
Identification of Conserved Pathways in Bacillus Strains Known for Plant Growth-Promoting Behavior Using a Multifaceted Computational Approach


Abstract
:1. Introduction
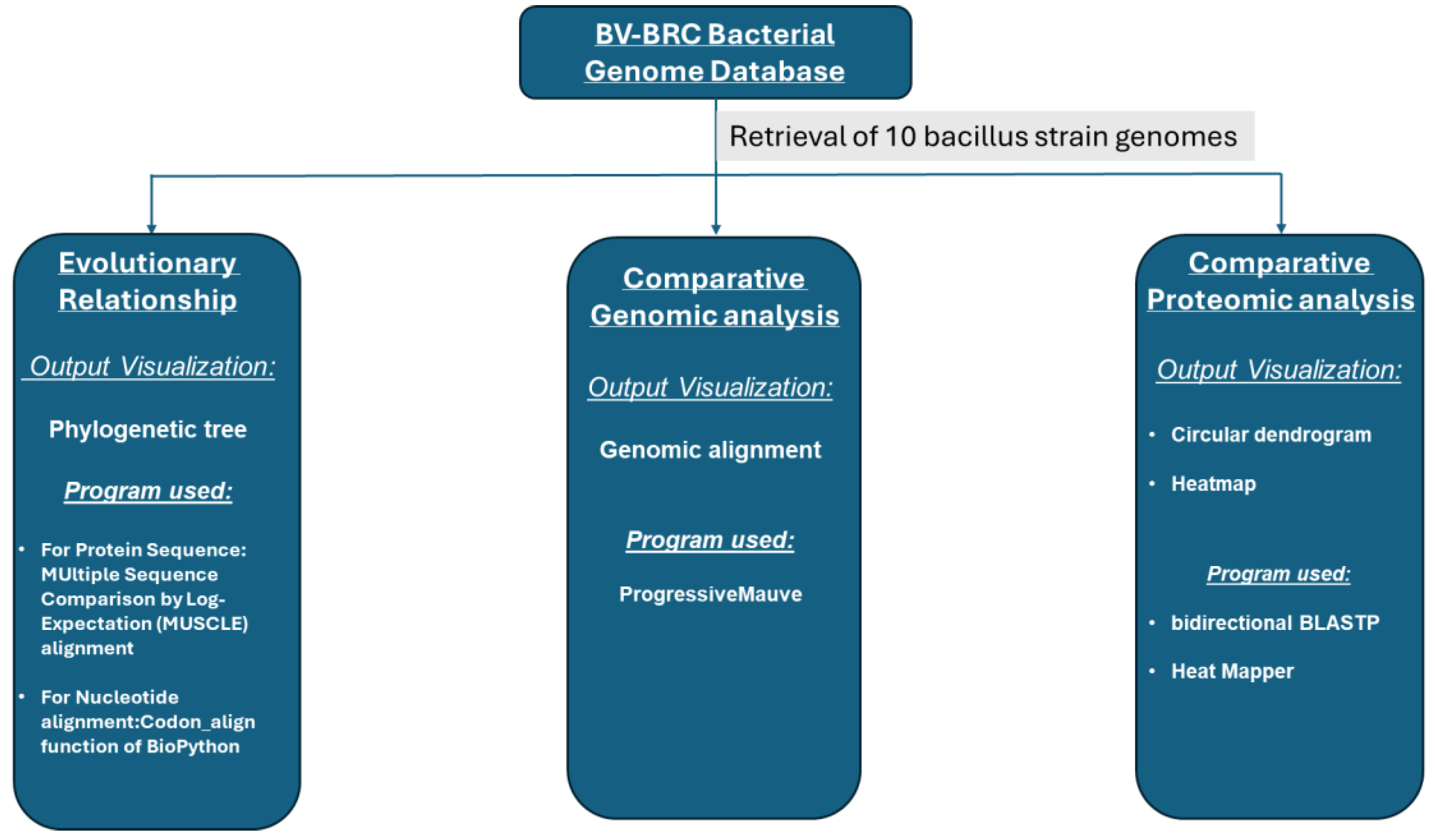
2. Materials and Methods
2.1. Genome Retrieval
2.2. Evaluation Using a Phylogenetic Tree
