Synthesis of Diatomite-Based Mesoporous Wool-Ball-Like Microspheres and Their Application for Toluene Total Oxidation Reaction

 , , and
, , and 
Abstract
:
1. Introduction
2. Experimental Details
2.1. Materials
2.2. Methods
2.2.1. Purification of Diatomite
2.2.2. Hydrothermal Treatment of DE
2.2.3. Preparation of Catalyst/DBM Samples
Ag/DBM Preparation
Mn/DBM Preparation
2.2.4. Material Characterization
2.2.5. Catalytic Activity Test
3. Results and Discussion
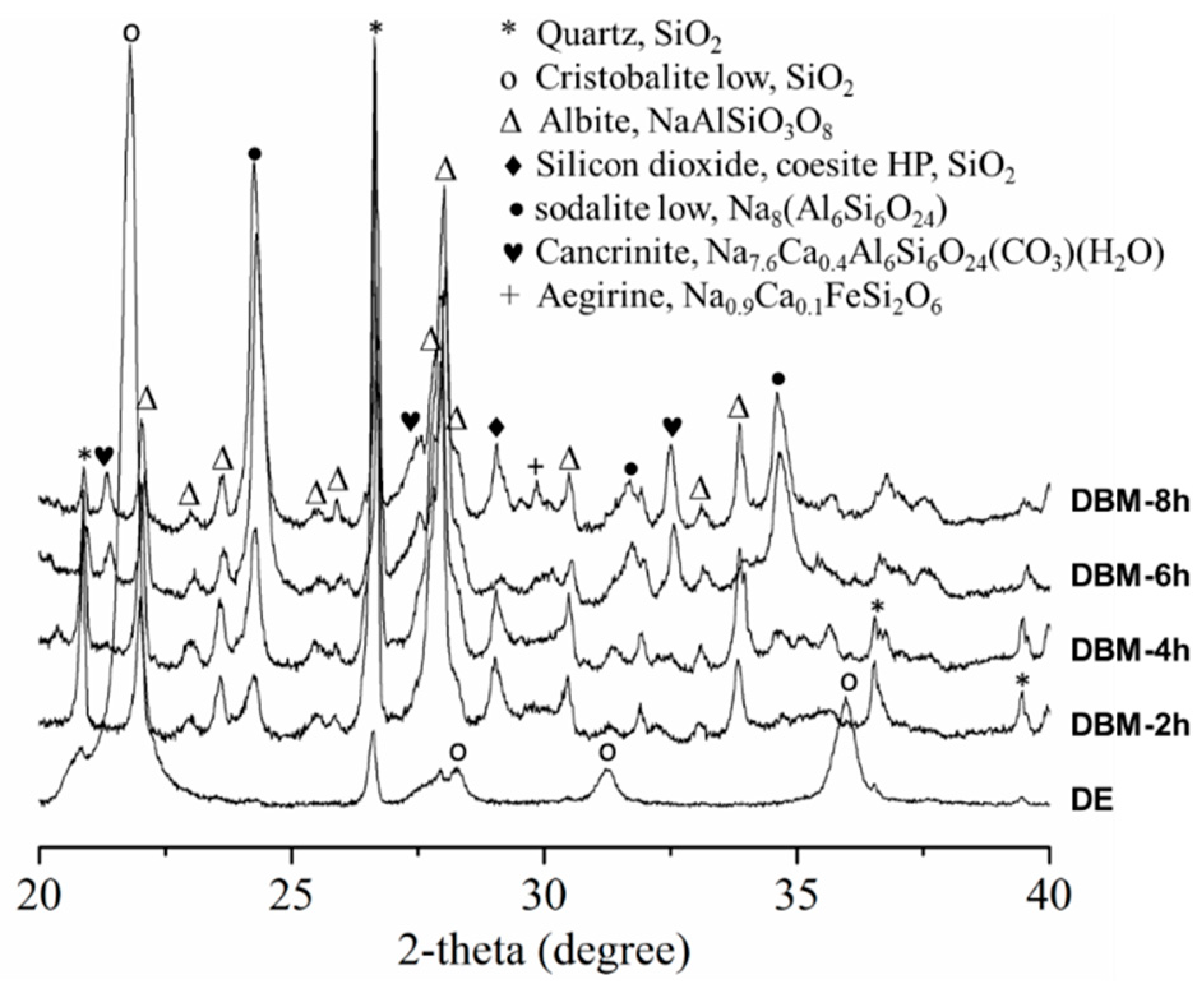
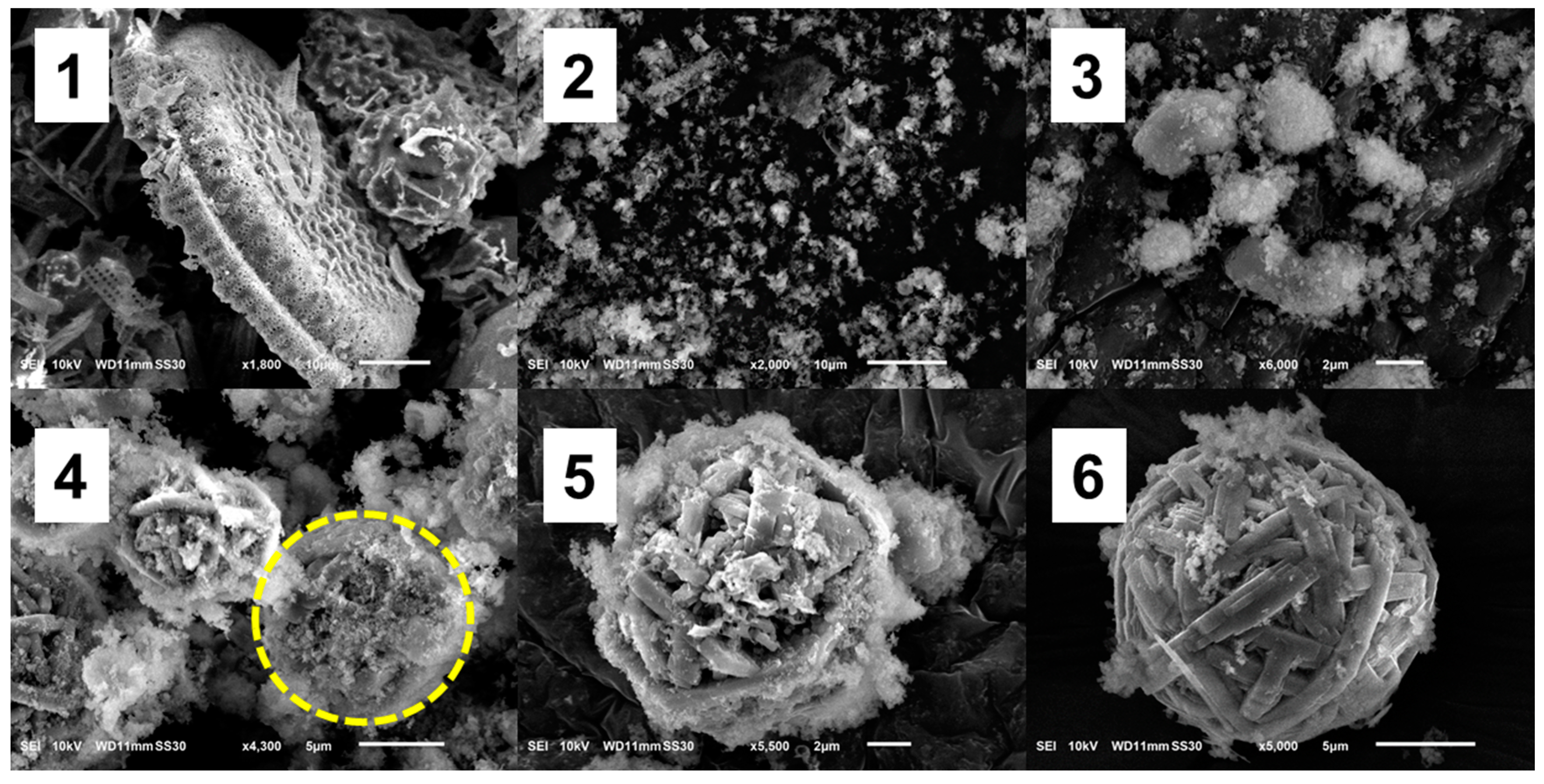
3.1. Structure and Formation Mechanism of DE WBL Microspheres
3.1.1. Formation Mechanism of WBL Microspheres
- DE dissolves under the attack of hydroxide ions.
- When the concentration of silicate species reaches supersaturation under the hydrothermal condition, mesophase structures (sheet-like) of silicates are formed and coalesced into clusters.
