
Fine-Tuning Quantitative Trait Loci Identified in Immortalized F2 Population Are Essential for Genomic Prediction of Hybrid Performance in Maize

 ,
,  ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Materials
2.2. Field Design and Trait Evaluation
2.3. Phenotypic Data Analyses
2.4. Genotyping and QTL Mapping
2.5. Genomic Prediction
3. Results
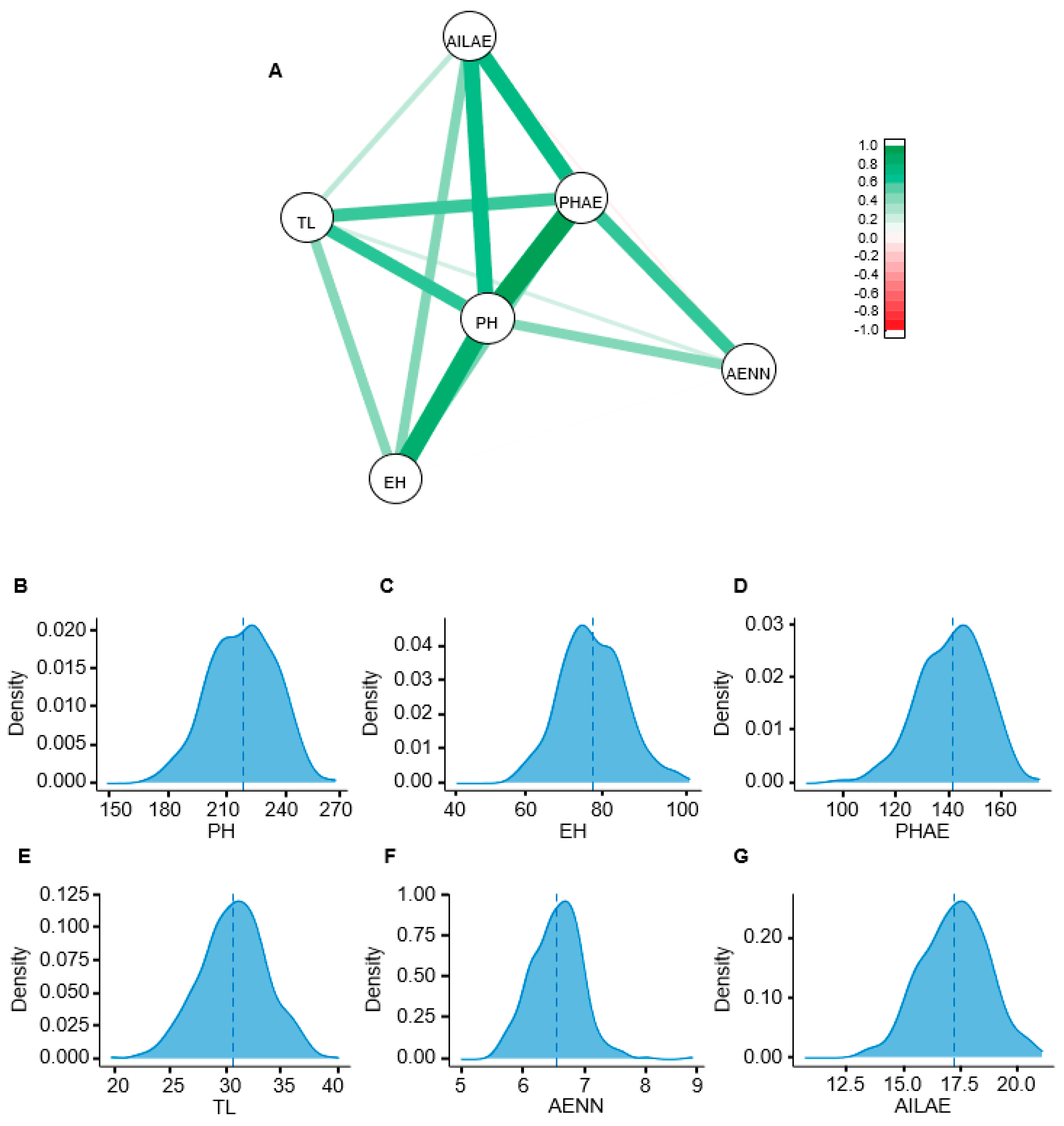
3.1. Phenotype Analysis under Multi-Environments
3.2. Additive Is the Main Contributor to All Tested Traits for the IMF2 Population
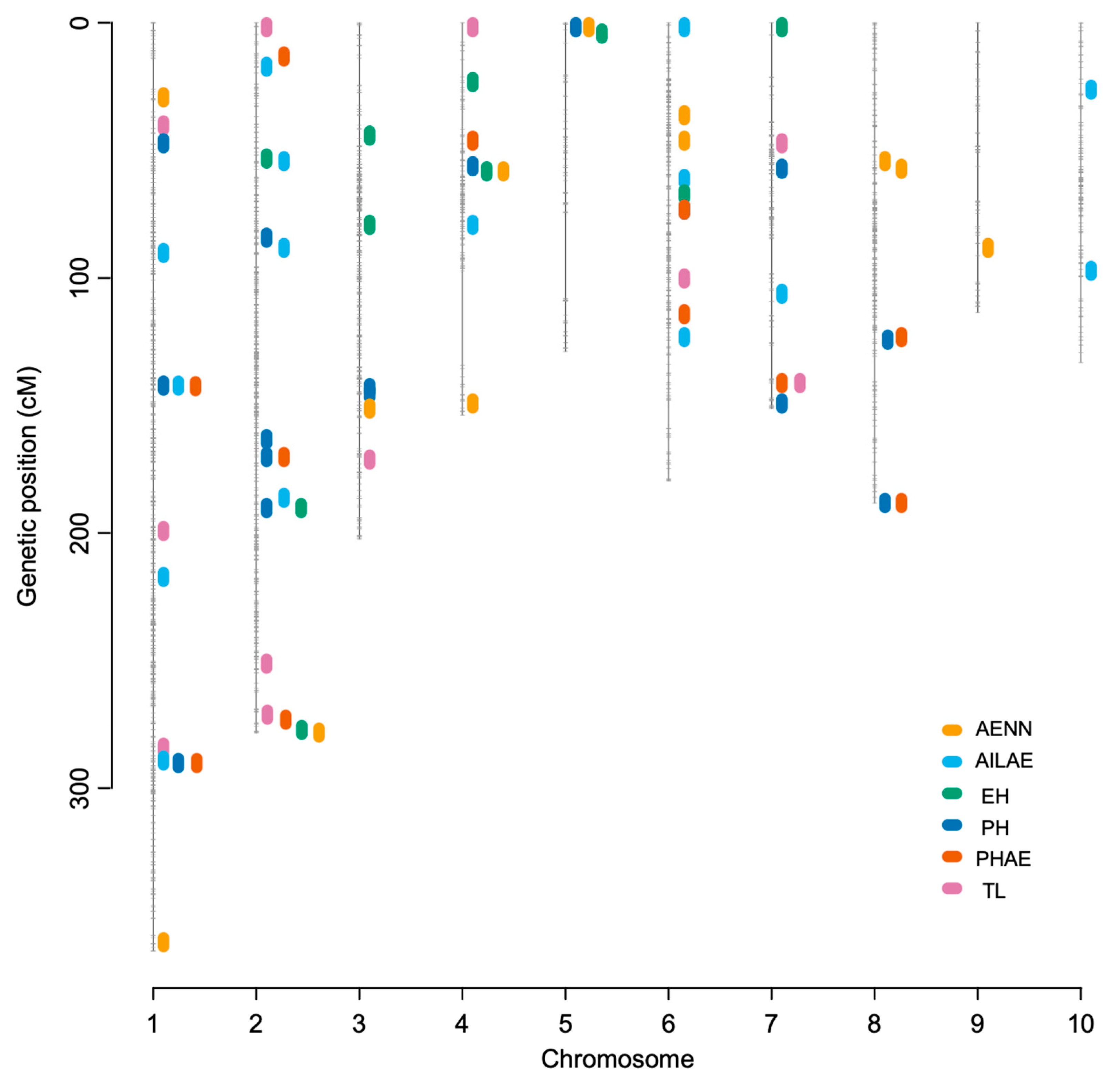
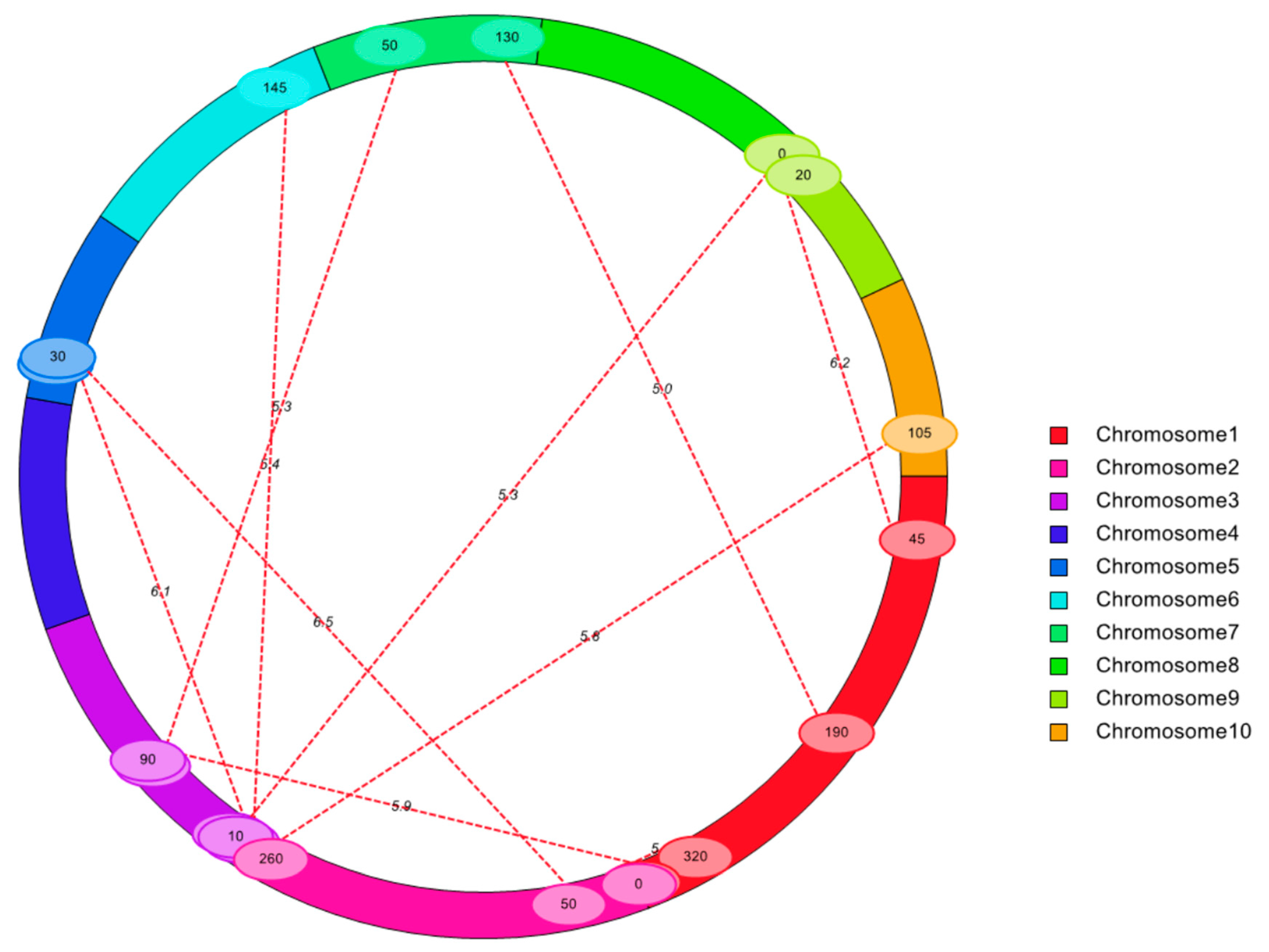
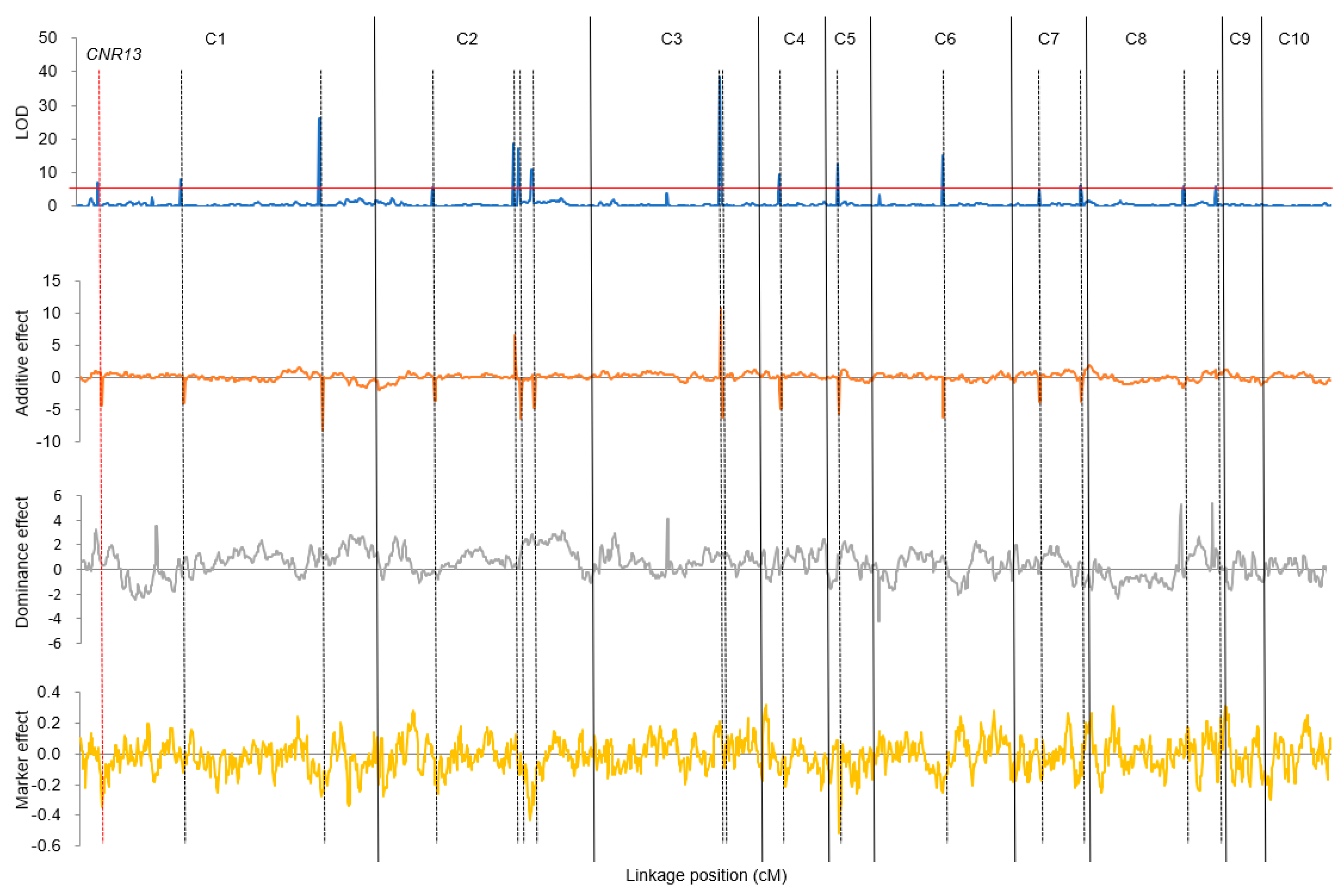
3.3. QTL Mapping Identified Pleiotropic Loci and Epistatic QTLs
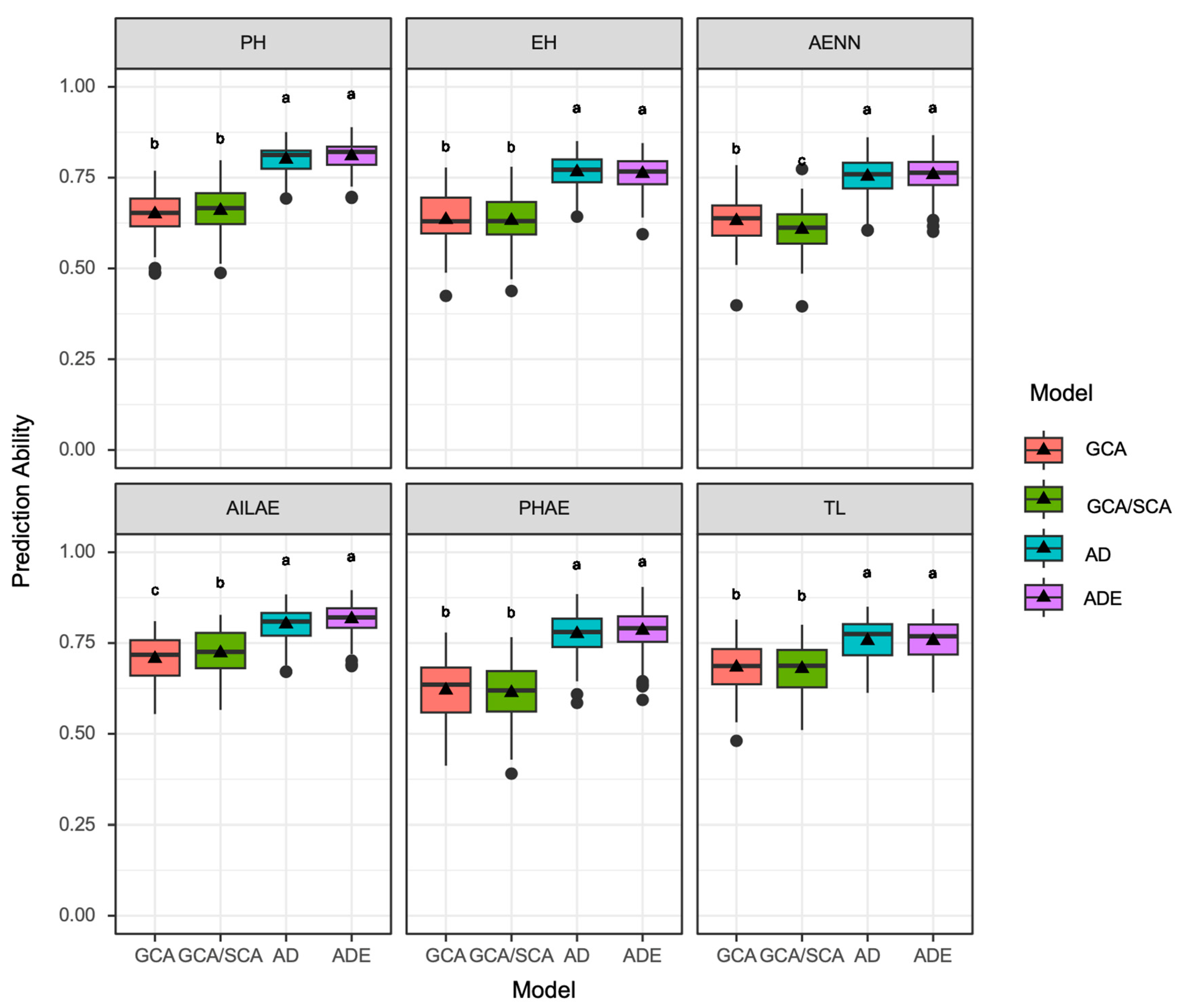
3.4. Comparison of Different Genomic Prediction Models
3.5. The Identified Fine-Tuning QTLs Are Essential for GP of Hybrid Performance
4. Discussion
4.1. Strong Additive Effects Contribute to Hybrid Performance
4.2. Fine-Tuning QTLs Could Improve the Prediction Accuracy
4.3. Models Fitting Different Genetic Factors Influence the Prediction Accuracy
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Xu, Y.; Skinner, D.J.; Wu, H.; Palacios-Rojas, N.; Araus, J.L.; Yan, J.; Gao, S.; Warburton, M.L.; Crouch, J.H. Advances in maize genomics and their value for enhancing genetic gains from breeding. Int. J. Plant Genom. 2009, 2009, 957602. [Google Scholar] [CrossRef]
- Hickey, L.T.; Hafeez, A.N.; Robinson, H.; Jackson, S.A.; Leal-Bertioli, S.C.M.; Tester, M.; Gao, C.; Godwin, I.D.; Hayes, B.J.; Wulff, B.B.H. Breeding crops to feed 10 billion. Nat. Biotechnol. 2019, 37, 744–754. [Google Scholar] [CrossRef] [PubMed]
- Tian, J.; Wang, C.; Xia, J.; Wu, L.; Xu, G.; Wu, W.; Li, D.; Qin, W.; Han, X.; Chen, Q.; et al. Teosinte ligule allele narrows plant architecture and enhances high-density maize yields. Science 2019, 365, 658–664. [Google Scholar] [CrossRef]
- Wang, P.; Yang, Y.; Li, D.; Xu, J.; Gu, R.; Zheng, J.; Fu, J.; Wang, J.; Zhang, H. Cloning of a new allele of ZmAMP1 and evaluation of its breeding value in hybrid maize. Crop J. 2023, 11, 157–165. [Google Scholar] [CrossRef]