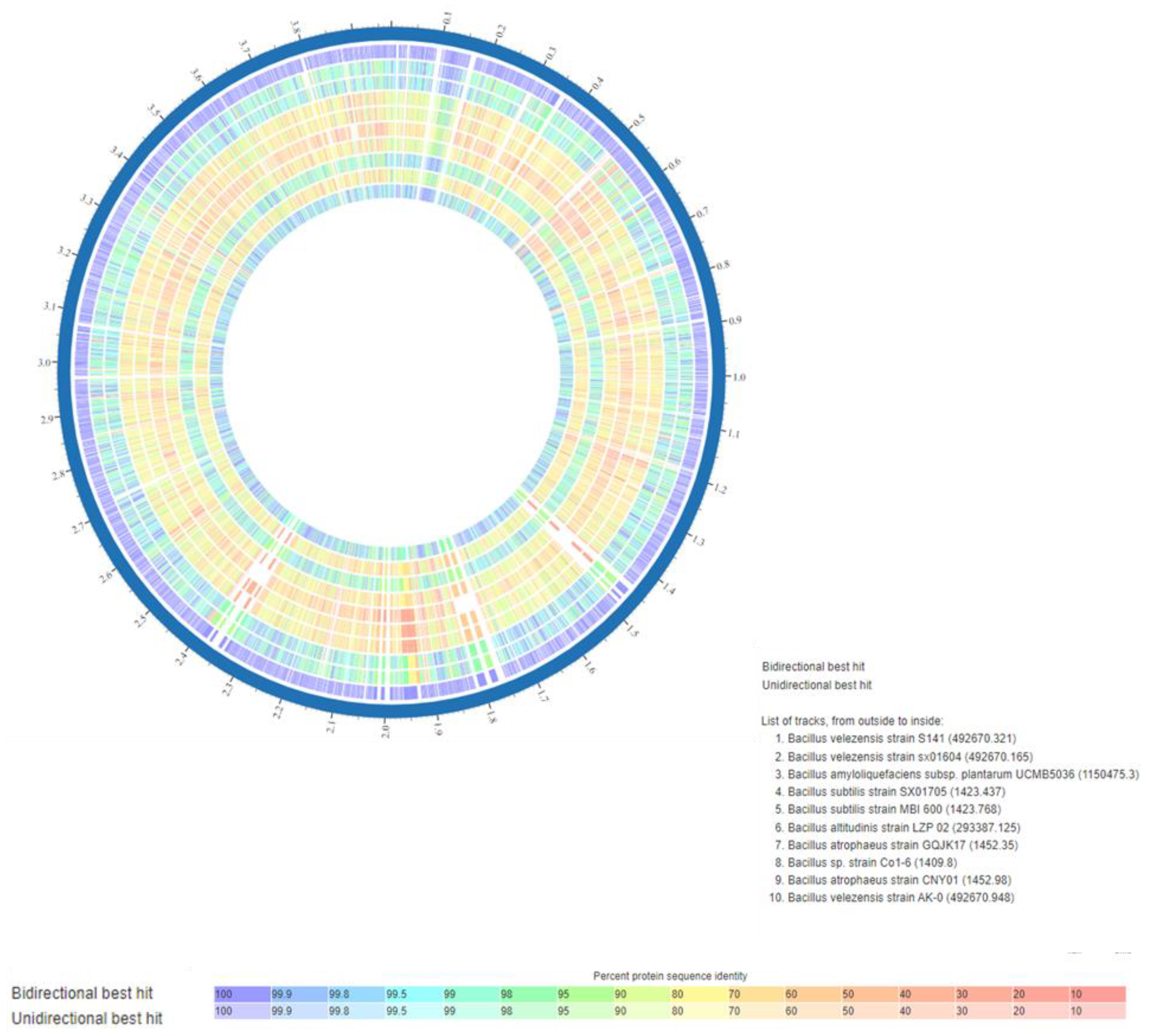
2.3. Comparative Genomic Analysis
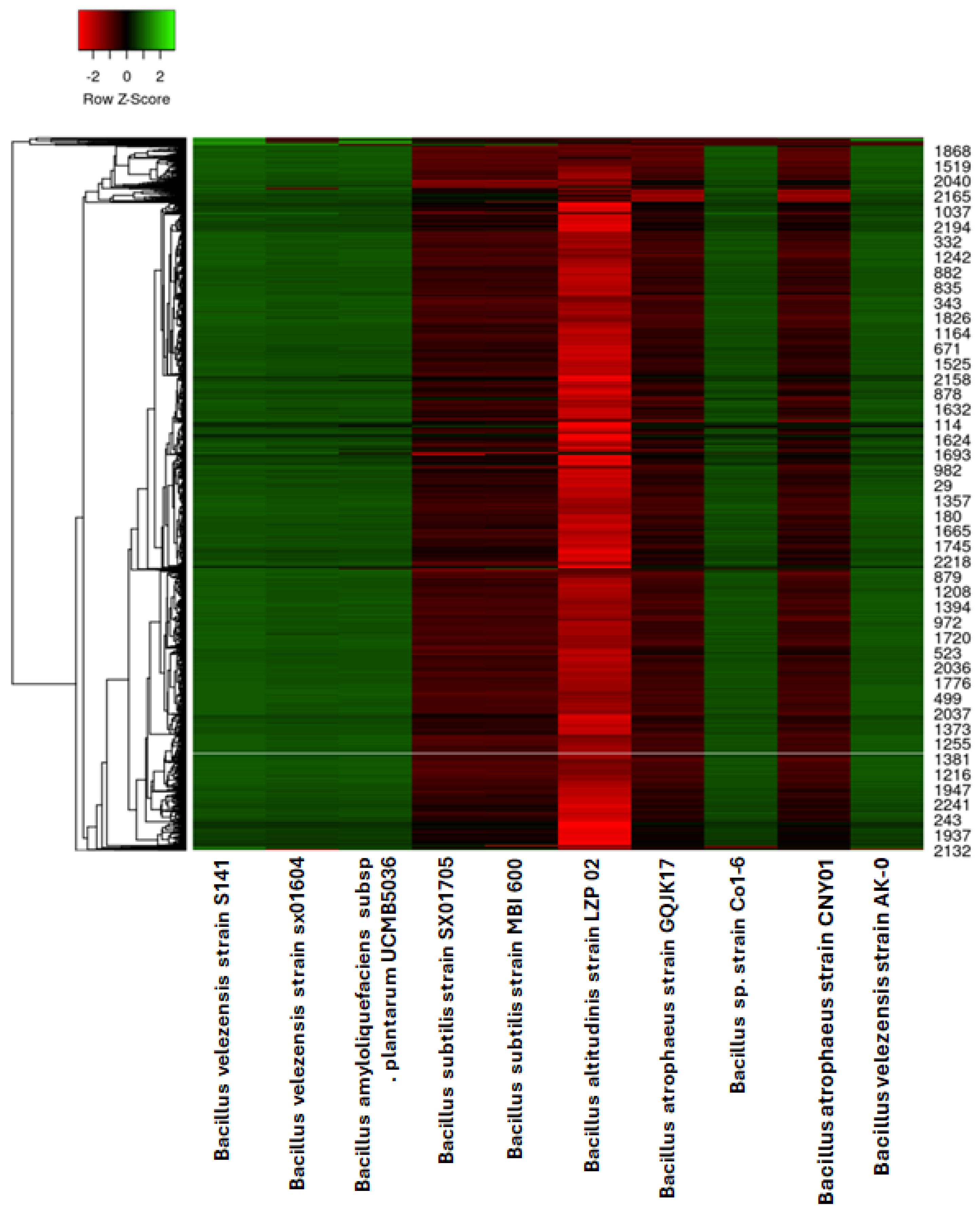
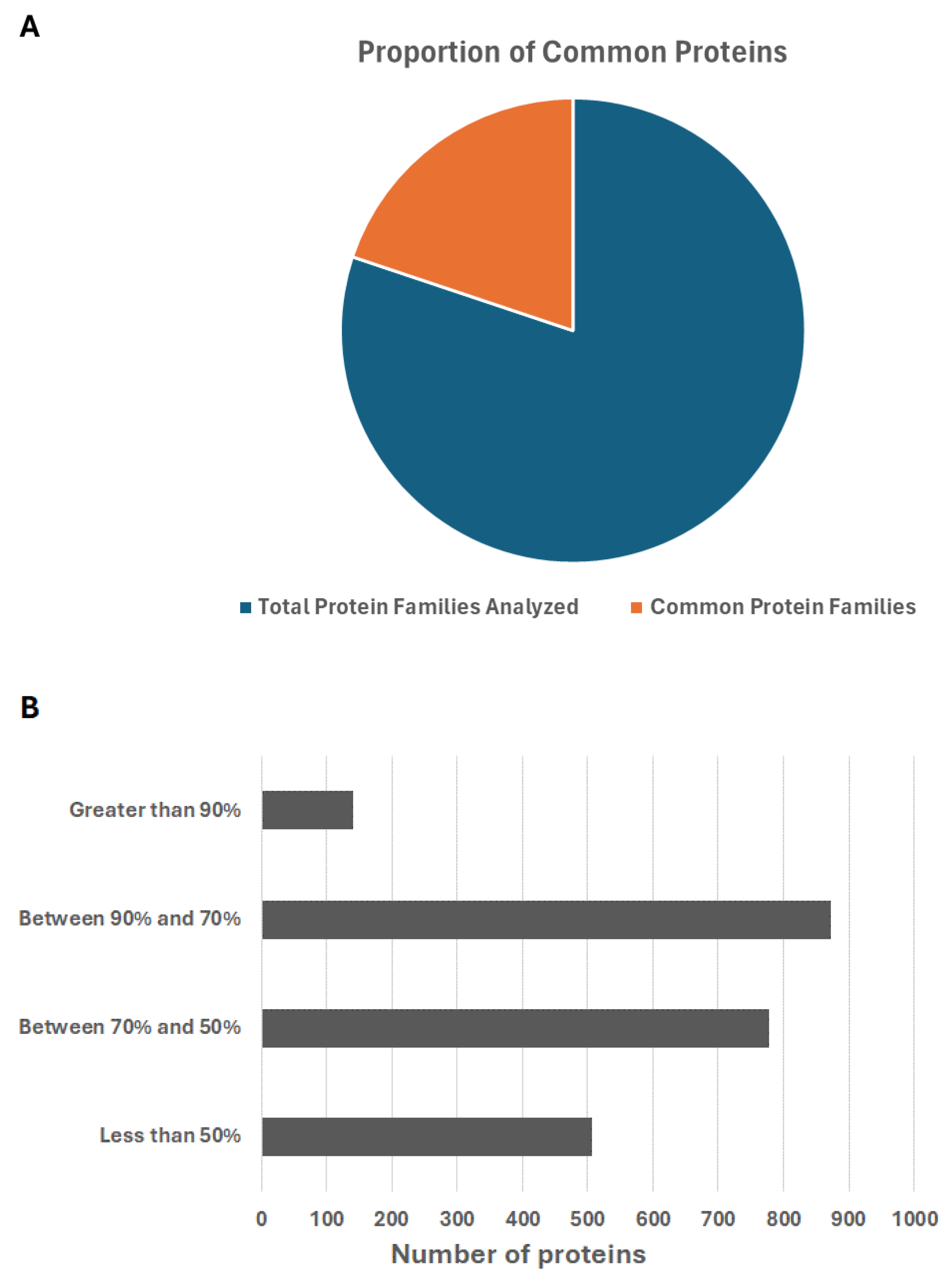
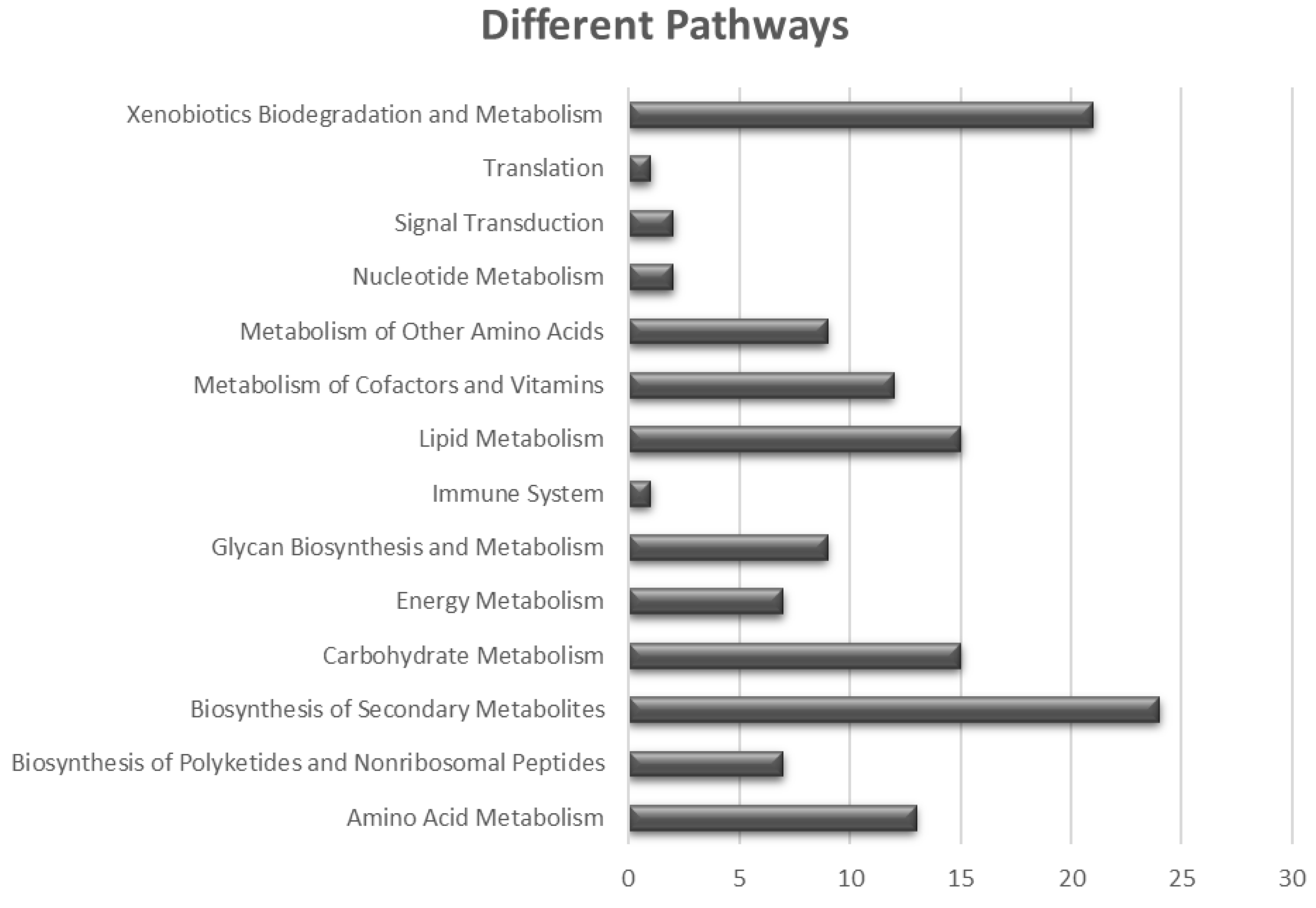
2.4. Comparative Proteomic Analysis
3. Results
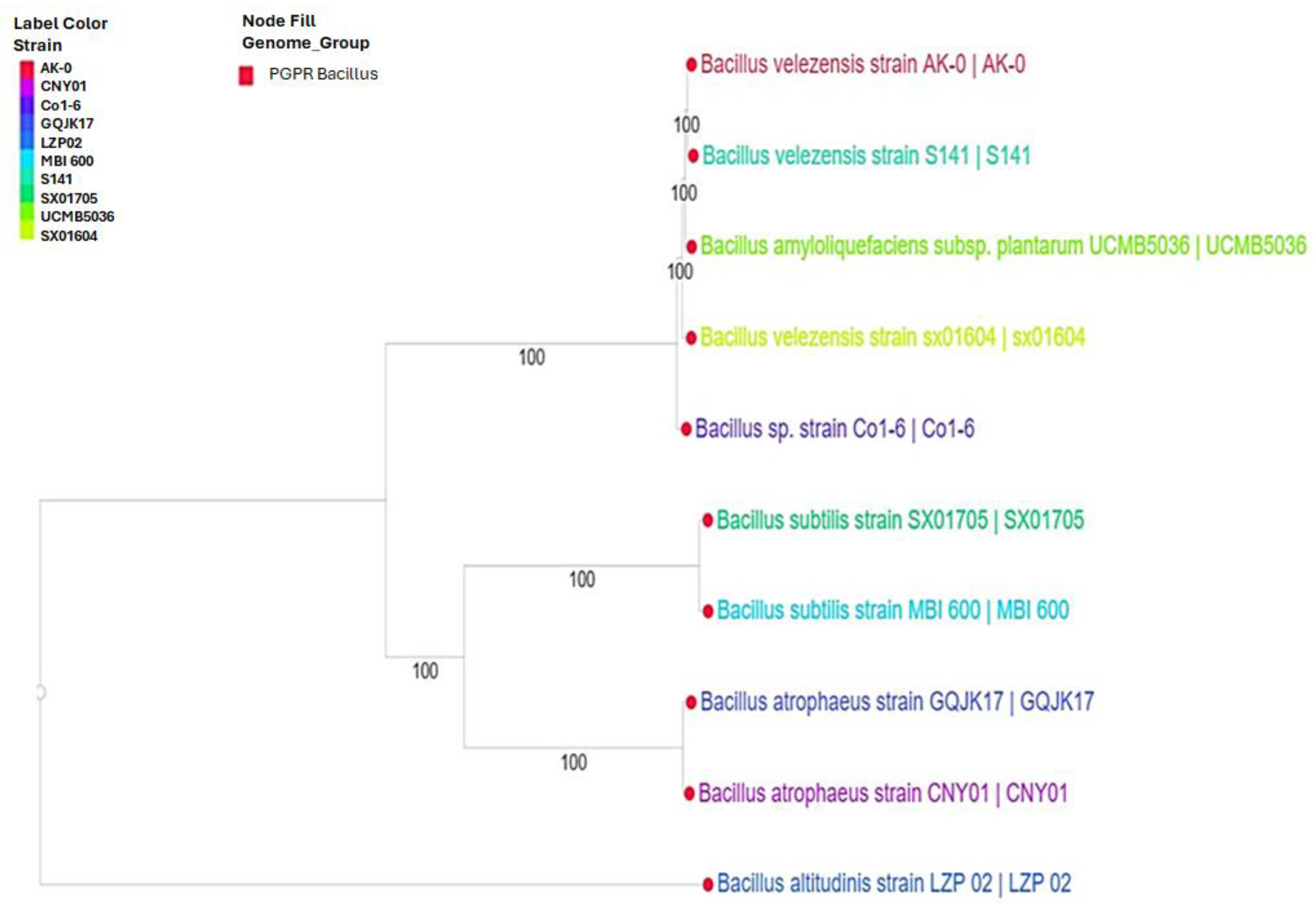
3.1. Phylogenetic Tree Showing the Evolutionary Relationships between Selected Bacillus Strains with a Positive Impact on Plant Growth
- Bacillus altitudinis group: This group consists of only Bacillus altitudinis strain LZP 02. As mentioned earlier, this strain is the most distantly related to all other strains in the tree.