- Crystallization begins at high temperature and pressure under hydrothermal conditions. With the preferential binding of CTA+, these mesophases favorably crystallize into microfibers. Then, these fibers curve to minimize the free energy and eventually form a WBL microsphere.
3.1.2. Effects of Cetyltrimethylammonium Bromide on DE-Based Materials Formation
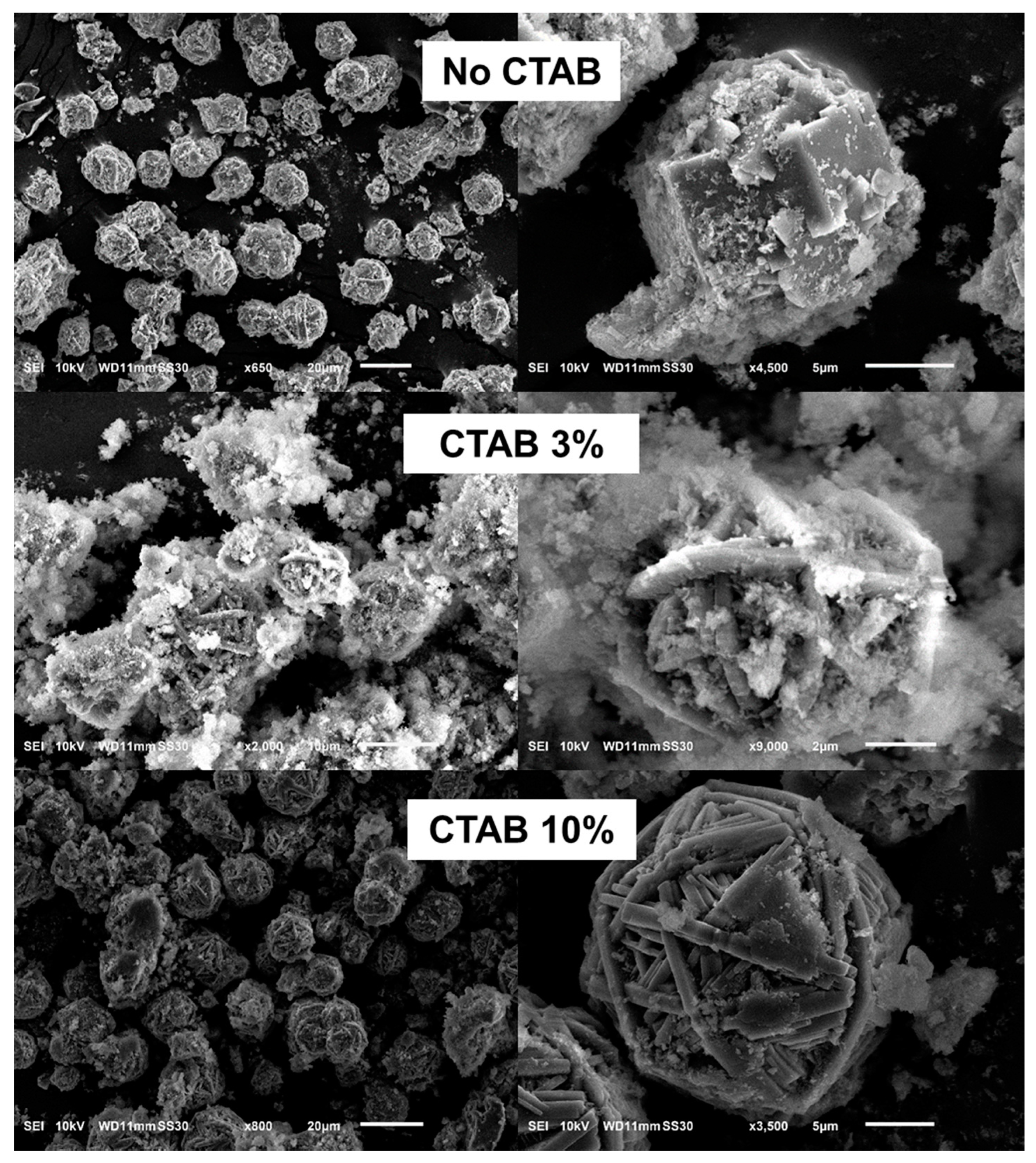
3.1.3. Effects of CTAB Concentration on Morphology and Crystal Structure
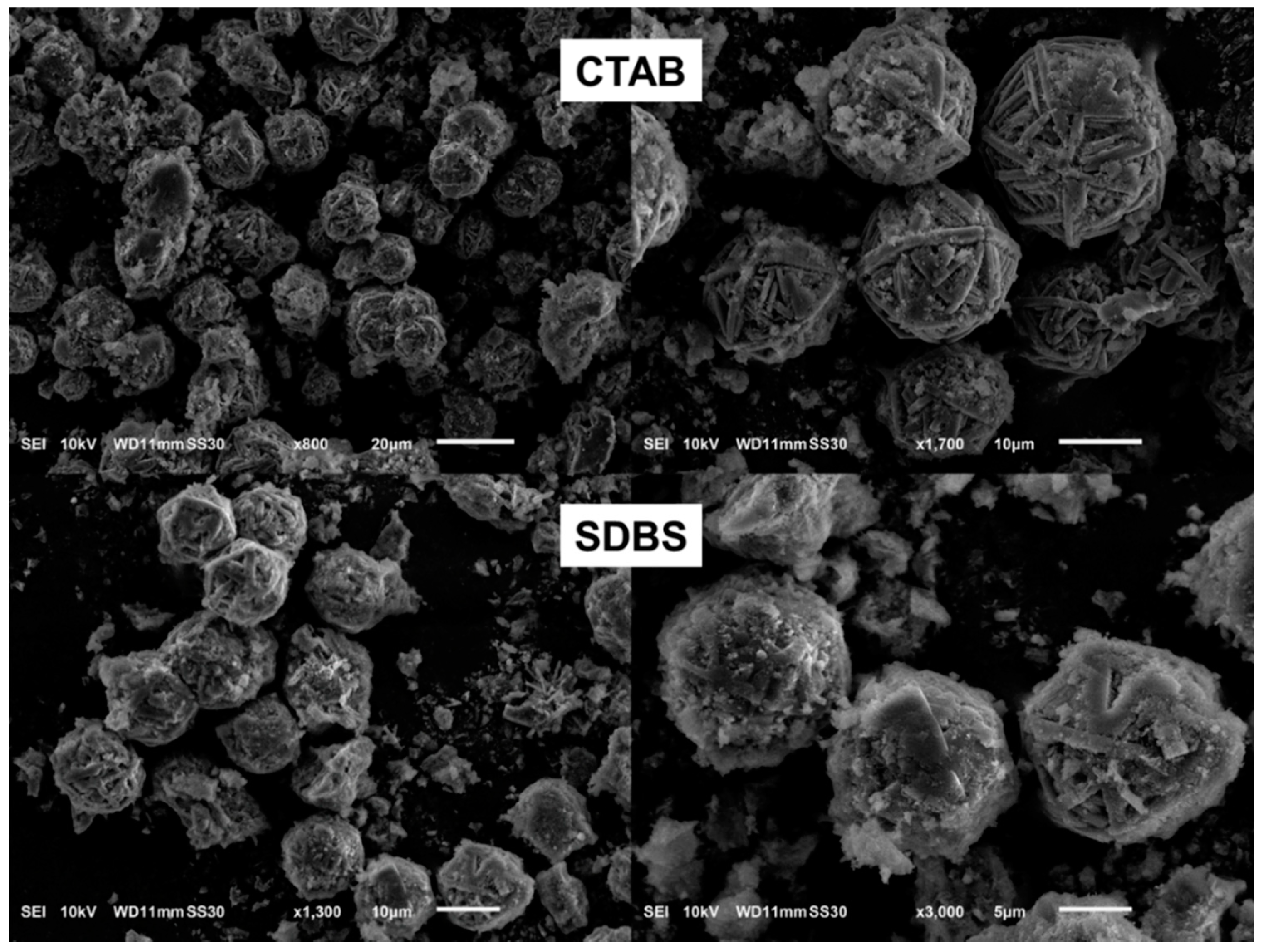
3.1.4. Effect of Surfactant Type (Cationic Versus Anionic)
3.1.5. Textural Properties
3.2. DE WBL Microspheres as Support for Metallic Catalyst
3.2.1. Crystalline Phase of Ag/DBM and Mn/DBM Catalysts
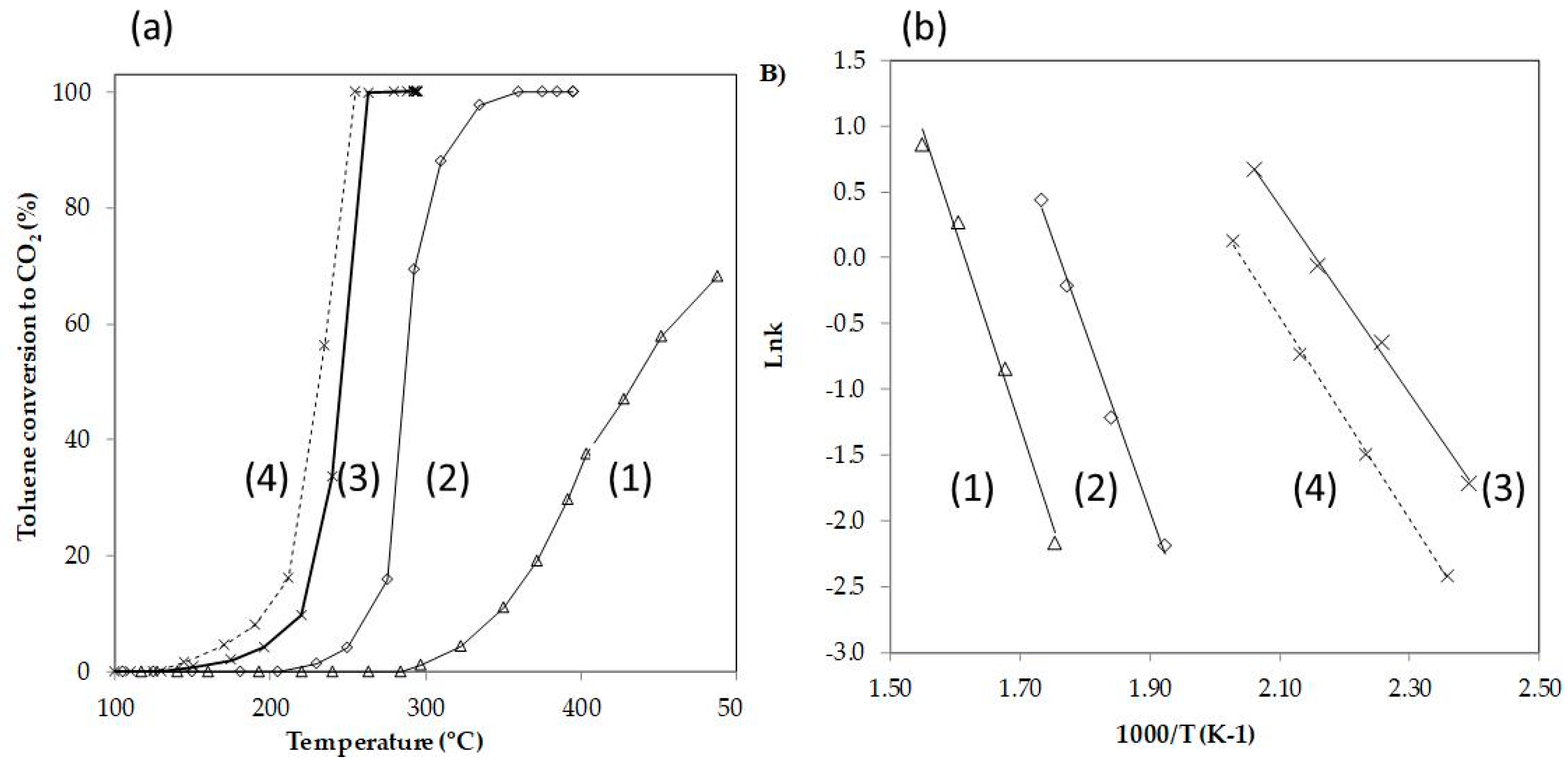
3.2.2. Catalytic Performance of Ag/DBM and Mn/DBM Catalysts
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Sandhage, K.H.; Dickerson, M.B.; Huseman, P.M.; Caranna, M.A.; Clifton, J.D.; Bull, T.A.; Heibel, T.J.; Overton, W.R.; Schoenwaelder, M.E.A. Novel, bioclastic route to self-assembled, 3D, chemically tailored meso/nanostructures: Shape-preserving reactive conversion of biosilica (Diatom) microshells. Adv. Mater. 2002, 14, 429–433. [Google Scholar] [CrossRef]
- Losic, D.; Mitchell, J.G.; Voelcker, N.H. Diatomaceous lessons in nanotechnology and advanced materials. Adv. Mater. 2009, 21, 2947–2958. [Google Scholar] [CrossRef]
- Li, Y.; Sun, H.; Feng, R.; Wang, Y.; Subhan, F.; Yan, Z.; Zhang, Z.; Liu, Z. Synthesis of ZSM-5 zeolite from diatomite for fluid catalytic cracking (FCC) application. Appl. Petrochem. Res. 2015, 5, 347–353. [Google Scholar] [CrossRef] [Green Version]
- Xia, P.; Wang, X.; Wang, X.; Zhang, J.; Wang, H.; Song, J.; Ma, R.; Wang, J.; Zhao, J. Synthesis and characterization of MgO modified diatomite for phosphorus recovery in eutrophic water. J. Chem. Eng. Data 2017, 62, 226–235. [Google Scholar] [CrossRef]
