
Effect of the Matrix and Target on the Accurate Quantification of Genomic and Plasmid DNA by Digital Polymerase Chain Reaction

Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. DNA Extraction
2.3. Sample Preparation
2.4. Primers and Probes
2.5. Real-Time Quantitative PCR
2.6. Droplet Digital PCR
3. Results
3.1. Effect of Method Sensitivity on dPCR Assays
3.2. Quantification of Low-Level Samples by qPCR
3.3. Quantification of Low-Level Samples by dPCR
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Vogelstein, B.; Kinzler, K.W. Digital PCR. Proc. Natl. Acad. Sci. USA 1999, 96, 9236–9241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Zhai, S.; Gao, H.; Xiao, F.; Li, Y.; Wu, G.; Wu, Y. Development and assessment of a duplex droplet digital PCR method for quantification of GM rice Kemingdao. Anal. Bioanal. Chem. 2021, 413, 4341–4351. [Google Scholar] [CrossRef] [PubMed]
- Bharuthram, A.; Paximadis, M.; Picton, A.C.; Tiemessen, C.T. Comparison of a quantitative real-time PCR assay and droplet digital PCR for copy number analysis of the CCL4L genes. Infec. Gen. Evol. 2014, 25, 28–35. [Google Scholar] [CrossRef] [PubMed]
- Hayden, R.T.; Gu, Z.; Ingersoll, J.; Abdul-Ali, D.; Shi, L.; Pounds, S.; Caliendo, A.M. Comparison of droplet digital PCR to real-time PCR for quantitative detection of cytomegalovirus. J. Clin. Microbiol. 2013, 51, 540–546. [Google Scholar] [CrossRef] [Green Version]
- The dMIQE Group. The digital MIQE guidelines update: Minimum information for publication of quantitative digital PCR experiments for 2020. Clin. Chem. 2020, 66, 1012–1029. [Google Scholar] [CrossRef]
- ISO. Biotechnology—Requirements for Evaluating the Performance of Quantification Methods for Nucleic Acid Target Sequences—qPCR and dPCR, ISO 20395:2019. Available online: https://www.iso.org/standard/67893.html (accessed on 30 March 2022).
- Huggett, J.F.; Foy, C.A.; Benes, V.; Emslie, K.; Garson, J.A.; Haynes, R.; Hellemans, J.; Kubista, M.; Mueller, R.D.; Nolan, T.; et al. The digital MIQE guidelines: Minimum information for publication of quantitative digital PCR experiments. Clin. Chem. 2013, 59, 892–902. [Google Scholar] [CrossRef] [Green Version]
- European Commission. JRC Technical Report, Guidance Document on Measurement Uncertainty for GMO Testing Laboratories 3rd Edition; EUR 30248 EN; Publications Office of the European Union: Luxembourg, 2020. [Google Scholar] [CrossRef]
- White, H.; Deprez, L.; Corbisier, P.; Hall, V.; Lin, F.; Mazoua, S.; Trapmann, S.; Aggerholm, A.; Andrikovics, H.; Akiki, S.; et al. A certified plasmid reference material for the standardisation of BCR-ABL1 mRNA quantification by real-time quantitative PCR. Leukemia 2015, 29, 369–376. [Google Scholar] [CrossRef] [Green Version]
- Liang, W.; Xu, L.; Sui, Z.; Li, Y.; Li, L.; Wen, Y.; Li, C.; Ren, S.; Liu, G. Quantification of plasmid DNA reference materials for Shiga toxin-producing Escherichia coli based on UV, HR-ICP-MS and digital PCR. Chem. Cent. J. 2016, 10, 55. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Li, X.; Wang, C.; Song, G.; Pi, L.; Zheng, L.; Zhang, D.; Yang, L. One novel multiple-target plasmid reference molecule targeting eight genetically modified canola events for genetically modified canola detection. J. Agric. Food Chem. 2017, 65, 8489–8500. [Google Scholar] [CrossRef]
- Block, A.; Schwarz, G. Validation of different genomic and cloned DNA calibration standards for construct-specific quantification of LibertyLink in rapeseed by real-time PCR. Eur. Food Res. Technol. 2003, 216, 421–427. [Google Scholar] [CrossRef]