- Bacillus atrophaeus group: This group consisted of Bacillus atrophaeus strains GQJK17 and CNY01. These two strains are more closely related to each other than any other strains in the tree.
- Bacillus velezensis/Bacillus subtilis group: This group is the largest and most diverse group in the tree. These strains included Bacillus velezensis strains sx01604, S141, and AK-04, Bacillus subtilis strains SX01705 and MBI 600, and Bacillus sp. strain Co1-6. The Bacillus velezensis and Bacillus subtilis strains are the most closely related strains in this group, while Bacillus sp. strain Co1-6 is more closely related to them than to the Bacillus atrophaeus group.
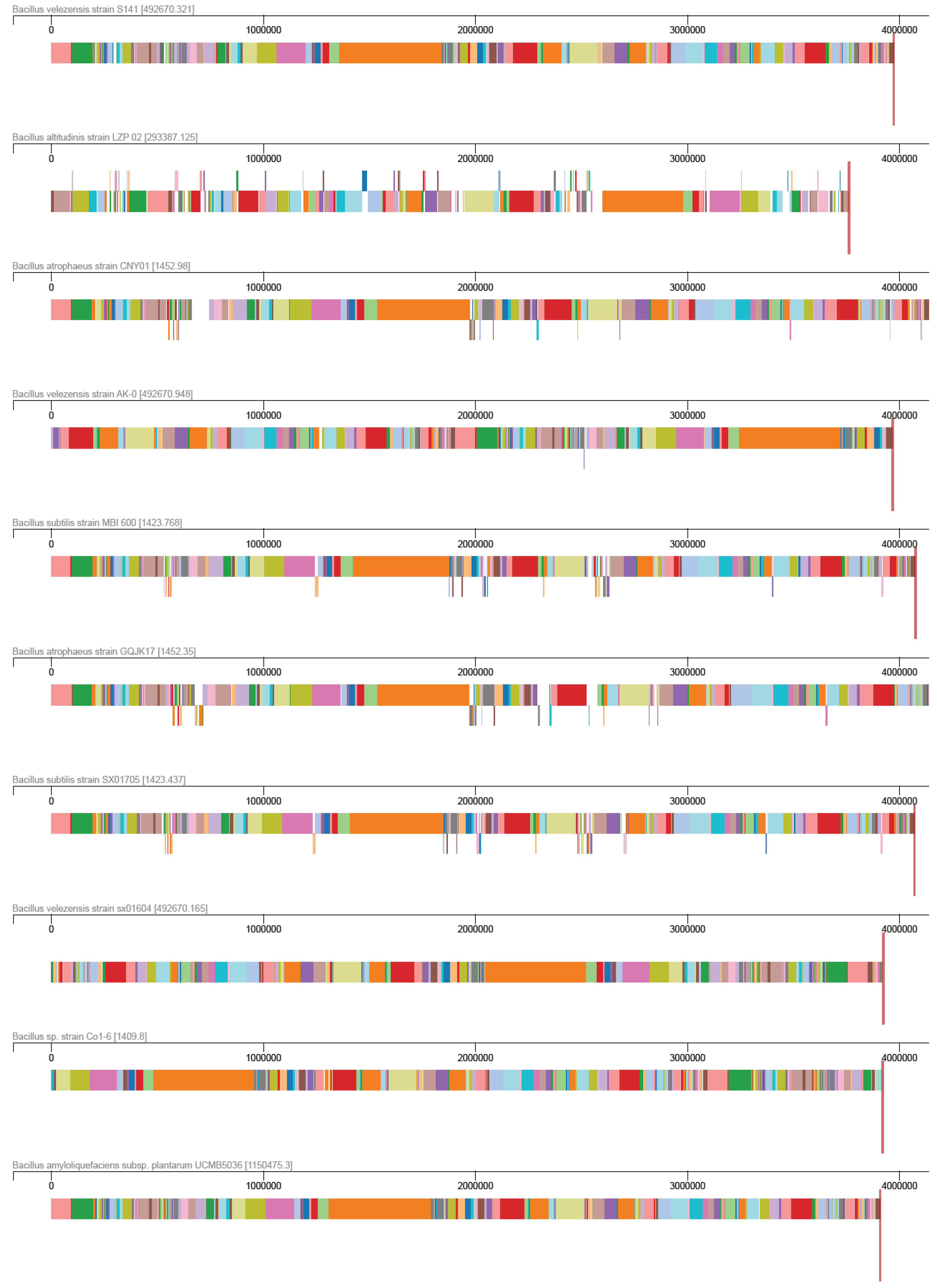
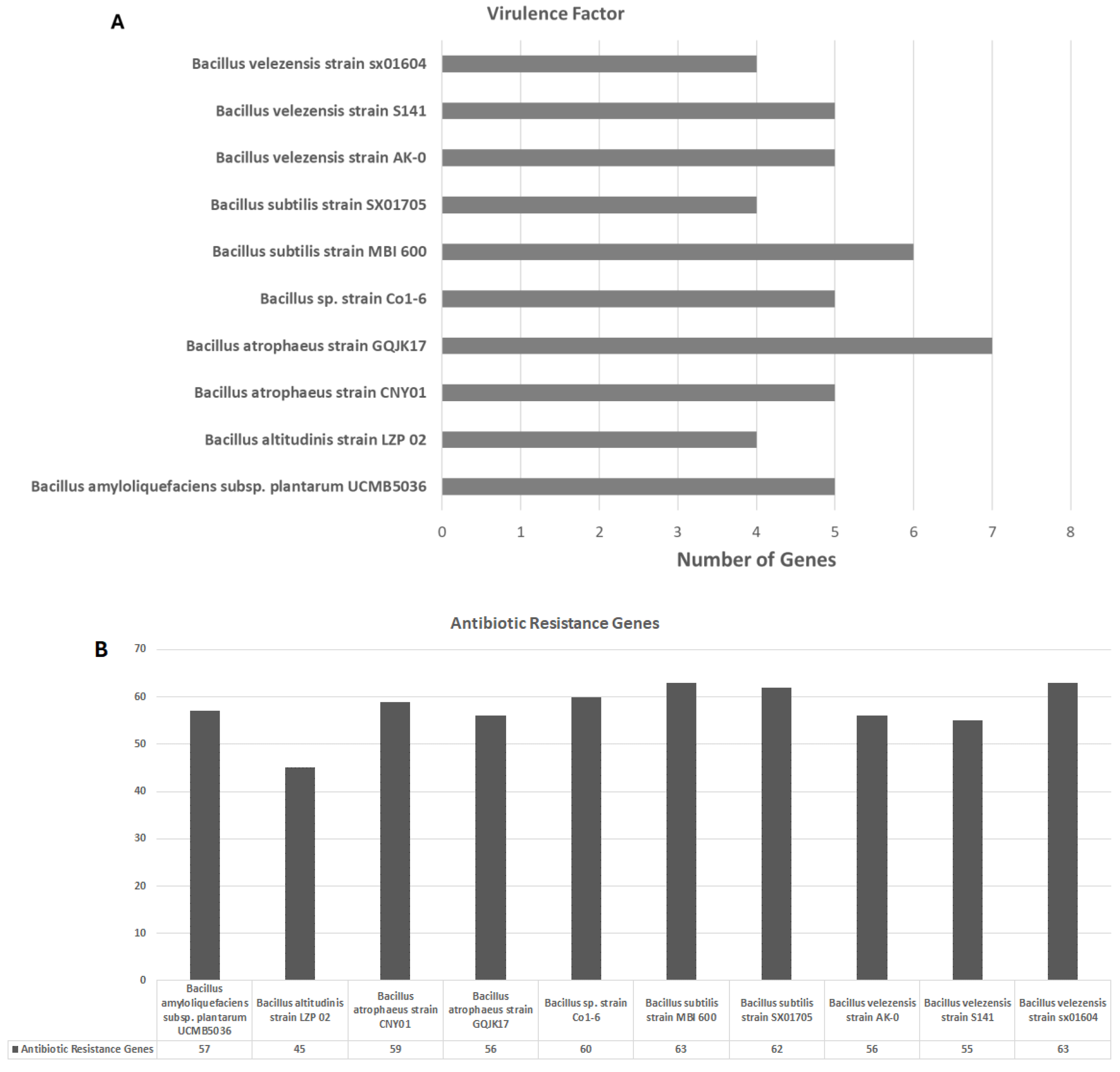
3.2. Comparative Genomic Analysis of the Ten Chosen Bacillus Strains
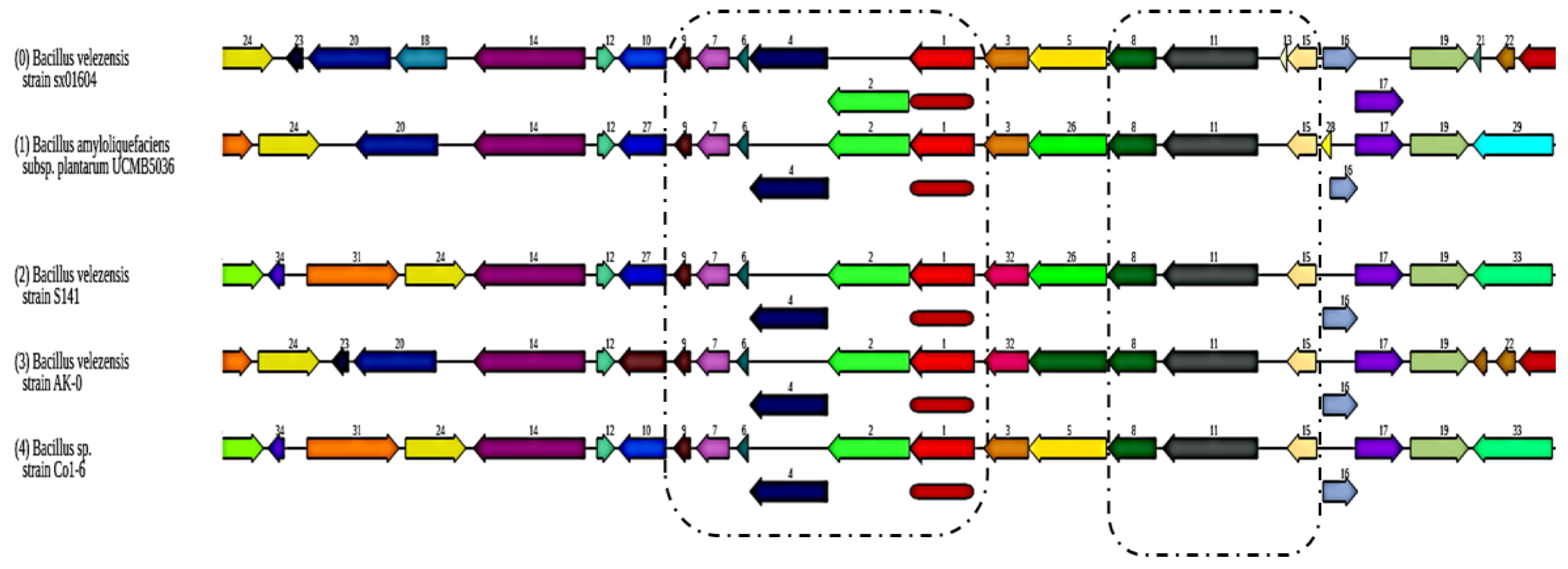
3.3. Comparative Proteomic Analysis of the Ten Chosen Bacillus Strains
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Upadhyay, S.K.; Srivastava, A.K.; Rajput, V.D.; Chauhan, P.K.; Bhojiya, A.A.; Jain, D.; Chaubey, G.; Dwivedi, P.; Sharma, B.; Minkina, T. Root Exudates: Mechanistic Insight of Plant Growth Promoting Rhizobacteria for Sustainable Crop Production. Front. Microbiol. 2022, 13, 916488. [Google Scholar] [CrossRef] [PubMed]
- Sumbul, A.; Ansari, A.R.; Rizvi, R.; Mahmood, I. Azotobacter: A potential bio-fertilizer for soil and plant health management. Saudi J. Biol. Sci. 2020, 27, 3634–3640. [Google Scholar] [CrossRef] [PubMed]
- Heo, A.Y.; Koo, Y.M.; Choi, H.W. Biological Control Activity of Plant Growth Promoting Rhizobacteria Burkholderia contaminans AY001 against Tomato Fusarium Wilt and Bacterial Speck Diseases. Biology 2022, 11, 619. [Google Scholar] [CrossRef] [PubMed]
- Romero, A.M.; Correa, O.S.; Mocia, S.; Rivas, J.G. Effect of Azospirillum-mediated plant growth promotion on the development of bacterial diseases on fresh-market and cherry tomato. J. Appl. Microbiol. 2003, 95, 832–838. [Google Scholar] [CrossRef] [PubMed]
- Zablotowicz, R.M.; Tipping, E.M.; Lifshitz, R.; Kloepper, J.W. Plant growth promotion mediated by bacterial rhizosphere colonizers. In The Rhizosphere and Plant Growth: Papers Presented at a Symposium Held May 8–11, 1989, at the Beltsville Agricultural Research Center (BARC); Maryland, B., Keister, D.L., Cregan, P.B., Eds.; Springer: Dordrecht, The Netherlands, 1991; pp. 315–326. [Google Scholar]
- Liu, M.; Philp, J.; Wang, Y.; Hu, J.; Wei, Y.; Li, J.; Ryder, M.; Toh, R.; Zhou, Y.; Denton, D.M.; et al. Plant growth-promoting rhizobacteria Burkholderia vietnamiensis B418 inhibits root-knot nematode on watermelon by modifying the rhizosphere microbial community. Sci. Rep. 2022, 12, 8381. [Google Scholar] [CrossRef] [PubMed]
- Shanmugam, V.; Thakur, H.; Gupta, S. Use of chitinolytic Bacillus atrophaeus strain S2BC-2 antagonistic to Fusarium spp. for control of rhizome rot of ginger. Ann. Microbiol. 2013, 63, 989–996. [Google Scholar] [CrossRef]
- Chen, E.; Yang, C.; Tao, W.; Li, S. Polysaccharides Produced by Plant Growth-Promoting Rhizobacteria Strain Burkholderia sp. BK01 Enhance Salt Stress Tolerance to Arabidopsis thaliana. Polymers 2024, 16, 145. [Google Scholar] [CrossRef]
- Vessey, J.K. Plant growth promoting rhizobacteria as biofertilizers. Plant Soil 2003, 255, 571–586. [Google Scholar] [CrossRef]
- Imran, A.; Mirza, M.S.; Shah, T.M.; Malik, K.A.; Hafeez, F.Y. Differential response of kabuli and desi chickpea genotypes toward inoculation with PGPR in different soils. Front. Microbiol. 2015, 6, 859. [Google Scholar] [CrossRef]
- Agbodjato, N.A.; Noumayo, P.A.; Adjanohoun, A.; Agbessi, L.; Moussa, L.B. Synergistic Effects of Plant Growth Promoting Rhizobacteria and Chitosan on In Vitro Seeds Germination, Greenhouse Growth, and Nutrient Uptake of Maize (Zea mays L.). Biotechnol. Res. Int. 2016, 2016, 7830182. [Google Scholar] [CrossRef]
- Abdelkrim, S.; Abid, G.; Chaieb, O.; Taamalli, W.; Mannai, K.; Louati, F.; Jebara, M.; Jebara, S.H. Plant growth promoting rhizobacteria modulates the antioxidant defense and the expression of stress-responsive genes providing Pb accumulation and tolerance of grass pea. Environ. Sci. Pollut. Res. Int. 2023, 30, 10789–10802. [Google Scholar] [CrossRef]
