
Identification of Hotspot Regions for Candidate Genes Associated with Peanut (Arachis hypogaea L.) Pod and Seed Size on Chromosome A05

Abstract
1. Introduction
2. Materials and Methods
2.1. Plant Materials and Development of RIL Population
2.2. Trait Measurements and Data Analysis
2.3. QTL Analysis
2.4. Further QTL Mapping Based on KASP Genotyping
2.5. Prediction of Candidate Genes in Hotspot Regions
3. Results
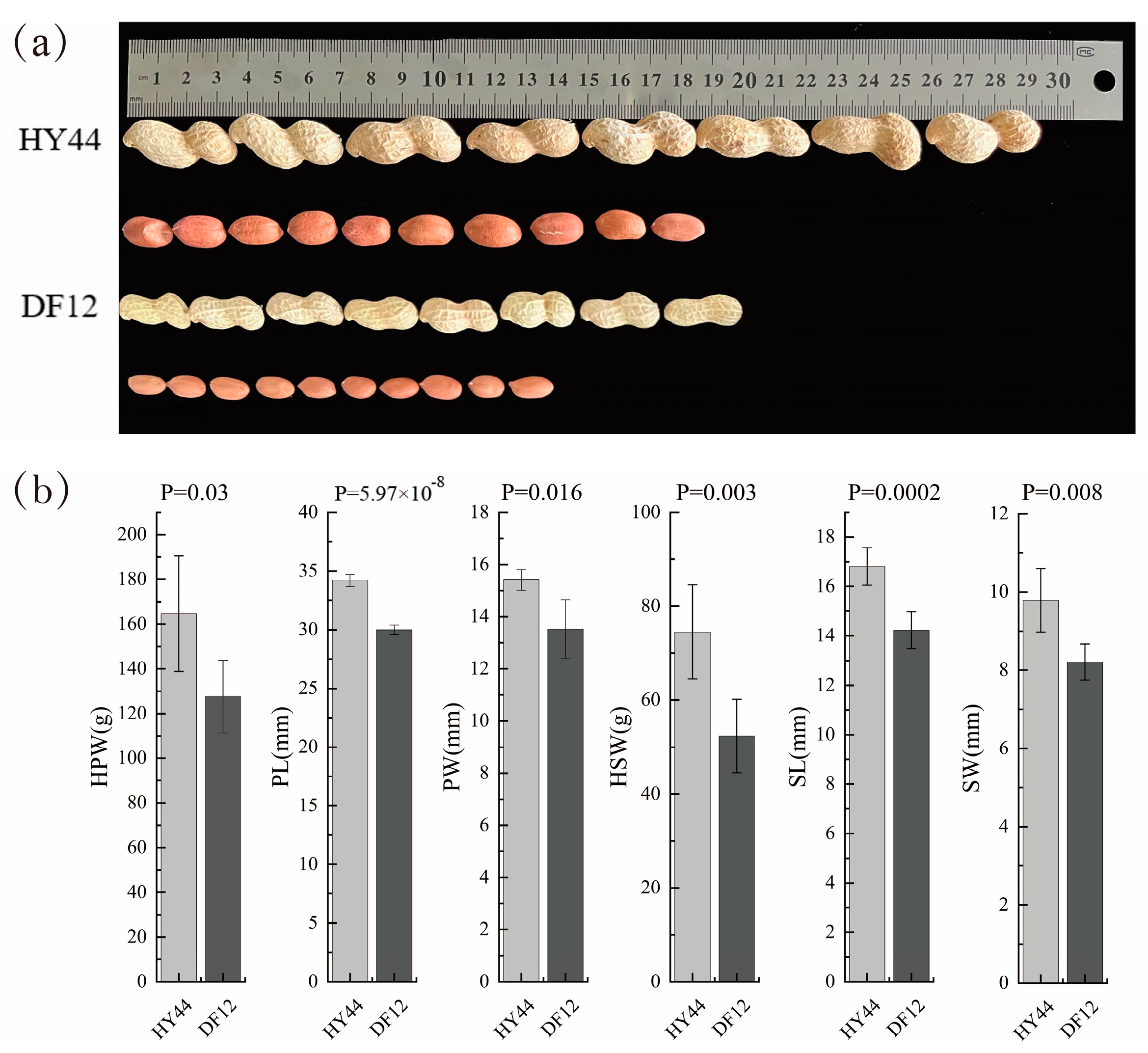
3.1. Agronomic Performance of the Parents and RIL Individuals
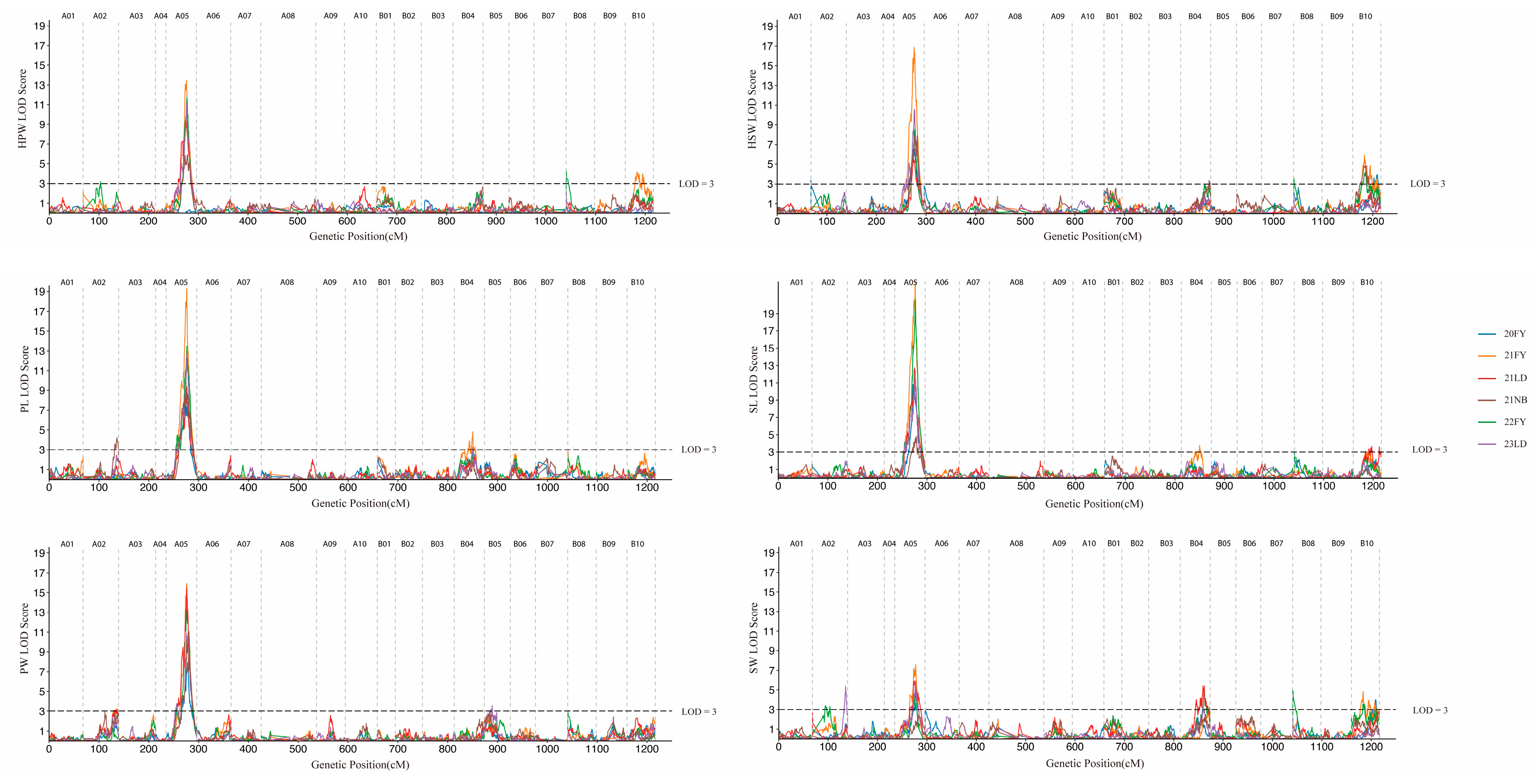
3.2. QTL Mapping for Traits Related to Pod and Seed Size
3.3. Further Identification of QTL Enrichment Regions on Chromosome A05
3.4. Confirmation and Identification of Candidate Gene Hotspot Regions on Chromosome A05
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
Abbreviation
| Abbreviation | Full Term |
| CIM | Composite Interval Mapping |
| KASP | Kompetitive Allele-Specific PCR |
| PVE | Phenotypic Variance Explained |
| QTL | Quantitative Trait Loci |
| RAD-seq | Restriction site-Associated DNA sequencing |
| RIL | Recombinant Inbred Line |
| SLAF-seq | Specific-Locus Amplified Fragment sequencing |
| SNP | Single Nucleotide Polymorphism |
References
- Guo, M.; Deng, L.; Gu, J.; Miao, J.; Yin, J.; Li, Y.; Fang, Y.; Huang, B.; Sun, Z.; Qi, F. Genome-wide association study and development of molecular markers for yield and quality traits in peanut (Arachis hypogaea L.). BMC Plant Biol. 2024, 24, 244. [Google Scholar] [CrossRef] [PubMed]
- Bertioli, D.J.; Cannon, S.B.; Froenicke, L.; Huang, G.; Farmer, A.D.; Cannon, E.K.; Liu, X.; Gao, D.; Clevenger, J.; Dash, S. The genome sequences of Arachis duranensis and Arachis ipaensis, the diploid ancestors of cultivated peanut. Nat. Genet. 2016, 48, 438–446. [Google Scholar] [CrossRef] [PubMed]
- Cuc, L.M.; Mace, E.S.; Crouch, J.H.; Quang, V.D.; Long, T.D.; Varshney, R.K. Isolation and characterization of novel microsatellite markers and their application for diversity assessment in cultivated groundnut (Arachis hypogaea). BMC Plant Biol. 2008, 8, 55. [Google Scholar] [CrossRef] [PubMed]
- Kochert, G.; Stalker, H.T.; Gimenes, M.; Galgaro, L.; Lopes, C.R.; Moore, K. RFLP and cytogenetic evidence on the origin and evolution of allotetraploid domesticated peanut, Arachis hypogaea (Leguminosae). Am. J. Bot. 1996, 83, 1282–1291. [Google Scholar] [CrossRef]
- Seijo, J.G.; Lavia, G.I.; Fernández, A.; Krapovickas, A.; Ducasse, D.; Moscone, E.A. Physical mapping of the 5S and 18S–25S rRNA genes by FISH as evidence that Arachis duranensis and A. ipaensis are the wild diploid progenitors of A. hypogaea (Leguminosae). Am. J. Bot. 2004, 91, 1294–1303. [Google Scholar] [CrossRef]
- Robledo, G.; Lavia, G.; Seijo, G. Species relations among wild Arachis species with the A genome as revealed by FISH mapping of rDNA loci and heterochromatin detection. Theor. Appl. Genet. 2009, 118, 1295–1307. [Google Scholar] [CrossRef]