- Taverniers, I.; Windels, P.; Vaitilingom, M.; Milcamps, A.; Van Bockstaele, E.; Van den Eede, G.; De Loose, M. Event-specific plasmid standards and real-time PCR methods for transgenic Bt11, Bt176, and GA21 maize and transgenic GT73 canola. J. Agric. Food Chem. 2005, 53, 3041–3052. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Zhang, X.; Wan, Y.; Jin, W. Development of a novel reference plasmid for accurate quantification of genetically modified Kefeng6 rice DNA in food and feed samples. Biomed. Res. Int. 2013, 2013, 134675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whale, A.S.; Cowen, S.; Foy, C.A.; Huggett, J.F. Methods for applying accurate digital PCR analysis on low copy DNA samples. PLoS ONE 2013, 8, e58177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Y.H.; Li, J.; Wang, Y.L.; Li, X.F.; Li, Y.J.; Zhu, L.; Li, J.; Wu, G. Development and application of a general plasmid reference material for GMO screening. Plasmid 2016, 87–88, 28–36. [Google Scholar] [CrossRef]
- Wu, Y.; Li, J.; Li, X.; Liang, J.; Li, Y.; Zeng, X.; Wu, G. Copy number and zygosity determination of transgenic rapeseed by droplet digital PCR. Oil Crop Sci. 2017, 2, 84–94. [Google Scholar]
- Li, J.; Wu, Y.; Li, X.; Wang, Y.; Zhang, L.; Li, Y.; Wu, G. Developing a matrix reference material for screening of transgenic rice. Anal. Bioanal. Chem. 2015, 407, 9153–9163. [Google Scholar] [CrossRef]
- Bhat, S.; Emslie, K.R. Digital polymerase chain PCR for characterization of DNA reference materials. Biomol. Detect. Quantif. 2016, 10, 47–49. [Google Scholar] [CrossRef] [Green Version]
- Dong, L.; Meng, Y.; Sui, Z.; Wang, J.; Wu, L.; Fu, B. Comparison of four digital PCR platforms for accurate quantification of DNA copy number of a certified plasmid DNA reference material. Sci. Rep. 2015, 5, 13174. [Google Scholar] [CrossRef] [Green Version]
- Pinheiro, L.B.; Coleman, V.A.; Hindson, C.M.; Herrmann, J.; Hindson, B.J.; Bhat, S.; Emslie, K.R. Evaluation of a droplet digital polymerase chain reaction format for DNA copy number quantification. Anal. Chem. 2012, 84, 1003–1011. [Google Scholar] [CrossRef]
- Yue, Y.; Wu, G.; Wu, Y.; Lu, C. Development of qualitative PCR method targeting marker genes in transgenic plants. Chin. J. Oil Crop Sci. 2011, 33, 280–289. (In Chinese) [Google Scholar]
- ISO/TS 21569-5; Horizontal Methods for Molecular Biomarker Analysis—Methods of Analysis for the Detection of Genetically Modified Organisms and Derived Products. Part 5: Real-time PCR Based Screening Method for the Detection of the FMV Promoter (P-FMV) DNA Sequence. ISO: Geneva, Switzerland, 2016. Available online: https://www.iso.org/standard/69339.html (accessed on 11 June 2020).
- Grohmann, L.; Brunen-Nieweler, C.; Nemeth, A.; Wai-blinger, H.U. Collaborative trial validation studies of real-time PCR-based GMO screening methods for detection of the bar gene and the ctp2-cp4epsps construct. J. Agric. Food Chem. 2009, 57, 8913–8920. [Google Scholar] [CrossRef] [PubMed]
- Reiting, R.; Broll, H.; Wablinger, H.U.; Grohmann, L. Collaborative study of a T-nos real-time PCR method for screening of genetically modified organisms in food products. J. Verbrauch Le-bensm 2007, 2, 116–121. [Google Scholar] [CrossRef]
- Fu, W.; Du, Z.; Wang, Q.; Wang, H.; Xu, W.; Wu, G.; Zhu, S.; Liu, X. Genetically Modified Organism Detection Method by Digital PCR. GB/T 33526-2017. 2017. Available online: https://www.antpedia.com/standard/1755877206.html (accessed on 11 June 2020).