- Han, L.; Zhang, H.; Bai, X.; Jiang, B. The peanut root exudate increases the transport and metabolism of nutrients and enhances the plant growth-promoting effects of Burkholderia pyrrocinia strain P10. BMC Microbiol. 2023, 23, 85. [Google Scholar] [CrossRef] [PubMed]
- Jiao, H.; Xu, W.; Chen, W.; Hu, Y.; Tian, R.; Wang, Z. Complete Genome Sequence Data of Bacillus altitudinis LZP02, a Bacterium from the Rice Rhizosphere, for Studying the Promotion of Plant Growth. Molecular Plant-Microbe Interact. 2022, 5, 428–431. [Google Scholar] [CrossRef] [PubMed]
- Sibponkrung, S.; Kondo, T.; Tanaka, K.; Tittabutr, P.; Boonkerd, N.; Teaumroong, N.; Yoshida, K.I. Genome Sequence of Bacillus velezensis S141, a New Strain of Plant Growth-Promoting Rhizobacterium Isolated from Soybean Rhizosphere. Genome Announc. 2017, 5, e01312-17. [Google Scholar] [CrossRef]
- Egorshina, A.A.; Khairullin, R.M.; Sakhabutdinova, R.M.; Luk’yantsev, M.A. Involvement of phytohormones in the development of interaction between wheat seedlings endophytic Bacillus subtilis strain, 11BM. Russ. J. Plant Physiol. 2012, 59, 134–140. [Google Scholar] [CrossRef]
- Sacristán-Pérez-Minayo, G.; Javier Lopez-Robles, D.; Rad, C.; Barroso, L.M. Microbial Inoculation for Productivity Improvements and Potential Biological Control in Sugar Beet Crops. Front. Plant Sci. 2020, 11. [Google Scholar] [CrossRef]
- Al-Turki, A.; Murali, M.; Omar, A.F.; Rehan, M.; Sayyed, R.Z. Recent advances in PGPR-mediated resilience toward interactive effects of drought and salt stress in plants. Front. Microbiol. 2023, 14, 1214845. [Google Scholar] [CrossRef]
- Ahmed, A.; Hasnain, S. Auxins as one of the factors of plant growth improvement by plant growth promoting rhizobacteria. Pol. J. Microbiol. 2014, 63, 261–266. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Q.; Xie, J.; Li, Y.; Gao, T.; Xu, C.; Wang, Q. Comparative genomic and functional analyses of four sequenced Bacillus cereus genomes reveal conservation of genes relevant to plant-growth-promoting traits. Sci. Rep. 2018, 8, 17009. [Google Scholar] [CrossRef]
- Hamdache, A.; Azarken, R.; Lamarti, A.; Aleu, J.; Collado, I.G. Comparative genome analysis of Bacillus spp. and its relationship with bioactive nonribosomal peptide production. Phytochem. Rev. 2013, 12, 685–716. [Google Scholar] [CrossRef]
- Danielsson, J.; Reva, O.; Meijer, J. Protection of oilseed rape (Brassica napus) toward fungal pathogens by strains of plant-associated Bacillus amyloliquefaciens. Microb. Ecol. 2007, 54, 134–140. [Google Scholar] [CrossRef]
- Niazi, A.; Manzoor, S.; Asari, S.; Bejai, S.; Meijer, J.; Bongcam-Rudloff, E. Genome analysis of Bacillus amyloliquefaciens Subsp. plantarum UCMB5113: A rhizobacterium that improves plant growth and stress management. PLoS ONE 2014, 9, e104651. [Google Scholar] [CrossRef] [PubMed]
- Sella, S.R.; Vandenberghe, L.; Soccol, C.R. Bacillus atrophaeus: Main characteristics and biotechnological applications—A review. Crit. Rev. Biotechnol. 2015, 35, 533–545. [Google Scholar] [CrossRef]
- Köberl, M.; White, R.A., III; Erschen, S.; Spanberger, N.; El-Arabi, T.F.; Jansson, J.K.; Berg, G. Complete Genome Sequence of Bacillus amyloliquefaciens Strain Co1-6, a Plant Growth-Promoting Rhizobacterium of Calendula officinalis. Am. Soc. Microbiol. Genome Announc. 2015, 3, e00862-15. [Google Scholar]
- Pham, H.-H.; Kim, D.-H.; Nguyen, T.L. Wide-genome selection of lactic acid bacteria harboring genes that promote the elimination of antinutritional factors. Front. Plant Sci. 2023, 14, 1145041. [Google Scholar] [CrossRef]
- Samaras, A.; Nikolaidis, M.; Antequera-Gomez, M.L.; Camara-Almiron, J.; Romero, D.; Moschakis, T.; Amoutzias, G.D.; Karaoglanidis, G.S. Whole Genome Sequencing and Root Colonization Studies Reveal Novel Insights in the Biocontrol Potential and Growth Promotion by Bacillus subtilis MBI 600 on Cucumber. Front. Microbiol. 2021, 11, 600393. [Google Scholar] [CrossRef] [PubMed]