- Liu, P.; He, H.; Wei, G.; Liu, D.; Liang, X.; Chen, T.; Zhu, J.; Zhu, R. An efficient catalyst of manganese supported on diatomite for toluene oxidation: Manganese species, catalytic performance, and structure-activity relationship. Microporous Mesoporous Mater. 2017, 239, 101–110. [Google Scholar] [CrossRef]
- Qian, T.; Li, J.; Deng, Y. Pore structure modified diatomite-supported PEG composites for thermal energy storage. Sci. Rep. 2016, 6, 32392. [Google Scholar] [CrossRef]
- Ruggiero, I.; Terracciano, M.; Martucci, N.M.; De Stefano, L.; Migliaccio, N.; Tatè, R.; Rendina, I.; Arcari, P.; Lamberti, A.; Rea, I. Diatomite silica nanoparticles for drug delivery. Nanoscale Res. Lett. 2014, 9, 329. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Tang, Y.; Dong, A.; Wang, X.; Ren, N.; Gao, Z. Zeolitization of diatomite to prepare hierarchical porous zeolite materials through a vapor-phase transport process. J. Mater. Chem. 2002, 12, 1812–1818. [Google Scholar] [CrossRef]
- Sanhueza, V.; Kelm, U.; Cid, R.; López-Escobar, L. Synthesis of ZSM-5 from diatomite: a case of zeolite synthesis from a natural material. J. Chem. Technol. Biotechnol. 2004, 79, 686–690. [Google Scholar] [CrossRef]
- Sanhueza, V.; Kelm, U.; Cid, R. Synthesis of mordenite from diatomite: a case of zeolite synthesis from natural material. J. Chem. Technol. Biotechnol. 2003, 78, 485–488. [Google Scholar] [CrossRef]
- Yao, G.; Lei, J.; Zhang, X.; Sun, Z.; Zheng, S.; Komarneni, S. Mechanism of zeolite X crystallization from diatomite. Mater. Res. Bull. 2018, 107, 132–138. [Google Scholar] [CrossRef]
- Hadjar, H.; Hamdi, B.; Jaber, M.; Brendlé, J.; Kessaïssia, Z.; Balard, H.; Donnet, J.B. Elaboration and characterisation of new mesoporous materials from diatomite and charcoal. Microporous Mesoporous Mater. 2008, 107, 219–226. [Google Scholar] [CrossRef]