- Debode, F.; Janssen, E.; Berben, G. Development of 10 new screening PCR assays for GMO detection targeting promoters (pFMV, pNOS, pSSuAra, pTA29, pUbi, pRice actin) and terminators (t35S, tE9, tOCS, tg7). Eur. Food Res. Technol. 2013, 236, 659–669. [Google Scholar] [CrossRef]
- Pansiot, J.; Chaouachi, M.; Cavellini, L.; Romaniuk, M.; Ayadi, M.; Bertheau, Y.; Laval, V. Development of two screening duplex PCR assays for genetically modified organism quantification using multiplex real-time PCR master mixes. Eur. Food Res. Technol. 2011, 232, 327–334. [Google Scholar] [CrossRef]
- Jacchia, S.; Sacco, M.G.; Savini, C.; Mazzara, M.; Emons, H. Event-specific Method for the Quantification of Oilseed Rape MS11 Using Real-time PCR—Validation Report. 2019. Available online: https://gmo-crl.jrc.ec.europa.eu/summaries/EURL-VL-03-16-VR.pdf (accessed on 11 June 2020).
- Mazzara, M.; Cordeil, S.; Van Den Eede, G. Report on the Verification of an Event-specific Detection Method for Identification of Rice GM-event LLRICE601 Using a Real-time PCR Assay. 2006. Available online: https://gmo-crl.jrc.ec.europa.eu/summaries/Verification%20Report%20LLRice601%20event.pdf (accessed on 11 June 2020).
- European Network of GMO Laboratories. Definition of Minimum Performance Requirements for Analytical Methods of GMO Testing. 2015. Available online: http://gmo-crl.jrc.ec.europa.eu/guidancedocs.htm (accessed on 11 June 2022).
- Caprioara-Buda, M.; Meyer, W.; Jeynov, B.; Corbisier, P.; Trapmann, S.; Emons, H. Evaluation of plasmid and genomic DNA calibrants used for the quantification of genetically modified organisms. Anal. Bioanal. Chem. 2012, 404, 29–42. [Google Scholar] [CrossRef]
- Lin, C.H.; Chen, Y.C.; Pan, T.M. Quantification bias caused by plasmid DNA conformation in quantitative real-time PCR assay. PLoS ONE 2011, 6, e29101. [Google Scholar] [CrossRef]