- Olson, R.D.; Assaf, R.; Brettin, T.; Conrad, N.; Cucinell, C.; Davis, J.J.; Dempsey, D.M.; Dickerman, A.; Dietrich, E.M.; Kenyon, R.W.; et al. Introducing the Bacterial Viral Bioinformatics Resource Center (BV-BRC): A resource combining, PATRIC, IRD, ViPR. Nucleic Acids Res. 2023, 51, D678–D689. [Google Scholar] [CrossRef] [PubMed]
- Wattam, A.R.; Davis, J.J.; Assaf, R.; Boisvert, S.; Brettin, T.; Bun, C.; Conrad, N.; Dietrich, E.M.; Disz, T.; Gabbard, J.L.; et al. Improvements to PATRIC, the all-bacterial Bioinformatics Database and Analysis Resource Center. Nucleic Acids Res. 2017, 45, D535–D542. [Google Scholar] [CrossRef]
- Babicki, S.; Arndt, D.; Marcu, A.; Liang, Y.; Grant, J.R.; Maciejewski, A.; Wishart, D.S. Heatmapper: Web-enabled heat mapping for all. Nucleic Acids Res. 2016, 44, W147–W153. [Google Scholar] [CrossRef]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef]
- Cock, P.J.; Antao, T.; Chang, J.T.; Chapman, B.A.; Cox, C.J.; Dalke, A.; Friedberg, I.; Hamelryck, T.; Kauff, F.; Wilczynski, B.; et al. Biopython: Freely available Python tools for computational molecular biology and bioinformatics. Bioinformatics 2009, 25, 1422–1423. [Google Scholar] [CrossRef] [PubMed]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef] [PubMed]
- Darling, A.E.; Mau, B.; Perna, N.T. progressiveMauve: Multiple genome alignment with gene gain, loss and rearrangement. PLoS ONE 2010, 5, e11147. [Google Scholar] [CrossRef] [PubMed]
- Boratyn, G.M.; Camacho, C.; Cooper, P.S.; Coulouris, G.; Fong, A.; Ma, N.; Madden, T.L.; Matten, W.T.; McGinnis, S.D.; Merezhuk, Y.; et al. BLAST: A more efficient report with usability improvements. Nucleic Acids Res. 2013, 41, W29–W33. [Google Scholar] [CrossRef] [PubMed]
- Overbeek, R.; Olson, R.; Pusch, G.D.; Olsen, G.J.; Davis, J.J.; Disz, T.; Edwards, R.A.; Gerdes, S.; Parello, B.; Shukla, M.; et al. The SEED and the Rapid Annotation of microbial genomes using Subsystems Technology (RAST). Nucleic Acids Res. 2014, 42, D206–D214. [Google Scholar] [CrossRef] [PubMed]
- Krzywinski, M.; Schein, J.; Birol, I.; Connors, J.; Gascoyne, R.; Horseman, D.; Jones, S.J.; Marra, M.A. Circos: An information aesthetic for comparative genomics. Genome Res. 2009, 19, 1639–1645. [Google Scholar] [CrossRef] [PubMed]
- Overbeek, R.; Begley, T.; Butler, R.M.; Choudhuri, J.V.; Chuang, H.Y.; Cohoon, M.; de Crécy-Lagard, V.; Diaz, N.; Disz, T.; Edwards, R.; et al. The subsystems approach to genome annotation and its use in the project to annotate 1000 genomes. Nucleic Acids Res. 2005, 33, 5691–5702. [Google Scholar] [CrossRef] [PubMed]
- Davis, J.J.; Gerdes, S.; Olsen, G.J.; Olson, R.; Pusch, G.D.; Shukla, M.; Vonstein, V.; Wattam, A.R.; Yoo, H. PATtyFams: Protein Families for the Microbial Genomes in the PATRIC Database. Front. Microbiol. 2016, 7, 118. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Chao, L.; Zheng, F.; He, C.; Zhang, X. [Cloning, expression and functional analysis of the dhbC gene from the siderophore producing bacterium Bacillus subtilis CAS15]. Sheng Wu Gong Cheng Xue Bao 2009, 25, 819–825. [Google Scholar]
- Spinks, R.R.; Spenkelink, L.M.; Stratmann, S.A.; Xu, Z.Q.; Stamford, N.P.J.; Brown, S.E.; Dixon, N.E.; Jergic, S.; van Oijen, A.M. DnaB helicase dynamics in bacterial DNA replication resolved by single-molecule studies. Nucleic Acids Res. 2021, 49, 6804–6816. [Google Scholar] [CrossRef]
- Behrmann, M.S.; Perera, H.M.; Hoang, J.M.; Venkat, T.A.; Visser, B.J.; Bates, D.; Trakselis, M.A. Targeted chromosomal Escherichia coli: dnaB exterior surface residues regulate DNA helicase behavior to maintain genomic stability and organismal fitness. PLoS Genet. 2021, 17, e1009886. [Google Scholar] [CrossRef] [PubMed]
- Kang, L.; Lai, M.D. [BTEB/KLF9 and its transcriptional regulation]. Yi Chuan 2007, 29, 515–522. [Google Scholar] [CrossRef] [PubMed]
- Gamba, P.; Hamoen, L.W.; Daniel, R.A. Cooperative Recruitment of FtsW to the Division Site of Bacillus subtilis. Front. Microbiol. 2016, 7, 1808. [Google Scholar] [CrossRef] [PubMed]