- Kresge, C.T.; Leonowicz, M.E.; Roth, W.J.; Vartuli, J.C.; Beck, J.S. Ordered mesoporous molecular sieves synthesized by a liquid-crystal template mechanism. Nature 1992, 359, 710–712. [Google Scholar] [CrossRef]
- Beck, J.S.; Vartuli, J.C.; Kennedy, G.J.; Kresge, C.T.; Roth, W.J.; Schramm, S.E. Molecular or supramolecular templating: Defining the role of surfactant chemistry in the formation of microporous and mesoporous molecular sieves. Chem. Mater. 1994, 6, 1816–1821. [Google Scholar] [CrossRef]
- Sachse, A.; García-Martínez, J. Surfactant-templating of zeolites: From design to application. Chem. Mater. 2017, 29, 3827–3853. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, S.; Wei, K.; Zhao, N.; Chen, J.; Wang, X. Hydrothermal synthesis of hydroxyapatite nanopowders using cationic surfactant as a template. Mater. Lett. 2006, 60, 1484–1487. [Google Scholar] [CrossRef]
- Monnier, A.; Schüth, F.; Huo, Q.; Kumar, D.; Margolese, D.; Maxwell, R.S.; Stucky, G.D.; Krishnamurty, M.; Petroff, P.; Firouzi, A.; et al. Cooperative formation of inorganic-organic interfaces in the synthesis of silicate mesostructures. Science 1993, 261, 1299–1303. [Google Scholar] [CrossRef]
- Kumari, L.; Li, W.; Kulkarni, S.; Wu, K.; Chen, W.; Wang, C.; Vannoy, C.H.; Leblanc, R.M. Effect of surfactants on the structure and morphology of magnesium borate hydroxide nanowhiskers synthesized by hydrothermal route. Nanoscale Res. Lett. 2009, 5, 149. [Google Scholar] [CrossRef] [Green Version]
- Lin, K.; Chang, J.; Cheng, R.; Ruan, M. Hydrothermal microemulsion synthesis of stoichiometric single crystal hydroxyapatite nanorods with mono-dispersion and narrow-size distribution. Mater. Lett. 2007, 61, 1683–1687. [Google Scholar] [CrossRef]
- Cui, X.; Mao, S.; Liu, M.; Yuan, H.; Du, Y. Mechanism of surfactant micelle formation. Langmuir 2008, 24, 10771–10775. [Google Scholar] [CrossRef]