- Whale, A.S.; Huggett, J.F.; Cowen, S.; Speirs, V.; Shaw, J.; Ellison, S.; Foy, C.A.; Scott, D.J. Comparison of microfluidic digital PCR and conventional quantitative PCR for measuring copy number variation. Nucleic Acids Res. 2012, 40, e82. [Google Scholar] [CrossRef]
- Pavšič, J.; Žel, J.; Milavec, M. Assessment of the real-time PCR and different digital PCR platforms for DNA quantification. Anal. Bioanal. Chem. 2016, 408, 107–121. [Google Scholar] [CrossRef]
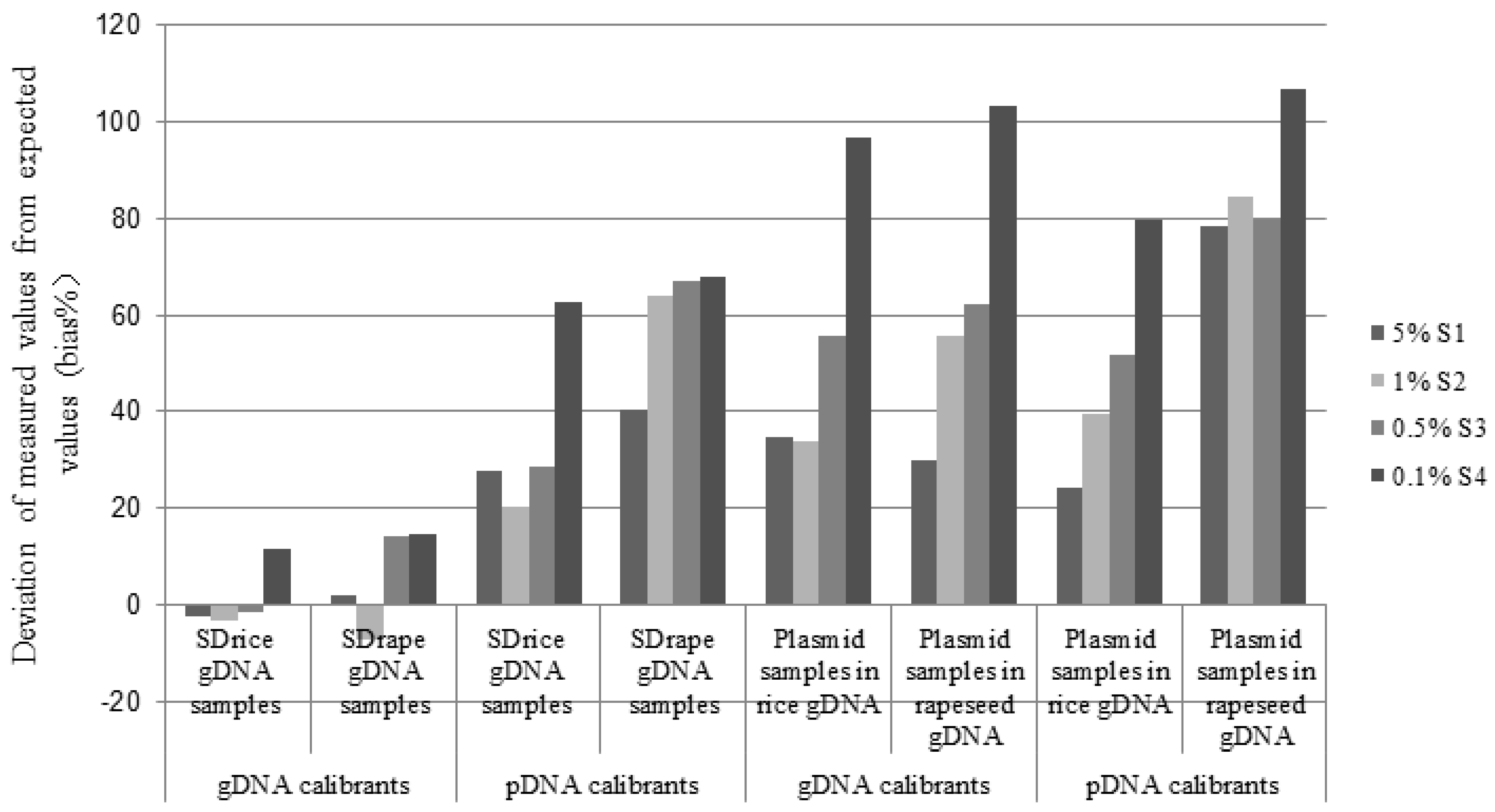


{kind=link}
{kind=link}
| Gene/Element | Primer/Probe | Sequence | Amplicon Size, bp | References |
|---|---|---|---|---|
| NPTII | qNPTⅡF63 | CTATGACTGGGCACAACAGACA | 101 | Lu et al., 2012 [22] |
| qNPTⅡR163 | CGGACAGGTCGGTCTTGACA | |||
| qNPTⅡFP90 | CTGCTCTGATGCCGCCGTGTTCCG | |||
| pFMV35S | pFMV35S-F | CAAAATAACGTGGAAAAGAGCT | 78 | ISO/TS 21569-5:2016 [23] |
| pFMV35S-R | TCTTTTGTGGTCGTCACTGC | |||
| pFMV35S-P | CTGACAGCCCACTCACTAATGC | |||
| Bar | RapB-F1 | ACAAGCACGGTCAACTTCC | 60 | Grohmann et al., 2009 [24] |
| RapB-R1 | GAGGTCGTCCGTCCACTC | |||
| RapB-S1 | TACCGAGCCGCAGGAACC | |||
| T-NOS | 180-F | CATGTAATGCATGACGTTATTTATG | 84 | Reiting et al., 2007 [25] |
| 180-R | TTGTTTTCTATCGCGTATTAAATGT | |||
| Tm-180 | ATGGGTTTTTATGATTAGAGTCCCGCAA | |||
| HPT | qHPTF286 | CAGGGTGTCACGTTGCAAGA | 110 | Lu et al., 2012 [26] |
| qHPTR395 | CCGCTCGTCTGGCTAAGATC | |||
| qHPTFP308 | TGCCTGAAACCGAACTGCCCGCTG | |||
| pCaMV35S | p35s-F | ATTGATGTGATATCTCCACTGACGT | 101 | Fu et al., 2017 [27] |
| p35s-R | CCTCTCCAAATGAAATGAACTTCCT | |||
| p35s-P | CCCACTATCCTTCGCAAGACCCTTCCT | |||
| PMI | PMIF240 | ACTGCCTTTCCTGTTCAAAGTATTAT | 96 | Lu et al., 2012 [22] |
| PMIR335 | TCTTTGGCAAAACCGATTTCAGAA | |||
| PMIP267 | CGCAGCACAGCCACTCTCCATTCAGG | |||
| TE9 | TE9-F | TGAGAATGAACAAAAGGACCATATCA | 87 | Debode et al., 2013 [27] |
| TE9-R | TTTTTATTCGGTTTTCGCTATCG | |||
| TE9-P | TCATTAACTCTTCTCCATCCATTTCCATTTCACAGT | |||
| Tg7 | Tg7-F | ATGCAAGTTTAAATTCAGAAATATTTCAA | 97 | Debode et al., 2013 [27] |