- Chu, Y.; Chee, P.; Isleib, T.G.; Holbrook, C.C.; Ozias-Akins, P. Major seed size QTL on chromosome A05 of peanut (Arachis hypogaea) is conserved in the US mini core germplasm collection. Mol. Breed. 2020, 40, 6. [Google Scholar] [CrossRef]
- Wu, Y.; Sun, Z.; Qi, F.; Tian, M.; Wang, J.; Zhao, R.; Wang, X.; Wu, X.; Shi, X.; Liu, H.; et al. Comparative transcriptomics analysis of developing peanut (Arachis hypogaea L.) pods reveals candidate genes affecting peanut seed size. Front. Plant Sci. 2022, 13, 958808. [Google Scholar] [CrossRef] [PubMed]
- Chavarro, C.; Chu, Y.; Holbrook, C.; Isleib, T.; Bertioli, D.; Hovav, R.; Butts, C.; Lamb, M.; Sorensen, R.; Jackson, S.A. Pod and seed trait QTL identification to assist breeding for peanut market preferences. G3 Genes Genomes Genet. 2020, 10, 2297–2315. [Google Scholar] [CrossRef]
- Xiang, D.-Q.; Cao, H.-H.; Cao, Y.-G.; Yang, J.-P.; Huang, L.-J.; Wang, S.-C.; Dai, J. Construction of a genetic map and location of quantitative trait loci for yield component traits in maize by SSR markers. Yi Chuan Xue Bao = Acta Genet. Sin. 2001, 28, 778–784. [Google Scholar]
- Hopkins, M.; Casa, A.; Wang, T.; Mitchell, S.; Dean, R.; Kochert, G.; Kresovich, S. Discovery and characterization of polymorphic simple sequence repeats (SSRs) in peanut. Crop Sci. 1999, 39, 1243–1247. [Google Scholar] [CrossRef]
- Varshney, R.; Bertioli, D.; Moretzsohn, M.d.C.; Vadez, V.; Krishnamurthy, L.; Aruna, R.; Nigam, S.; Moss, B.; Seetha, K.; Ravi, K. The first SSR-based genetic linkage map for cultivated groundnut (Arachis hypogaea L.). Theor. Appl. Genet. 2009, 118, 729–739. [Google Scholar] [CrossRef] [PubMed]
- Ravi, K.; Vadez, V.; Isobe, S.; Mir, R.; Guo, Y.; Nigam, S.; Gowda, M.; Radhakrishnan, T.; Bertioli, D.; Knapp, S. Identification of several small main-effect QTL and a large number of epistatic QTL for drought tolerance related traits in groundnut (Arachis hypogaea L.). Theor. Appl. Genet. 2011, 122, 1119–1132. [Google Scholar] [CrossRef]
- Shirasawa, K.; Koilkonda, P.; Aoki, K.; Hirakawa, H.; Tabata, S.; Watanabe, M.; Hasegawa, M.; Kiyoshima, H.; Suzuki, S.; Kuwata, C. In silico polymorphism analysis for the development of simple sequence repeat and transposon markers and construction of linkage map in cultivated peanut. BMC Plant Biol. 2012, 12, 80. [Google Scholar] [CrossRef] [PubMed]
- Fonceka, D.; Tossim, H.-A.; Rivallan, R.; Vignes, H.; Faye, I.; Ndoye, O.; Moretzsohn, M.C.; Bertioli, D.J.; Glaszmann, J.-C.; Courtois, B. Fostered and left behind alleles in peanut: Interspecific QTL mapping reveals footprints of domestication and useful natural variation for breeding. BMC Plant Biol. 2012, 12, 26. [Google Scholar] [CrossRef]
- Huang, L.; He, H.; Chen, W.; Ren, X.; Chen, Y.; Zhou, X.; Xia, Y.; Wang, X.; Jiang, X.; Liao, B. Quantitative trait locus analysis of agronomic and quality-related traits in cultivated peanut (Arachis hypogaea L.). Theor. Appl. Genet. 2015, 128, 1103–1115. [Google Scholar] [CrossRef]