- Duvick, D.N. The contribution of breeding to yield advances in maize (Zea mays L.). Adv. Agron. 2005, 86, 83–145. [Google Scholar] [CrossRef]
- Assefa, Y.; Prasad, P.V.V.; Carter, P.; Hinds, M.; Bhalla, G.; Schon, R.; Jeschke, M.; Paszkiewicz, S.; Ciampitti, I.A. Yield responses to planting density for US modern corn hybrids: A synthesis–analysis. Crop. Sci. 2016, 56, 2802–2817. [Google Scholar] [CrossRef]
- Liang, Y.; Liu, H.-J.; Yan, J.; Tian, F. Natural variation in crops: Realized understanding, continuing promise. Annu. Rev. Plant Biol. 2021, 72, 357–385. [Google Scholar] [CrossRef] [PubMed]
- Peng, J.; Richards, D.E.; Hartley, N.M.; Murphy, G.P.; Devos, K.M.; Flintham, J.E.; Beales, J.; Fish, L.J.; Worland, A.J.; Pelica, F.; et al. ‘Green revolution’ genes encode mutant gibberellin response modulators. Nature 1999, 400, 256–261. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, A.; Ashikari, M.; Ueguchi-Tanaka, M.; Itoh, H.; Nishimura, A.; Swapan, D.; Ishiyama, K.; Saito, T.; Kobayashi, M.; Khush, G.S.; et al. A mutant gibberellin-synthesis gene in rice. Nature 2002, 416, 701–702. [Google Scholar] [CrossRef]
- Yano, M.; Katayose, Y.; Ashikari, M.; Yamanouchi, U.; Monna, L.; Fuse, T.; Baba, T.; Yamamoto, K.; Umehara, Y.; Nagamura, Y.; et al. Hd1, a major photoperiod sensitivity quantitative trait locus in rice, is closely related to the Arabidopsis flowering time gene CONSTANS. Plant Cell 2000, 12, 2473. [Google Scholar] [CrossRef]
- Zhang, Z.H.; Wang, K.; Guo, L.; Zhu, Y.J.; Fan, Y.Y.; Cheng, S.H.; Zhuang, J.Y. Pleiotropism of the photoperiod-insensitive allele of Hd1 on heading date, plant height and yield traits in rice. PLoS ONE 2012, 7, e52538. [Google Scholar] [CrossRef]
- Wojciechowski, T.; Gooding, M.; Ramsay, L.; Gregory, P. The effects of dwarfing genes on seedling root growth of wheat. J. Exp. Bot. 2009, 60, 2565–2573. [Google Scholar] [CrossRef] [PubMed]
- Salvi, S.; Sponza, G.; Morgante, M.; Tomes, D.; Niu, X.; Fengler, K.A.; Meeley, R.; Ananiev, E.V.; Svitashev, S.; Bruggemann, E.; et al. Conserved noncoding genomic sequences associated with a flowering-time quantitative trait locus in maize. Proc. Natl. Acad. Sci. USA 2007, 104, 11376–11381. [Google Scholar] [CrossRef] [PubMed]