| Tg7-R | ATGTATTACACATAATATCGCACTCAGTCT | |||
| Tg7-P | ACTGATTATATCAGCTGGTACATTGCCGTAGATGA | |||
| T35S | T35SM-F | CCCTTAGTATGTATTTGTATTTGTAAAATACTTC | 83 | Pansiot et al., 2011 [28] |
| T35SM-R | GGATTTTAGTACTGGATTTTGGTTTTAG | |||
| T35S-P | TATCAATAAAATTTCTAATTC | |||
| P-NOS | P-NOS-F | GTGACCTTAGGCGACTTTTGAAC | 79 | Debode et al., 2013 [27] |
| P-NOS-R | CGCGGGTTTCTGGAGTTTAA | |||
| P-NOS-P | CGCAATAATGGTTTCTGACGTATGTGCTTAGC | |||
| CruA | qCruAF | GGCCAGGGCTTCCGTGAT | 101 | Jacchia et al., 2009 [29] |
| qCruAR | CCGTCGTTGTAGAACCATTGG | |||
| qCruFP | AGTCCTTATGTGCTCCACTTTCTGGTGCA | |||
| PLD | KVM-159 | TGGTGAGCGTTTTGCAGTCT | 68 | Mazzara et al., 2006 [30] |
| KVM-160 | CTGATCCACTAGCAGGAGGTCC | |||
| TM-013 | TGTTGTGCTGCCAATGTGGCCTG |
| Sample | Variation Source | Sum of Squares (SS) | Degrees of Freedom (df) | Mean of Squares (MS) | p Value | F value | F0.05(10,22) | |
|---|---|---|---|---|---|---|---|---|
| Type | No. | |||||||
| SDrape gDNA in non-GM rapeseed gDNA | S1-G-rape | Within-dPCR | 0.482 | 10 | 0.048 | 0.097 | 1.919 | 2.297 |
| Between-dPCR | 0.553 | 22 | 0.025 | |||||
| S2-G-rape | Within-dPCR | 0.052 | 10 | 0.005 | 0.529 | 0.925 | 2.297 | |
| Between-dPCR | 0.123 | 22 | 0.006 | |||||
| S3-G-rape | Within-dPCR | 0.011 | 10 | 0.0011 | 0.517 | 0.942 | 2.297 | |
| Between-dPCR | 0.026 | 22 | 0.0012 | |||||
| S4-G-rape | Within-dPCR | 0.003 | 10 | 0.0003 | 0.289 | 1.302 | 2.297 | |
| Between-dPCR | 0.006 | 22 | 0.0003 | |||||
| plasmid in non-GM rapeseed gDNA | S1-P-rape | Within-dPCR | 0.898 | 10 | 0.090 | 0.067 | 2.133 | 2.297 |
| Between-dPCR | 0.926 | 22 | 0.042 | |||||
| S2-P-rape | Within-dPCR | 0.059 | 10 | 0.006 | 0.082 | 2.017 | 2.297 | |
| Between-dPCR | 0.064 | 22 | 0.003 | |||||
| S3-P-rape | Within-dPCR | 0.016 | 10 | 0.002 | 0.922 | 0.419 | 2.297 | |
| Between-dPCR | 0.083 | 22 | 0.004 | |||||
| S4-P-rape | Within-dPCR | 0.004 | 10 | 0.0004 | 0.435 | 1.054 | 2.297 | |
| Between-dPCR | 0.008 | 22 | 0.0004 | |||||