- Emami, K.; Guyet, A.; Kawai, Y.; Devi, J.; Wu, L.J.; Allenby, N.; Daniel, R.A.; Errington, J. RodA as the missing glycosyltransferase in Bacillus subtilis and antibiotic discovery for the peptidoglycan polymerase pathway. Nat. Microbiol. 2017, 2, 16253. [Google Scholar] [CrossRef]
- Henriques, A.O.; Glaser, P.; Piggot, P.J.; Moran Jr, C.P. Control of cell shape and elongation by the rodA gene in Bacillus subtilis. Mol. Microbiol. 1998, 28, 235–247. [Google Scholar] [CrossRef] [PubMed]
- Merrikh, C.N.; Merrikh, H. Gene inversion potentiates bacterial evolvability and virulence. Nat. Commun. 2018, 9, 4662. [Google Scholar] [CrossRef] [PubMed]
- Imataka, H.; Nakayama, K.; Yasumoto, K.; Mizuno, A.; Fuji-Kuriyama, Y.; Hayami, M. Cell-specific translational control of transcription factor BTEB expression. The role of an upstream AUG in the 5‘-untranslated region. J. Biol. Chem. 1994, 269, 20668–20673. [Google Scholar] [CrossRef] [PubMed]
- van de Putte, P.; Goosen, N. DNA inversions in phages and bacteria. Trends Genet. 1992, 8, 457–462. [Google Scholar] [CrossRef] [PubMed]
- Huynh, T.; Vörö, M.; Kedves, O.; Turbat, A.; Sipos, G.; Leitgeb, B.; Kredics, L.; Vágvölgy, C.; Szekeres, A. Discrimination between the Two Closely Related Species of the Operational Group B. amyloliquefaciens Based on Whole-Cell Fatty Acid Profiling. Microorganisms 2022, 10, 418. [Google Scholar] [CrossRef]
- Gibbons, H.S.; Broomall, S.M.; McNew, L.A.; Daligault, H.; Chapman, C.; Bruce, D.; Karavis, M.; Krepps, M.; McGreegor, P.A.; Hong, C.; et al. Genomic signatures of strain selection and enhancement in Bacillus atrophaeus var. globigii, a historical biowarfare simulant. PLoS ONE 2011, 6, e17836. [Google Scholar] [CrossRef]
- Lang, K.S.; Merrikh, H. The Clash of Macromolecular Titans: Replication-Transcription Conflicts in Bacteria. Annu. Rev. Microbiol. 2018, 72, 71–88. [Google Scholar] [CrossRef] [PubMed]
- Lang, K.S.; Hall, A.N.; Merrikh, C.N.; Ragheb, M.; Tabakh, H.; Pollock, A.J.; Woodward, J.J.; Dreifus, J.E.; Merrikh, H. Replication-Transcription Conflicts Generate R-Loops that Orchestrate Bacterial Stress Survival and Pathogenesis. Cell 2017, 170, 787–799.e18. [Google Scholar] [CrossRef] [PubMed]
- Dimude, J.U.; Midgley-Smith, S.L.; Rudolph, C.J. Replication-transcription conflicts trigger extensive DNA degradation in Escherichia coli cells lacking RecBCD. DNA Repair. 2018, 70, 37–48. [Google Scholar] [CrossRef] [PubMed]
- Paul, S.; Million-Weaver, S.; Chattopadhyay, S.; Sokurenko, E.; Merrikh, H. Accelerated gene evolution through replication-transcription conflicts. Nature 2013, 495, 512–515. [Google Scholar] [CrossRef] [PubMed]
- Merrikh, H. Spatial and Temporal Control of Evolution through Replication-Transcription Conflicts. Trends Microbiol. 2017, 25, 515–521. [Google Scholar] [CrossRef] [PubMed]
- Olanrewaju, O.S.; Ayangbenro, A.S.; Glick, B.R.; Babalola, O.O. Plant health: Feedback effect of root exudates-rhizobiome interactions. Appl. Microbiol. Biotechnol. 2018, 103, 1155–1166. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Prajapati, V.; Prajapati, S.; Bais, H.; Lu, J. Comparative Genome Analysis of Bacillus amyloliquefaciens Focusing on Phylogenomics, Functional Traits, and Prevalence of Antimicrobial and Virulence Genes. Front. Genet. 2021, 12, 724217. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Yang, D.; Kendall, J.R.A.; Borriss, R.; Druzhinina, I.S.; Kubicek, C.P.; Shen, Q.; Zhang, R. Comparative Genomic Analysis of Bacillus amyloliquefaciens and Bacillus subtilis Reveals Evolutional Traits for Adaptation to Plant-Associated Habitats. Front. Microbiol. 2016, 7, 2039. [Google Scholar] [CrossRef]
- Shen, X.; Hu, H.; Peng, H.; Wang, W.; Zhang, X. Comparative genomic analysis of four representative plant growth-promoting rhizobacteria in Pseudomonas. BMC Genom. 2013, 14, 271. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | Strain | Plasmids | Contigs | Size | GC Content | Contig L50 | Contig N50 | TRNA | RRNA | CDS |
|---|---|---|---|---|---|---|---|---|---|---|
| Bacillus amyloliquefaciens | UCMB5036 | 1 | 3,910,324 | 46.6 | 1 | 3,910,324 | 89 | 30 | 3914 | |