- Teng, F.; Zhai, L.; Liu, R.; Bai, W.; Wang, L.; Huo, D.; Tao, Y.; Zheng, Y.; Zhang, Z. ZmGA3ox2, a candidate gene for a major QTL, qPH3.1, for plant height in maize. Plant J. 2013, 73, 405–416. [Google Scholar] [CrossRef] [PubMed]
- Xing, A.; Gao, Y.; Ye, L.; Zhang, W.; Cai, L.; Ching, A.; Llaca, V.; Johnson, B.; Liu, L.; Yang, X.; et al. A rare SNP mutation in Brachytic2 moderately reduces plant height and increases yield potential in maize. J. Exp. Bot. 2015, 66, 3791–3802. [Google Scholar] [CrossRef] [PubMed]
- Wei, L.; Zhang, X.; Zhang, Z.; Liu, H.; Lin, Z. A new allele of the Brachytic2 gene in maize can efficiently modify plant architecture. Heredity 2018, 121, 75–86. [Google Scholar] [CrossRef]
- Wang, F.; Yu, Z.; Zhang, M.; Wang, M.; Lu, X.; Liu, X.; Li, Y.; Zhang, X.; Tan, B.; Li, C.; et al. ZmTE1 promotes plant height by regulating intercalary meristem formation and internode cell elongation in maize. Plant Biotechnol. J. 2021, 20, 526–537. [Google Scholar] [CrossRef]
- Lv, H.; Zheng, J.; Wang, T.; Fu, J.; Huai, J.; Min, H.; Zhang, X.; Tian, B.; Shi, Y.; Wang, G. The maize d2003, a novel allele of VP8, is required for maize internode elongation. Plant Mol. Biol. 2013, 84, 243–257. [Google Scholar] [CrossRef]
- Birchler, J.A.; Yao, H.; Chudalayandi, S. Unraveling the genetic basis of hybrid vigor. Proc. Natl. Acad. Sci. USA 2006, 103, 12957–12958. [Google Scholar] [CrossRef]
- Lippman, Z.B.; Zamir, D. Heterosis: Revisiting the magic. Trends Genet. 2007, 23, 60–66. [Google Scholar] [CrossRef]
- Wang, H.; Xu, C.; Liu, X.; Guo, Z.; Xu, X.; Wang, S.; Xie, C.; Li, W.-X.; Zou, C.; Xu, Y. Development of a multiple-hybrid population for genome-wide association studies: Theoretical consideration and genetic mapping of flowering traits in maize. Sci. Rep. 2017, 7, 40239. [Google Scholar] [CrossRef] [PubMed]
- Hua, J.; Xing, Y.; Wu, W.; Xu, C.; Sun, X.; Yu, S.; Zhang, Q. Single-locus heterotic effects and dominance by dominance interactions can adequately explain the genetic basis of heterosis in an elite rice hybrid. Proc. Natl. Acad. Sci. USA 2003, 100, 2574–2579. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.; Ma, X.; Teng, W.; Yan, J.; Wu, W.; Dai, J.; Li, J. Detection of quantitative trait loci and heterotic loci for plant height using an immortalized F2 population in maize. Chin. Sci. Bull. 2007, 52, 477–483. [Google Scholar] [CrossRef]