- Liu, Y.; Tourbin, M.; Lachaize, S.; Guiraud, P. Silica nanoparticles separation from water: Aggregation by cetyltrimethylammonium bromide (CTAB). Chemosphere 2013, 92, 681–687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coppola, L.; Gianferri, R.; Nicotera, I.; Oliviero, C.; Antonio Ranieri, G. Structural changes in CTAB/H2O mixtures using a rheological approach. Phys. Chem. Chem. Phys. 2004, 6, 2364–2372. [Google Scholar] [CrossRef]
- Krishnaswamy, R.; Ghosh, S.K.; Lakshmanan, S.; Raghunathan, V.A.; Sood, A.K. Phase behavior of concentrated aqueous solutions of cetyltrimethylammonium bromide (CTAB) and sodium hydroxy naphthoate (SHN). Langmuir 2005, 21, 10439–10443. [Google Scholar] [CrossRef]
- Zhao, D.; Yang, P.; Margolese, D.I.; Stucky, G.D. Synthesis of continuous mesoporous silica thin films with three-dimensional accessible pore structures. Chem. Commun. 1998, 22, 2499–2500. [Google Scholar] [CrossRef]
- Zhao, D.; Huo, Q.; Feng, J.; Chmelka, B.F.; Stucky, G.D. Nonionic triblock and star diblock copolymer and oligomeric surfactant syntheses of highly ordered, hydrothermally stable, mesoporous silica structures. J. Am. Chem. Soc. 1998, 120, 6024–6036. [Google Scholar] [CrossRef]
- Corma, A. From microporous to mesoporous molecular sieve materials and their use in catalysis. Chem. Rev. 1997, 97, 2373–2420. [Google Scholar] [CrossRef]
- Goren, R.; Baykara, T.; Marsoglu, M. A study on the purification of diatomite in hydrochloric acid. Scand. J. Metall. 2002, 31, 115–119. [Google Scholar] [CrossRef]
- Martin, T.; Galarneau, A.; Di Renzo, F.; Fajula, F.; Plee, D. Morphological control of MCM-41 by pseudomorphic synthesis. Angew. Chem. Int. Ed. 2002, 41, 2590–2592. [Google Scholar] [CrossRef]
- Garcia, G.; Cardenas, E.; Cabrera, S.; Hedlund, J.; Mouzon, J. Synthesis of zeolite Y from diatomite as silica source. Microporous Mesoporous Mater. 2016, 219, 29–37. [Google Scholar] [CrossRef]