- Chen, W.; Jiao, Y.; Cheng, L.; Huang, L.; Liao, B.; Tang, M.; Ren, X.; Zhou, X.; Chen, Y.; Jiang, H. Quantitative trait locus analysis for pod-and kernel-related traits in the cultivated peanut (Arachis hypogaea L.). BMC Genet. 2016, 17, 25. [Google Scholar] [CrossRef]
- Luo, H.; Ren, X.; Li, Z.; Xu, Z.; Li, X.; Huang, L.; Zhou, X.; Chen, Y.; Chen, W.; Lei, Y. Co-localization of major quantitative trait loci for pod size and weight to a 3.7 cM interval on chromosome A05 in cultivated peanut (Arachis hypogaea L.). BMC Genom. 2017, 18, 58. [Google Scholar] [CrossRef]
- Zhuang, W.; Chen, H.; Yang, M.; Wang, J.; Pandey, M.K.; Zhang, C.; Chang, W.-C.; Zhang, L.; Zhang, X.; Tang, R. The genome of cultivated peanut provides insight into legume karyotypes, polyploid evolution and crop domestication. Nat. Genet. 2019, 51, 865–876. [Google Scholar] [CrossRef]
- Chen, X.; Lu, Q.; Liu, H.; Zhang, J.; Hong, Y.; Lan, H.; Li, H.; Wang, J.; Liu, H.; Li, S. Sequencing of cultivated peanut, Arachis hypogaea, yields insights into genome evolution and oil improvement. Mol. Plant 2019, 12, 920–934. [Google Scholar] [CrossRef]
- Bertioli, D.J.; Jenkins, J.; Clevenger, J.; Dudchenko, O.; Gao, D.; Seijo, G.; Leal-Bertioli, S.C.; Ren, L.; Farmer, A.D.; Pandey, M.K. The genome sequence of segmental allotetraploid peanut Arachis hypogaea. Nat. Genet. 2019, 51, 877–884. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Hu, X.; Miao, H.; Chu, Y.; Cui, F.; Yang, W.; Wang, C.; Shen, Y.; Xu, T.; Zhao, L. QTL identification for seed weight and size based on a high-density SLAF-seq genetic map in peanut (Arachis hypogaea L.). BMC Plant Biol. 2019, 19, 537. [Google Scholar] [CrossRef]
- Alyr, M.H.; Pallu, J.; Sambou, A.; Nguepjop, J.R.; Seye, M.; Tossim, H.-A.; Djiboune, Y.R.; Sane, D.; Rami, J.-F.; Fonceka, D. Fine-mapping of a wild genomic region involved in pod and seed size reduction on chromosome A07 in peanut (Arachis hypogaea L.). Genes 2020, 11, 1402. [Google Scholar] [CrossRef]
- Yang, Y.; Su, Q.; Li, Y.; Cheng, Z.; Song, Y.; Jin, X.; Wang, J. Fine mapping of a major QTL qHYF_B06 for peanut yield. Crop J. 2023, 11, 1533–1540. [Google Scholar] [CrossRef]
- Lv, Z.; Lan, G.; Bai, B.; Yu, P.; Wang, C.; Zhang, H.; Zhong, C.; Zhao, X.; Yu, H. Identification of candidate genes associated with peanut pod length by combined analysis of QTL-seq and RNA-seq. Genomics 2024, 116, 110835. [Google Scholar] [CrossRef]
- Yang, H.; Luo, L.; Li, Y.; Li, H.; Zhang, X.; Zhang, K.; Zhu, S.; Li, X.; Li, Y.; Wan, Y. Fine mapping of qAHPS07 and functional studies of AhRUVBL2 controlling pod size in peanut (Arachis hypogaea L.). Plant Biotechnol. J. 2023, 21, 1785–1798. [Google Scholar] [CrossRef] [PubMed]
- Zhao, K.; Wang, L.; Qiu, D.; Cao, Z.; Wang, K.; Li, Z.; Wang, X.; Wang, J.; Ma, Q.; Cao, D. PSW1, an LRR receptor kinase, regulates pod size in peanut. Plant Biotechnol. J. 2023, 21, 2113–2124. [Google Scholar] [CrossRef]