- Li, D.; Zhou, Z.; Lu, X.; Jiang, Y.; Li, G.; Li, J.; Wang, H.; Chen, S.; Li, X.; Würschum, T.; et al. Genetic dissection of hybrid performance and heterosis for yield-related traits in maize. Front. Plant Sci. 2021, 12, 774478. [Google Scholar] [CrossRef] [PubMed]
- Meuwissen, T.H.; Hayes, B.J.; E Goddard, M. Prediction of total genetic value using genome-wide dense marker maps. Genetics 2001, 157, 1819–1829. [Google Scholar] [CrossRef] [PubMed]
- Bernardo, R.; Yu, J. Prospects for genome wide selection for quantitative traits in maize. Crop. Sci. 2007, 47, 1082–1090. [Google Scholar] [CrossRef]
- Hickey, J.M.; Chiurugwi, T.; Mackay, I.; Powell, W. Genomic prediction unifies animal and plant breeding programs to form platforms for biological discovery. Nat. Genet. 2017, 49, 1297–1303. [Google Scholar] [CrossRef]
- Xu, Y.; Liu, X.; Fu, J.; Wang, H.; Wang, J.; Huang, C.; Prasanna, B.M.; Olsen, M.S.; Wang, G.; Zhang, A. Enhancing genetic gain through genomic selection: From livestock to plants. Plant Commun. 2019, 1, 100005. [Google Scholar] [CrossRef]
- Xu, S.; Zhu, D.; Zhang, Q. Predicting hybrid performance in rice using genomic best linear unbiased prediction. Proc. Natl. Acad. Sci. USA 2014, 111, 12456–12461. [Google Scholar] [CrossRef]
- Covarrubias-Pazaran, G. Genome-assisted prediction of quantitative traits using the R package sommer. PLoS ONE 2016, 11, e0156744. [Google Scholar] [CrossRef]
- Li, D.; Li, G.; Wang, H.; Guo, Y.; Wang, M.; Lu, X.; Luo, Z.; Zhu, X.; Weiß, T.M.; Roller, S.; et al. Genetic dissection of phosphorus use efficiency and genotype-by-environment interaction in maize. Int. J. Mol. Sci. 2022, 23, 13943. [Google Scholar] [CrossRef]
- Bernal-Vasquez, A.-M.; Utz, H.-F.; Piepho, H.-P. Outlier detection methods for generalized lattices: A case study on the transition from ANOVA to REML. Theor. Appl. Genet. 2016, 129, 787–804. [Google Scholar] [CrossRef]
