- Takegahara, N.; Kim, H.; Choi, Y. RANKL biology. Bone 2022, 159, 116353. [Google Scholar] [CrossRef]
- Anderson, D.M.; Maraskovsky, E.; Billingsley, W.L.; Dougall, W.C.; Tometsko, M.E.; Roux, E.R.; Teepe, M.C.; DuBose, R.F.; Cosman, D.; Galibert, L.J. A homologue of the TNF receptor and its ligand enhance T-cell growth and dendritic-cell function. Nature 1997, 390, 175–179. [Google Scholar] [CrossRef]
- Fernandez-Canon, J.M.; Granadino, B.; Beltran-Valero de Bernabe, D.; Renedo, M.; Fernandez-Ruiz, E.; Penalva, M.A.; Rodriguez de Cordoba, S. The molecular basis of alkaptonuria. Nat. Genet. 1996, 14, 19–24. [Google Scholar] [CrossRef]
- Ranganath, L.R.; Jarvis, J.C.; Gallagher, J.A. Recent advances in management of alkaptonuria (invited review; best practice article). J. Clin. Pathol. 2013, 66, 367–373. [Google Scholar] [CrossRef]
- Singh, R.; Gupta, S.; Dastidar, S.; Ray, A. Cysteinyl Leukotrienes and Their Receptors: Molecular and Functional Characteristics. Pharmacology 2010, 85, 336–349. [Google Scholar] [CrossRef]
- Wu, X.; Hong, H.; Zuo, K.; Han, M.; Li, J.; Wen, W.; Xu, G.; Miao, B.; Li, H. Expression of leukotriene and its receptors in eosinophilic chronic rhinosinusitis with nasal polyps. Int. Forum Allergy Rhinol. 2015, 6, 75–81. [Google Scholar] [CrossRef] [PubMed]
- Fukai, H.; Ogasawara, Y.; Migita, O.; Koga, M.; Ichikawa, K.; Shibasaki, M.; Arinami, T.; Noguchi, E. Association between a polymorphism in cysteinyl leukotriene receptor 2 on chromosome 13q14 and atopic asthma. Pharmacogenetics 2004, 14, 683–690. [Google Scholar] [CrossRef] [PubMed]
- Dewulf, J.P.; Wiame, E.; Dorboz, I.; Elmaleh-Bergès, M.; Imbard, A.; Dumitriu, D.; Rak, M.; Bourillon, A.; Helaers, R.; Malla, A.; et al. SLC13A3 variants cause acute reversible leukoencephalopathy and α-ketoglutarate accumulation. Ann. Neurol. 2019, 85, 385–395. [Google Scholar] [CrossRef] [PubMed]
- Bai, X.; Chen, X.; Feng, Z.; Hou, K.; Zhang, P.; Fu, B.; Shi, S. Identification of basolateral membrane targeting signal of human sodium-dependent dicarboxylate transporter 3. J. Cell. Physiol. 2005, 206, 821–830. [Google Scholar] [CrossRef]
- Bergeron, M.; Clémençon, B.; Hediger, M.; Markovich, D. SLC13 family of Na+-coupled di- and tri-carboxylate/sulfate transporters. Mol. Asp. Med. 2013, 34, 299–312. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Luan, Y.; Fang, Y.; Jiang, L.; Ma, Y.; Wu, S.; Zhou, J.; Pu, Y.; Zhao, Q.; He, X. Landscape of Global Gene Expression Reveals Distinctive Tissue Characteristics in Bactrian Camels (Camelus bactrianus). Agriculture 2022, 12, 958. https://doi.org/10.3390/agriculture12070958
Luan Y, Fang Y, Jiang L, Ma Y, Wu S, Zhou J, Pu Y, Zhao Q, He X. Landscape of Global Gene Expression Reveals Distinctive Tissue Characteristics in Bactrian Camels (Camelus bactrianus). Agriculture. 2022; 12(7):958. https://doi.org/10.3390/agriculture12070958
Chicago/Turabian StyleLuan, Yuanyuan, Yan Fang, Lin Jiang, Yuehui Ma, Shangjie Wu, Junwen Zhou, Yabin Pu, Qianjun Zhao, and Xiaohong He. 2022. "Landscape of Global Gene Expression Reveals Distinctive Tissue Characteristics in Bactrian Camels (Camelus bactrianus)" Agriculture 12, no. 7: 958. https://doi.org/10.3390/agriculture12070958