- Zhang, X.; Zhang, X.; Wang, L.; Liu, Q.; Liang, Y.; Zhang, J.; Xue, Y.; Tian, Y.; Zhang, H.; Li, N. Fine mapping of a QTL and identification of candidate genes associated with cold tolerance during germination in peanut (Arachis hypogaea L.) on chromosome B09 using whole genome re-sequencing. Front. Plant Sci. 2023, 14, 1153293. [Google Scholar] [CrossRef]
- Arends, D.; Prins, P.; Jansen, R.C.; Broman, K.W. R/qtl: High-throughput multiple QTL mapping. Bioinformatics 2010, 26, 2990–2992. [Google Scholar] [CrossRef]
- Li, L.; Yang, X.; Cui, S.; Meng, X.; Mu, G.; Hou, M.; He, M.; Zhang, H.; Liu, L.; Chen, C.Y. Construction of high-density genetic map and mapping quantitative trait loci for growth habit-related traits of peanut (Arachis hypogaea L.). Front. Plant Sci. 2019, 10, 745. [Google Scholar] [CrossRef]
- Wang, Z.; Yan, L.; Chen, Y.; Wang, X.; Huai, D.; Kang, Y.; Jiang, H.; Liu, K.; Lei, Y.; Liao, B. Detection of a major QTL and development of KASP markers for seed weight by combining QTL-seq, QTL-mapping and RNA-seq in peanut. Theor. Appl. Genet. 2022, 135, 1779–1795. [Google Scholar] [CrossRef] [PubMed]
- Luo, H.; Guo, J.; Ren, X.; Chen, W.; Huang, L.; Zhou, X.; Chen, Y.; Liu, N.; Xiong, F.; Lei, Y. Chromosomes A07 and A05 associated with stable and major QTL for pod weight and size in cultivated peanut (Arachis hypogaea L.). Theor. Appl. Genet. 2018, 131, 267–282. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Huai, D.; Zhang, Z.; Cheng, K.; Kang, Y.; Wan, L.; Yan, L.; Jiang, H.; Lei, Y.; Liao, B. Development of a high-density genetic map based on specific length amplified fragment sequencing and its application in quantitative trait loci analysis for yield-related traits in cultivated peanut. Front. Plant Sci. 2018, 9, 827. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Ren, X.; Zheng, Y.; Zhou, X.; Huang, L.; Yan, L.; Jiao, Y.; Chen, W.; Huang, S.; Wan, L. Genetic mapping of yield traits using RIL population derived from Fuchuan Dahuasheng and ICG6375 of peanut (Arachis hypogaea L.). Mol. Breed. 2017, 37, 17. [Google Scholar] [CrossRef]
- Ma, M.; Wang, Q.; Li, Z.; Cheng, H.; Li, Z.; Liu, X.; Song, W.; Appels, R.; Zhao, H. Expression of Ta CYP 78A3, a gene encoding cytochrome P450 CYP 78A3 protein in wheat (Triticum aestivum L.), affects seed size. Plant J. 2015, 83, 312–325. [Google Scholar] [CrossRef]
- Chakrabarti, M.; Zhang, N.; Sauvage, C.; Munos, S.; Blanca, J.; Cañizares, J.; Diez, M.J.; Schneider, R.; Mazourek, M.; McClead, J. A cytochrome P450 regulates a domestication trait in cultivated tomato. Proc. Natl. Acad. Sci. USA 2013, 110, 17125–17130. [Google Scholar] [CrossRef]
- Zhou, X.; Zhang, H.; Wang, P.; Liu, Y.; Zhang, X.; Song, Y.; Wang, Z.; Ali, A.; Wan, L.; Yang, G. BnaC7. ROT3, the causal gene of cqSL-C7, mediates silique length by affecting cell elongation in Brassica napus. J. Exp. Bot. 2022, 73, 154–167. [Google Scholar] [CrossRef]