- Cullis, B.R.; Smith, A.B.; Coombes, N.E. On the design of early generation variety trials with correlated data. J. Agric. Biol. Environ. Stat. 2006, 11, 381–393. [Google Scholar] [CrossRef]
- Butler, D.G.; Cullis, B.R.; Gilmour, A.R.; Gogel, B.J.; Thompson, R. ASReml-R Reference Manual Version 4; VSN International Ltd.: Hemel Hempstead, UK, 2017. [Google Scholar]
- Doyle, J.J.; Doyle, J.L. A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem. Bull. 1987, 19, 11–15. [Google Scholar]
- Baurley, J.W.; Edlund, C.K.; Pardamean, C.I.; Conti, D.V.; Bergen, A.W. Smokescreen: A targeted genotyping array for addiction research. BMC Genom. 2016, 17, 145. [Google Scholar] [CrossRef]
- Furuta, T.; Ashikari, M.; Jena, K.K.; Doi, K.; Reuscher, S. Adapting genotyping-by-sequencing for rice F2 populations. G3 Genes|Genomes|Genet. 2017, 7, 881–893. [Google Scholar] [CrossRef]
- Meng, L.; Li, H.; Zhang, L.; Wang, J. QTL IciMapping: Integrated software for genetic linkage map construction and quantitative trait locus mapping in biparental populations. Crop J. 2015, 3, 269–283. [Google Scholar] [CrossRef]
- Pérez, P.; de los Campos, G. Genome-wide regression and prediction with the BGLR statistical package. Genetics 2014, 198, 483–495. [Google Scholar] [CrossRef]
- Wang, B.; Hou, M.; Shi, J.; Ku, L.; Song, W.; Li, C.; Ning, Q.; Li, X.; Li, C.; Zhao, B.; et al. De novo genome assembly and analyses of 12 founder inbred lines provide insights into maize heterosis. Nat. Genet. 2023, 55, 312–323. [Google Scholar] [CrossRef]
- Yi, Q.; Liu, Y.; Hou, X.; Zhang, X.; Li, H.; Zhang, J.; Liu, H.; Hu, Y.; Yu, G.; Li, Y.; et al. Genetic dissection of yield-related traits and mid-parent heterosis for those traits in maize (Zea mays L.). BMC Plant Biol. 2019, 19, 392. [Google Scholar] [CrossRef]
- Mayer, M.; Hölker, A.C.; González-Segovia, E.; Bauer, E.; Presterl, T.; Ouzunova, M.; Melchinger, A.E.; Schön, C.-C. Discovery of beneficial haplotypes for complex traits in maize landraces. Nat. Commun. 2020, 11, 4954. [Google Scholar] [CrossRef]
- Tang, J.; Tian, X.; Mei, E.; He, M.; Gao, J.; Yu, J.; Xu, M.; Liu, J.; Song, L.; Li, X.; et al. WRKY53 negatively regulates rice cold tolerance at the booting stage by fine-tuning anther gibberellin levels. Plant Cell 2022, 34, 4495–4515. [Google Scholar] [CrossRef]