- Sanhueza, V.; López-Escobar, L.; Kelm, U.; Cid, R. Synthesis of a mesoporous material from two natural sources. J. Chem. Technol. Biotechnol. 2006, 81, 614–617. [Google Scholar] [CrossRef]
- Zhang, Y.; Jing, Z.; Kameda, T.; Yoshioka, T. Hydrothermal synthesis of hardened diatomite-based adsorbents with analcime formation for methylene blue adsorption. RSC Adv. 2016, 6, 26765–26774. [Google Scholar] [CrossRef]
- Janicke, M.T.; Landry, C.C.; Christiansen, S.C.; Kumar, D.; Stucky, G.D.; Chmelka, B.F. Aluminum incorporation and interfacial structures in MCM-41 mesoporous molecular sieves. J. Am. Chem. Soc. 1998, 120, 6940–6951. [Google Scholar] [CrossRef]
- Einicke, W.-D.; Enke, D.; Dvoyashkin, M.; Valiullin, R.; Gläser, R. The mechanism of pseudomorphic transformation of spherical silica gel into MCM-41 studied by PFG NMR diffusometry. Materials 2013, 6, 3688–3709. [Google Scholar] [CrossRef] [PubMed]
- Yu, Q.; Hui, J.; Wang, P.; Xu, B.; Zhuang, J.; Wang, X. Hydrothermal synthesis of mesoporous silica spheres: effect of the cooling process. Nanoscale 2012, 4, 7114–7120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, K.; Na, K.; Kim, J.; Terasaki, O.; Ryoo, R. Zeolite synthesis using hierarchical structure-directing surfactants: Retaining porous structure of initial synthesis gel and precursors. Chem. Mater. 2012, 24, 2733–2738. [Google Scholar] [CrossRef]
- Du, G.; LÜ, G.; He, X. Apparent dissolution kinetics of diatomite in alkaline solution. Chin. J. Chem. Eng. 2013, 21, 736–741. [Google Scholar] [CrossRef]
- Niibori, Y.; Kunita, M.; Tochiyama, O.; Chida, T. Dissolution rates of amorphous silica in highly alkaline solution. J. Nucl. Sci. Technol. 2000, 37, 349–357. [Google Scholar] [CrossRef]
- Tamada, O.; Gibbs, G.V.; Boisen, M.B., Jr.; Rimstidt, J.D. Silica dissolution catalyzed by NaOH: Reaction kinetics and energy barriers simulated by quantum mechanical strategies. J. Mineral. Petrol. Sci. 2012, 107, 87–98. [Google Scholar] [CrossRef] [Green Version]