- Adamski, N.M.; Anastasiou, E.; Eriksson, S.; O’Neill, C.M.; Lenhard, M. Local maternal control of seed size by KLUH/CYP78A5-dependent growth signaling. Proc. Natl. Acad. Sci. USA 2009, 106, 20115–20120. [Google Scholar] [CrossRef]
- Fang, W.; Wang, Z.; Cui, R.; Li, J.; Li, Y. Maternal control of seed size by EOD3/CYP78A6 in Arabidopsis thaliana. Plant J. 2012, 70, 929–939. [Google Scholar] [CrossRef]
- Liu, Y.; Yi, C.; Liu, Q.; Wang, C.; Wang, W.; Han, F.; Hu, X. Multi-omics profiling identifies candidate genes controlling seed size in peanut. Plants 2022, 11, 3276. [Google Scholar] [CrossRef]
- Tao, Y.; Wang, J.; Miao, J.; Chen, J.; Wu, S.; Zhu, J.; Zhang, D.; Gu, H.; Cui, H.; Shi, S. The spermine synthase OsSPMS1 regulates seed germination, grain size, and yield. Plant Physiol. 2018, 178, 1522–1536. [Google Scholar] [CrossRef] [PubMed]
- Ren, J.; Li, C.; Xiu, Q.; Xu, M.; Liu, H. Overexpression of wheat spermidine synthase gene enhances wheat resistance to Fusarium head blight. Phytopathol. Res. 2024, 6, 24. [Google Scholar] [CrossRef]
- Leviczky, T.; Molnár, E.; Papdi, C.; Őszi, E.; Horváth, G.V.; Vizler, C.; Nagy, V.; Pauk, J.; Bögre, L.; Magyar, Z. E2FA and E2FB transcription factors coordinate cell proliferation with seed maturation. Development 2019, 146, dev179333. [Google Scholar] [CrossRef] [PubMed]
- Gangurde, S.S.; Wang, H.; Yaduru, S.; Pandey, M.K.; Fountain, J.C.; Chu, Y.; Isleib, T.; Holbrook, C.C.; Xavier, A.; Culbreath, A.K. Nested-association mapping (NAM)-based genetic dissection uncovers candidate genes for seed and pod weights in peanut (Arachis hypogaea). Plant Biotechnol. J. 2020, 18, 1457–1471. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Zhang, X.; Zhao, K.; Zhao, K.; Qu, C.; Gao, G.; Gong, F.; Ma, X.; Yin, D. Comprehensive transcriptome analyses reveal candidate genes for variation in seed size/weight during peanut (Arachis hypogaea L.) domestication. Front. Plant Sci. 2021, 12, 666483. [Google Scholar] [CrossRef]
- Huang, J.; Lu, G.; Liu, L.; Raihan, M.S.; Xu, J.; Jian, L.; Zhao, L.; Tran, T.M.; Zhang, Q.; Liu, J. The kernel size-related quantitative trait locus qKW9 encodes a pentatricopeptide repeat protein that affects photosynthesis and grain filling. Plant Physiol. 2020, 183, 1696–1709. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Trait | Env. | Parents | RIL Population | ||||||
|---|---|---|---|---|---|---|---|---|---|
| DF12 | HY44 | Max | Min | Mean ± SD | CV/% | Kurt | Skew | ||
| HPW | 20FY | 132.64 | 158.65 | 356.15 | 77.80 | 140.62 ± 35.51 | 25.25 | 14.93 | 3.24 |
| (g) | 21FY | 162.97 | 192.86 | 203.23 | 65.50 | 149.30 ± 24.69 | 16.54 | 0.02 | −0.36 |
| 21LD | 127.86 | 154.54 | 220.92 | 36.10 | 126.57 ± 26.24 | 20.73 | 1.48 | −0.02 | |
| 21NB | 112.37 | 140.68 | 180.24 | 62.71 | 123.27 ± 21.10 | 17.11 | 0.03 | −0.12 | |
| 22FY | 94.01 | 126.99 | 179.81 | 77.10 | 126.52 ± 19.51 | 15.43 | 0.33 | 0.06 | |
| 23LD | 135.66 | 213.91 | 236.88 | 111.26 | 166.74 ± 23.77 | 14.25 | 0.02 | 0.32 | |