- Yue, Z.L.; Liu, N.; Deng, Z.P.; Zhang, Y.; Wu, Z.M.; Zhao, J.L.; Sun, Y.; Wang, Z.Y.; Zhang, S.W. The receptor kinase OsWAK11 monitors cell wall pectin changes to fine-tune brassinosteroid signaling and regulate cell elongation in rice. Curr. Biol. 2022, 32, 2454–2466. [Google Scholar] [CrossRef]
- Li, H.; Zhang, Y.; Wu, C.; Bi, J.; Chen, Y.; Jiang, C.; Cui, M.; Chen, Y.; Hou, X.; Yuan, M.; et al. Fine--tuning OsCPK18/OsCPK4 activity via genome editing of phosphorylation motif improves rice yield and immunity. Plant Biotechnol. J. 2022, 20, 2258–2271. [Google Scholar] [CrossRef]
- Louis, J.; Basu, S.; Varsani, S.; Castano-Duque, L.; Jiang, V.; Williams, W.P.; Felton, G.W.; Luthe, D.S. Ethylene contributes to maize insect resistance1-mediated maize defense against the phloem sap-sucking corn leaf aphid. Plant Physiol. 2015, 169, 313–324. [Google Scholar] [CrossRef]
- Khadka, K.; Raizada, M.N.; Navabi, A. Recent progress in germplasm evaluation and gene mapping to enable breeding of drought-tolerant wheat. Front. Plant Sci. 2020, 11, 1149. [Google Scholar] [CrossRef]
- Xu, S. Mapping quantitative trait loci by controlling polygenic background effects. Genetics 2013, 195, 1209–1222. [Google Scholar] [CrossRef]
- Jiang, Y.; Schmidt, R.H.; Zhao, Y.; Reif, J.C. A quantitative genetic framework highlights the role of epistatic effects for grain-yield heterosis in bread wheat. Nat. Genet. 2017, 49, 1741–1746. [Google Scholar] [CrossRef]
- Zhao, Y.; Li, Z.; Liu, G.; Jiang, Y.; Maurer, H.P.; Würschum, T.; Mock, H.-P.; Matros, A.; Ebmeyer, E.; Schachschneider, R.; et al. Genome-based establishment of a high-yielding heterotic pattern for hybrid wheat breeding. Proc. Natl. Acad. Sci. USA 2015, 112, 15624–15629. [Google Scholar] [CrossRef]
- Sprague, G.F.; Tatum, L.A. General vs. specific combining ability in single crosses of corn. Agron. J. 1942, 34, 923–932. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Traits | Mean | Min | Max | SD | CV (%) | σ2G | σ2G×E | NE | h2 |
|---|---|---|---|---|---|---|---|---|---|
| PH | 220.0 | 173.0 | 267.9 | 17.06 | 7.75 | 271.76 ** | 28.13 ** | 3 | 0.94 |