| Bacillus sp. | Co1-6 | 1 | 3,922,431 | 46.6 | 1 | 3,922,431 | 86 | 27 | 3997 | |
| Bacillus subtilis | SX01705 | 2 | 3 | 4,169,021 | 43.7 | 1 | 4,072,531 | 86 | 30 | 4365 |
| Bacillus subtilis | MBI 600 | 1 | 4,076,736 | 43.8 | 1 | 4,076,736 | 86 | 20 | 4271 | |
| Bacillus atrophaeus | GQJK17 | 1 | 4,325,818 | 43.3 | 1 | 4,325,818 | 84 | 12 | 4507 | |
| Bacillus atrophaeus | CNY01 | 1 | 4,144,521 | 43.5 | 1 | 4,144,521 | 82 | 24 | 4332 | |
| Bacillus altitudinis | LZP 02 | 1 | 3,763,082 | 41.4 | 1 | 3,763,082 | 81 | 23 | 3911 | |
| Bacillus velezensis | sx01604 | 1 | 3,926,520 | 46.5 | 1 | 3,926,520 | 86 | 14 | 3987 | |
| Bacillus velezensis | S141 | 1 | 3,974,582 | 46.5 | 1 | 3,974,582 | 87 | 27 | 4028 | |
| Bacillus velezensis | AK-0 | 1 | 3,969,447 | 46.5 | 1 | 3,969,447 | 86 | 27 | 4017 |
| Genes Reported to Support Plant Growth | Plant Growth Promoting Property | B. amyloliquefaciens subsp. plantarum UCMB5036 | B. sp. strain Co1-6 | B. subtilis strain SX01705 | B. subtilis strain MBI 600 | B. atrophaeus strain GQJK17 | B. atrophaeus strain CNY01 | B. velezensis strain sx01604 | B. velezensis strain S141 | B. velezensis strain AK-0 | B. altitudinis strain LZP 02 |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Trilactone hydrolase [bacillibactin] siderophore | Improves Iron availability | + | + | + | + | + | + | + | + | + | + |
| 2,3-dihydro-2,3-dihydroxybenzoate dehydrogenase (Siderphore biosynthesis) | Siderophore Biosynthesis | + | + | + | + | + | + | + | + | + | - |
| Nitrite-sensitive transcriptional repressor (NsrR) | stress resistance | + | + | + | + | + | + | + | + | + | + |
| Superoxide Dimutase | stress resistance | + | + | + | + | + | + | + | + | + | + |
| betaine aldehyde dehydrogenase | stress resistance | + | + | + | + | + | + | + | + | + | + |
| copC | copper resistance genes | + | + | + | + | + | + | + | + | + | + |
| copD | copper resistance genes | + | + | + | + | + | + | + | + | + | + |
| Fosfomycin | antibiotic resistance genes | + | + | + | + | + | - | + | + | + | - |
| ykkD | antibiotic resistance genes | + | + | + | + | + | + | + | + | + | + |
| ykkC | antibiotic resistance genes | + | + | + | + | + | + | + | + | + | - |
| norD | Nitrogen Metabolism | + | + | + | + | + | + | + | + | + | + |
| norQ | Nitrogen Metabolism | + | + | + | + | + | + | + | + | + | + |
| Tpx and related | Sulfate Metabolism | + | + | + | + | + | + | + | + | + | + |
| phoP | phosphate metabolism | + | + | + | + | + | + | + | + | + | + |
| tryptophan synthase | auxin biosynthesis | + | + | + | + | + | + | + | + | + | + |
| anthranilate phosphoribosyltransferase (trpD) | auxin biosynthesis | + | + | + | + | + | + | + | + | + | + |
| phosphoribosylanthranilate isomerase | auxin biosynthesis | + | + | + | + | + | + | + | + | + | + |
| gabR | Ɣ-Aminobutyric Acid (GABA) Metabolism | + | + | + | + | + | + | + | + | + | + |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Das, V.A.; Gautam, B.; Yadav, P.K.; Singh, S. Identification of Conserved Pathways in Bacillus Strains Known for Plant Growth-Promoting Behavior Using a Multifaceted Computational Approach. Agriculture 2024, 14, 838. https://doi.org/10.3390/agriculture14060838
Das VA, Gautam B, Yadav PK, Singh S. Identification of Conserved Pathways in Bacillus Strains Known for Plant Growth-Promoting Behavior Using a Multifaceted Computational Approach. Agriculture. 2024; 14(6):838. https://doi.org/10.3390/agriculture14060838
Chicago/Turabian StyleDas, Vandana Apurva, Budhayash Gautam, Pramod Kumar Yadav, and Satendra Singh. 2024. "Identification of Conserved Pathways in Bacillus Strains Known for Plant Growth-Promoting Behavior Using a Multifaceted Computational Approach" Agriculture 14, no. 6: 838. https://doi.org/10.3390/agriculture14060838
APA StyleDas, V. A., Gautam, B., Yadav, P. K., & Singh, S. (2024). Identification of Conserved Pathways in Bacillus Strains Known for Plant Growth-Promoting Behavior Using a Multifaceted Computational Approach. Agriculture, 14(6), 838. https://doi.org/10.3390/agriculture14060838