| PL | 20FY | 29.81 | 33.44 | 43.85 | 26.62 | 34.40 ± 3.08 | 8.96 | −0.17 | 0.10 |
| (mm) | 21FY | 31.22 | 34.65 | 39.40 | 27.06 | 33.44 ± 2.80 | 8.36 | −0.73 | −0.14 |
| 21LD | 29.85 | 34.78 | 41.99 | 15.70 | 32.15 ± 3.58 | 11.12 | 1.98 | −0.63 | |
| 21NB | 29.77 | 33.69 | 41.77 | 22.40 | 33.10 ± 3.46 | 10.44 | −0.31 | −0.18 | |
| 22FY | 29.49 | 34.75 | 40.04 | 25.27 | 31.94 ± 2.64 | 8.26 | −0.10 | 0.09 | |
| 23LD | 29.78 | 33.87 | 37.72 | 25.49 | 32.25 ± 2.52 | 7.83 | −0.29 | −0.26 | |
| PW | 20FY | 12.48 | 14.85 | 19.04 | 11.70 | 14.91 ± 1.15 | 7.74 | 0.94 | 0.48 |
| (mm) | 21FY | 15.98 | 16.29 | 18.07 | 12.25 | 14.98 ± 1.34 | 8.95 | −0.81 | 0.21 |
| 21LD | 14.45 | 15.46 | 17.90 | 10.70 | 14.54 ± 1.26 | 8.69 | 0.06 | 0.23 | |
| 21NB | 12.25 | 14.77 | 17.46 | 11.89 | 14.59 ± 1.07 | 7.31 | 0.23 | 0.42 | |
| 22FY | 12.57 | 15.67 | 17.84 | 11.56 | 14.48 ± 1.23 | 8.49 | −0.23 | 0.26 | |
| 23LD | 13.37 | 15.43 | 17.24 | 11.88 | 14.59 ± 0.97 | 6.62 | −0.10 | 0.19 | |
| HSW | 20FY | 54.15 | 74.36 | 77.46 | 33.15 | 56.45 ± 8.58 | 15.20 | −0.40 | −0.05 |
| (g) | 21FY | 60.34 | 75.87 | 85.82 | 33.26 | 65.07 ± 8.69 | 13.36 | 0.22 | −0.20 |
| 21LD | 48.12 | 85.85 | 92.30 | 32.30 | 61.50 ± 12.26 | 19.93 | −0.39 | 0.04 | |
| 21NB | 62.57 | 76.45 | 81.93 | 29.80 | 55.49 ± 9.43 | 17.00 | 0.02 | −0.04 | |
| 22FY | 33.19 | 44.69 | 66.09 | 10.29 | 46.48 ± 8.12 | 17.48 | 1.46 | −0.38 | |
| 23LD | 55.70 | 89.94 | 98.24 | 44.38 | 68.54 ± 10.00 | 14.60 | −0.10 | 0.29 | |
| SL | 20FY | 14.92 | 16.24 | 18.55 | 10.89 | 15.27 ± 1.27 | 8.30 | 0.05 | −0.15 |
| (mm) | 21FY | 15.47 | 16.60 | 18.75 | 12.34 | 15.81 ± 1.21 | 7.65 | −0.43 | −0.17 |
| 21LD | 13.79 | 18.11 | 18.41 | 10.58 | 15.47 ± 1.46 | 9.44 | −0.15 | −0.30 | |
| 21NB | 13.91 | 17.79 | 18.13 | 11.68 | 15.22 ± 1.30 | 8.53 | −0.53 | −0.23 | |
| 22FY | 12.90 | 15.35 | 17.37 | 11.83 | 14.48 ± 1.14 | 7.85 | −0.37 | 0.04 | |
| 23LD | 14.52 | 16.79 | 18.01 | 12.85 | 15.50 ± 1.10 | 7.10 | −0.47 | −0.23 | |
| SW | 20FY | 8.43 | 10.01 | 9.75 | 6.32 | 8.62 ± 0.54 | 6.28 | 1.08 | −0.56 |
| (mm) | 21FY | 8.81 | 10.32 | 10.81 | 7.54 | 9.31 ± 0.51 | 5.44 | 0.49 | 0.04 |
| 21LD | 8.18 | 10.22 | 10.49 | 6.55 | 8.78 ± 0.70 | 7.99 | 0.14 | −0.22 | |
| 21NB | 8.12 | 9.35 | 10.92 | 6.76 | 8.73 ± 0.64 | 7.35 | 0.53 | −0.17 | |
| 22FY | 6.96 | 7.80 | 9.35 | 6.47 | 8.06 ± 0.55 | 6.83 | 0.05 | −0.24 | |
| 23LD | 8.77 | 11.04 | 11.10 | 7.87 | 9.54 ± 0.63 | 6.63 | −0.30 | 0.18 | |
| Environment | Trait | HPW | PL | PW | HSW | SL | SW |
|---|---|---|---|---|---|---|---|
| 20FY | HPW | 1 | |||||
| PL | 0.20 ** | 1 | |||||
| PW | 0.37 *** | 0.64 *** | 1 | ||||
| HSW | 0.36 *** | 0.39 *** | 0.44 *** | 1 | |||
| SL | 0.28 *** | 0.70 *** | 0.52 *** | 0.76 *** | 1 | ||
| SW | 0.28 *** | 0.045 | 0.20 ** | 0.83 *** | 0.38 *** | 1 | |
| 21FY | HPW | 1 | |||||
| PL | 0.66 *** | 1 | |||||
| PW | 0.65 *** | 0.65 *** | 1 | ||||
| HSW | 0.85 *** | 0.62 *** | 0.61 *** | 1 | |||