| SDrice gDNA in non-GM rice gDNA | S1-G-rice | Within-dPCR | 1.467 | 10 | 0.147 | 0.063 | 2.161 | 2.297 |
| Between-dPCR | 1.493 | 22 | 0.068 | |||||
| S2-G-rice | Within-dPCR | 0.110 | 10 | 0.011 | 0.059 | 2.201 | 2.297 | |
| Between-dPCR | 0.110 | 22 | 0.005 | |||||
| S3-G-rice | Within-dPCR | 0.039 | 10 | 0.004 | 0.112 | 1.839 | 2.297 | |
| Between-dPCR | 0.047 | 22 | 0.002 | |||||
| S4-G-rice | Within-dPCR | 0.005 | 10 | 0.0005 | 0.282 | 1.317 | 2.297 | |
| Between-dPCR | 0.008 | 22 | 0.0004 | |||||
| plasmid in non-GM rice gDNA | S1-P-rice | Within-dPCR | 0.589 | 10 | 0.059 | 0.069 | 2.110 | 2.297 |
| Between-dPCR | 0.614 | 22 | 0.028 | |||||
| S2-P-rice | Within-dPCR | 0.055 | 10 | 0.006 | 0.502 | 0.960 | 2.297 | |
| Between-dPCR | 0.126 | 22 | 0.006 | |||||
| S3-P-rice | Within-dPCR | 0.013 | 10 | 0.001 | 0.465 | 1.012 | 2.297 | |
| Between-dPCR | 0.029 | 22 | 0.001 | |||||
| S4-P-rice | Within-dPCR | 0.001 | 10 | 0.0001 | 0.514 | 0.945 | 2.297 | |
| Between-dPCR | 0.003 | 22 | 0.0002 | |||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Si, N.; Li, J.; Gao, H.; Li, Y.; Zhai, S.; Xiao, F.; Zhang, L.; Wu, G.; Wu, Y. Effect of the Matrix and Target on the Accurate Quantification of Genomic and Plasmid DNA by Digital Polymerase Chain Reaction. Agriculture 2023, 13, 127. https://doi.org/10.3390/agriculture13010127
Si N, Li J, Gao H, Li Y, Zhai S, Xiao F, Zhang L, Wu G, Wu Y. Effect of the Matrix and Target on the Accurate Quantification of Genomic and Plasmid DNA by Digital Polymerase Chain Reaction. Agriculture. 2023; 13(1):127. https://doi.org/10.3390/agriculture13010127
Chicago/Turabian StyleSi, Nengwu, Jun Li, Hongfei Gao, Yunjing Li, Shanshan Zhai, Fang Xiao, Li Zhang, Gang Wu, and Yuhua Wu. 2023. "Effect of the Matrix and Target on the Accurate Quantification of Genomic and Plasmid DNA by Digital Polymerase Chain Reaction" Agriculture 13, no. 1: 127. https://doi.org/10.3390/agriculture13010127