| EH | 78.6 | 58.8 | 104.1 | 8.36 | 10.64 | 63.13 ** | 8.37 ** | 3 | 0.91 |
| PHAE | 141.6 | 97.2 | 174.8 | 12.70 | 8.97 | 148.54 ** | 17.17 ** | 3 | 0.93 |
| TL | 30.7 | 18.7 | 40.3 | 3.34 | 10.89 | 9.41 ** | 2.23 ** | 3 | 0.85 |
| AENN | 6.7 | 5.7 | 8.9 | 0.42 | 6.28 | 0.15 ** | 0.01 ** | 3 | 0.86 |
| AILAE | 17.3 | 13.2 | 21.1 | 1.45 | 8.41 | 1.85 ** | 0.22 ** | 3 | 0.89 |
| Env. | Trait | Chr. | Pos. | Left Marker | Right Marker | LOD | PVE (%) | Add. | Dom. |
|---|---|---|---|---|---|---|---|---|---|
| BLUE | PH | 1 | 141 | S1_110002889 | S1_143444026 | 7.9 | 2.4 | −4.04 | 0.84 |
| BLUE | PHAE | 1 | 141 | S1_110002889 | S1_143444026 | 9.9 | 6.4 | −4.15 | 0.48 |
| BLUE | AILAE | 1 | 141 | S1_110002889 | S1_143444026 | 14.5 | 6.6 | −0.50 | 0.13 |
| 21XT | PH | 1 | 141 | S1_110002889 | S1_143444026 | 72.4 | 24.3 | −21.76 | 0.65 |
| 21XT | PHAE | 1 | 141 | S1_110002889 | S1_143444026 | 13.2 | 6.7 | −6.69 | 0.53 |
| 21XT | AILAE | 1 | 141 | S1_110002889 | S1_143444026 | 4.2 | 3.2 | −0.35 | 0.12 |
| BLUE | PH | 1 | 289 | S1_269329879 | S1_270329585 | 26.2 | 9.3 | −8.23 | 0.91 |
| BLUE | PHAE | 1 | 289 | S1_269329879 | S1_270329585 | 20.2 | 13.7 | −6.08 | 1.53 |
| 21XT | PH | 1 | 289 | S1_269329879 | S1_270329585 | 16.9 | 3.3 | −8.20 | 1.13 |
| 21XT | PHAE | 1 | 289 | S1_269329879 | S1_270329585 | 38.5 | 23.8 | −12.84 | 1.78 |
| BLUE | TL | 1 | 290 | S1_273002635 | S1_273836450 | 26.5 | 15.8 | −1.99 | 0.28 |
| 21XT | TL | 1 | 290 | S1_273002635 | S1_273836450 | 12.5 | 8.0 | −1.55 | 0.21 |
| BLUE | AILAE | 1 | 292 | S1_273608504 | S1_275164590 | 10.9 | 4.7 | −0.43 | 0.11 |
| 21LY | PH | 1 | 292 | S1_273608504 | S1_275164590 | 9.7 | 8.4 | −6.71 | 2.30 |
| 21LY | PHAE | 1 | 292 | S1_273608504 | S1_275164590 | 5.4 | 5.3 | −4.04 | 0.75 |
| 21LY | TL | 1 | 295 | S1_279919454 | S1_279982555 | 6.0 | 3.0 | −1.24 | 0.50 |
| 21LY | PH | 2 | 184 | S2_190810969 | S2_191462633 | 9.0 | 7.9 | −6.68 | 2.63 |
| 21LY | PHAE | 2 | 184 | S2_190810969 | S2_191462633 | 6.1 | 6.3 | −4.46 | 1.26 |
| BLUE | AILAE | 2 | 185 | S2_190810969 | S2_191462633 | 8.5 | 3.7 | −0.41 | −0.03 |
| 21XT | AILAE | 2 | 188 | S2_193721872 | S2_193814824 | 6.6 | 5.0 | −0.46 | 0.19 |