| SL | 0.70 *** | 0.86 *** | 0.65 *** | 0.77 *** | 1 | ||
| SW | 0.68 *** | 0.26 *** | 0.44 *** | 0.83 *** | 0.41 *** | 1 | |
| 21LD | HPW | 1 | |||||
| PL | 0.71 *** | 1 | |||||
| PW | 0.68 *** | 0.68 *** | 1 | ||||
| HSW | 0.83 *** | 0.55 *** | 0.57 *** | 1 | |||
| SL | 0.69 *** | 0.77 *** | 0.59 *** | 0.78 *** | 1 | ||
| SW | 0.65 *** | 0.19 ** | 0.42 *** | 0.84 *** | 0.42 *** | 1 | |
| 21NB | HPW | 1 | |||||
| PL | 0.58 *** | 1 | |||||
| PW | 0.64 *** | 0.65 *** | 1 | ||||
| HSW | 0.82 *** | 0.51 *** | 0.57 *** | 1 | |||
| SL | 0.44 *** | 0.63 *** | 0.44 *** | 0.52 *** | 1 | ||
| SW | 0.46 *** | 0.07 | 0.27 *** | 0.61 *** | 0.25 *** | 1 | |
| 22FY | HPW | 1 | |||||
| PL | 0.65 *** | 1 | |||||
| PW | 0.62 *** | 0.72 *** | 1 | ||||
| HSW | 0.90 *** | 0.53 *** | 0.48 *** | 1 | |||
| SL | 0.74 *** | 0.85 *** | 0.64 *** | 0.71 *** | 1 | ||
| SW | 0.77 *** | 0.23 *** | 0.35 *** | 0.81 *** | 0.43 *** | 1 | |
| 23LD | HPW | 1 | |||||
| PL | 0.54 *** | 1 | |||||
| PW | 0.63 *** | 0.69 *** | 1 | ||||
| HSW | 0.95 *** | 0.44 *** | 0.55 *** | 1 | |||
| SL | 0.63 *** | 0.82 *** | 0.63 *** | 0.61 *** | 1 | ||
| SW | 0.79 *** | 0.093 | 0.37 *** | 0.87 *** | 0.29 *** | 1 |
| Trait | QTL | Chr. | Range (cm) | LOD | Env. | Additive | PVE% |
|---|---|---|---|---|---|---|---|
| HPW | qHPWA05.1 | A05 | 29.91–49.52 | 13.45 | 21FY | 12.80 | 26.63 |
| qHPWA05.2 | A05 | 34.81–49.52 | 11.71 | 22FY | 9.52 | 23.64 | |
| qHPWA05.3 | A05 | 29.91–49.42 | 9.36 | 21LD | 11.77 | 19.39 | |
| qHPWA05.4 | A05 | 30.17–48.99 | 11.16 | 23LD | 11.41 | 22.66 | |
| qHPWA05.5 | A05 | 35.34–48.72 | 5.95 | 21NB | 7.68 | 12.81 | |
| qHPWB08 | B08 | 0–0.80 | 4.18 | 22FY | −5.94 | 9.18 | |
| qHPWB10 | B10 | 23.66–24.76 | 4.17 | 21FY | 7.50 | 9.16 | |
| PL | qPLA02 | A02 | 67.60–68.37 | 4.21 | 21NB | 1.05 | 9.24 |
| qPLA05.1 | A05 | 24.34–48.19 | 7.88 | 20FY | 1.26 | 16.59 | |
| qPLA05.2 | A05 | 25.14–52.32 | 19.28 | 21FY | 1.68 | 35.85 | |
| qPLA05.3 | A05 | 21.43–49.52 | 13.48 | 22FY | 1.37 | 26.82 | |
| qPLA05.4 | A05 | 27.78–48.19 | 9.41 | 21LD | 1.61 | 19.49 | |
| qPLA05.5 | A05 | 27.78–49.52 | 12.62 | 23LD | 1.28 | 25.22 | |
| qPLA05.6 | A05 | 26.73–52.32 | 9.36 | 21NB | 1.55 | 19.38 | |
| qPLB04 | B04 | 35.65–37.53 | 4.83 | 21FY | −0.91 | 10.52 | |
| PW | qPWA05.1 | A05 | 34.02–48.19 | 7.88 | 20FY | 0.47 | 16.60 |
| qPWA05.2 | A05 | 29.91–49.52 | 15.89 | 21FY | 1.37 | 30.65 | |
| qPWA05.3 | A05 | 30.96–49.52 | 13.26 | 22FY | 0.63 | 26.31 | |
| qPWA05.4 | A05 | 25.14–52.32 | 15.51 | 21LD | 0.70 | 30.04 | |
| qPWA05.5 | A05 | 35.87–49.52 | 10.91 | 23LD | 0.46 | 22.22 | |
| qPWA05.6 | A05 | 34.02–52.32 | 10.26 | 21NB | 0.49 | 21.04 | |
| HSW | qHSWA05.1 | A05 | 34.81–46.33 | 6.59 | 20FY | 3.29 | 14.08 |
| qHSWA05.2 | A05 | 27.8–49.52 | 16.88 | 21FY | 4.97 | 32.20 | |
| qHSWA05.3 | A05 | 35.34–47.39 | 8.49 | 22FY | 3.44 | 17.75 | |
| qHSWA05.4 | A05 | 29.91–49.52 | 10.60 | 23LD | 4.70 | 21.66 | |
| qHSWA05.5 | A05 | 34.02–49.52 | 7.92 | 21NB | 3.90 | 16.68 | |
| qHSWB10.1 | B10 | 20.64–35.72 | 5.95 | 21FY | 3.13 | 12.82 | |
| qHSWB10.2 | B10 | 23.12–26.91 | 4.39 | 22FY | 2.48 | 9.61 | |
| qHSWB10.3 | B10 | 20.64–30.13 | 4.83 | 21NB | 3.10 | 10.52 | |
| SL | qSLA05.1 | A05 | 30.17–52.32 | 10.83 | 20FY | 0.60 | 22.08 |
| qSLA05.2 | A05 | 21.17–52.32 | 22.72 | 21FY | 0.77 | 40.73 | |
| qSLA05.3 | A05 | 19.84–52.32 | 20.66 | 22FY | 0.70 | 37.86 | |
| qSLA05.4 | A05 | 23.82–49.52 | 12.72 | 21LD | 0.74 | 25.39 | |
| qSLA05.5 | A05 | 21.17–48.19 | 12.10 | 23LD | 0.54 | 24.31 | |
| qSLA05.6 | A05 | 38.22–44.25 | 4.84 | 21NB | 0.42 | 10.54 | |
| SW | qSWA02 | A02 | 66.49–68.37 | 5.35 | 23LD | −0.21 | 11.60 |
| qSWA05.1 | A05 | 30.70–46.59 | 7.57 | 21FY | 0.20 | 16.00 | |
| qSWA05.2 | A05 | 36.92–43.73 | 5.18 | 22FY | 0.18 | 11.25 | |
| qSWA05.3 | A05 | 37.18–48.99 | 5.88 | 21LD | 0.25 | 12.67 | |
| qSWA05.4 | A05 | 38.75–45.55 | 5.90 | 23LD | 0.23 | 12.69 | |
| qSWB04.1 | B04 | 47.79–51.78 | 4.73 | 22FY | 0.18 | 10.31 | |
| qSWB04.2 | B04 | 32.43–51.25 | 5.40 | 21LD | 0.24 | 11.70 | |
| qSWB08 | B08 | 0–1.07 | 4.92 | 22FY | −0.18 | 10.71 | |
| qSWB10 | B10 | 20.64–35.45 | 4.84 | 21FY | 0.17 | 10.54 |
| Trait | Chromosome | Marker Interval | LOD | Add | PVE(%) |
|---|---|---|---|---|---|
| qHPWA05.6 | A05 | Ah900023–Ah900027 | 11.31 | −9.89 | 16.69 |
| qPLA05.7 | A05 | Ah900023–Ah900027 | 16.82 | −1.32 | 23.90 |
| qPWA05.7 | A05 | Ah900023–Ah900027 | 12.43 | −0.56 | 18.06 |
| qHSWA05.6 | A05 | Ah900027–Ah900026 | 20.26 | −0.65 | 27.92 |
| qSLA05.7 | A05 | Ah900023–Ah900027 | 12.15 | −4.10 | 18.36 |
| qSWA05.5 | A05 | Ah900023–Ah900027 | 7.08 | −0.20 | 10.75 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, X.; Wang, L.; Liu, Q.; Zhang, X.; Tian, Y.; Xue, Y.; Zhang, H.; Li, N.; Zhang, X.; Bai, D. Identification of Hotspot Regions for Candidate Genes Associated with Peanut (Arachis hypogaea L.) Pod and Seed Size on Chromosome A05. Agriculture 2024, 14, 1634. https://doi.org/10.3390/agriculture14091634
Zhang X, Wang L, Liu Q, Zhang X, Tian Y, Xue Y, Zhang H, Li N, Zhang X, Bai D. Identification of Hotspot Regions for Candidate Genes Associated with Peanut (Arachis hypogaea L.) Pod and Seed Size on Chromosome A05. Agriculture. 2024; 14(9):1634. https://doi.org/10.3390/agriculture14091634
Chicago/Turabian StyleZhang, Xiaoji, Luhuan Wang, Qimei Liu, Xiaoyu Zhang, Yuexia Tian, Yunyun Xue, Huiqi Zhang, Na Li, Xin Zhang, and Dongmei Bai. 2024. "Identification of Hotspot Regions for Candidate Genes Associated with Peanut (Arachis hypogaea L.) Pod and Seed Size on Chromosome A05" Agriculture 14, no. 9: 1634. https://doi.org/10.3390/agriculture14091634
APA StyleZhang, X., Wang, L., Liu, Q., Zhang, X., Tian, Y., Xue, Y., Zhang, H., Li, N., Zhang, X., & Bai, D. (2024). Identification of Hotspot Regions for Candidate Genes Associated with Peanut (Arachis hypogaea L.) Pod and Seed Size on Chromosome A05. Agriculture, 14(9), 1634. https://doi.org/10.3390/agriculture14091634