| BLUE | EH | 2 | 189 | S2_194070269 | S2_194953435 | 9.5 | 7.3 | −2.94 | 1.29 |
| 21XT | PH | 2 | 190 | S2_194070269 | S2_194953435 | 20.9 | 4.2 | −9.24 | 3.24 |
| 21XX | EH | 2 | 190 | S2_194070269 | S2_194953435 | 6.5 | 4.9 | −2.25 | 0.63 |
| BLUE | PH | 2 | 191 | S2_194953435 | S2_195895088 | 11.2 | 3.4 | −4.89 | 2.16 |
| 21XX | TL | 2 | 271 | S2_241126960 | S2_241790578 | 4.7 | 3.8 | −1.14 | −0.12 |
| BLUE | TL | 2 | 272 | S2_241126960 | S2_241790578 | 7.5 | 4.0 | −0.97 | −0.37 |
| BLUE | PHAE | 2 | 273 | S2_241126960 | S2_241790578 | 10.3 | 6.8 | −4.45 | −0.45 |
| 21XT | TL | 2 | 274 | S2_241126960 | S2_241790578 | 8.9 | 5.5 | −1.31 | −0.32 |
| 21XX | PHAE | 2 | 274 | S2_241126960 | S2_241790578 | 8.4 | 6.5 | −4.53 | 1.31 |
| BLUE | EH | 2 | 277 | S2_242359991 | S2_243177289 | 11.2 | 8.9 | 3.24 | 1.31 |
| 21XT | EH | 2 | 277 | S2_242359991 | S2_243177289 | 7.8 | 3.4 | 3.42 | 1.42 |
| BLUE | AENN | 2 | 278 | S2_243537180 | S2_244109746 | 12.6 | 2.0 | −0.17 | −0.03 |
| 21LY | AENN | 2 | 278 | S2_243537180 | S2_244109746 | 5.6 | 1.2 | −0.14 | −0.01 |
| 21XT | AENN | 2 | 278 | S2_243537180 | S2_244109746 | 13.9 | 3.0 | −0.18 | −0.04 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, P.; Ma, X.; Jin, X.; Wu, X.; Zhang, X.; Zhang, H.; Wang, H.; Zhang, H.; Fu, J.; Xie, Y.; et al. Fine-Tuning Quantitative Trait Loci Identified in Immortalized F2 Population Are Essential for Genomic Prediction of Hybrid Performance in Maize. Agriculture 2024, 14, 340. https://doi.org/10.3390/agriculture14030340
Wang P, Ma X, Jin X, Wu X, Zhang X, Zhang H, Wang H, Zhang H, Fu J, Xie Y, et al. Fine-Tuning Quantitative Trait Loci Identified in Immortalized F2 Population Are Essential for Genomic Prediction of Hybrid Performance in Maize. Agriculture. 2024; 14(3):340. https://doi.org/10.3390/agriculture14030340
Chicago/Turabian StyleWang, Pingxi, Xingye Ma, Xining Jin, Xiangyuan Wu, Xiaoxiang Zhang, Huaisheng Zhang, Hui Wang, Hongwei Zhang, Junjie Fu, Yuxin Xie, and et al. 2024. "Fine-Tuning Quantitative Trait Loci Identified in Immortalized F2 Population Are Essential for Genomic Prediction of Hybrid Performance in Maize" Agriculture 14, no. 3: 340. https://doi.org/10.3390/agriculture14030340