- Belton, D.J.; Deschaume, O.; Perry, C.C. An overview of the fundamentals of the chemistry of silica with relevance to biosilicification and technological advances. FEBS J. 2012, 279, 1710–1720. [Google Scholar] [CrossRef]
- Byrappa, K.; Yoshimura, M. 5 - Hydrothermal growth of some selected crystals. In Handbook of Hydrothermal Technology; Byrappa, K., Yoshimura, M., Eds.; William Andrew Publishing: Norwich, NY, USA, 2001. [Google Scholar]
- Sun, X.M.; Chen, X.; Deng, Z.X.; Li, Y.D. A CTAB-assisted hydrothermal orientation growth of ZnO nanorods. Mater. Chem. Phys. 2003, 78, 99–104. [Google Scholar] [CrossRef]
- Lei, F.; Yan, B.; Chen, H.-H.; Zhang, Q.; Zhao, J.-T. Surfactant-assisted hydrothermal synthesis, physical characterization, and photoluminescence of PbWO4. Cryst. Growth Des. 2009, 9, 3730–3736. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, L.; Xu, L.; Meng, C.; Zhu, W. Ag/TiO2 nanofiber heterostructures: Highly enhanced photocatalysts under visible light. J. Appl. Phys. 2013, 113, 174311. [Google Scholar] [CrossRef]
- Attard, G.S.; Glyde, J.C.; Göltner, C.G. Liquid-crystalline phases as templates for the synthesis of mesoporous silica. Nature 1995, 378, 366–368. [Google Scholar] [CrossRef]
- Chen, X.; Huang, L.; Li, Q. Hydrothermal transformation and characterization of porous silica templated by surfactants. J. Phys. Chem. B 1997, 101, 8460–8467. [Google Scholar] [CrossRef]
- Alvarez, R.; Sparks, D.L. Polymerization of silicate anions in solutions at low concentrations. Nature 1985, 318, 649–651. [Google Scholar] [CrossRef]
- Walker, S.A.; Zasadzinski, J.A. Self-Assembly of Silicate/Surfactant Mesophases. MRS Proceedings 1994, 371, 93. [Google Scholar] [CrossRef]
- Park, K.; Drummy, L.F.; Wadams, R.C.; Koerner, H.; Nepal, D.; Fabris, L.; Vaia, R.A. Growth mechanism of gold nanorods. Chem. Mater. 2013, 25, 555–563. [Google Scholar] [CrossRef]
- Bricha, M.; Belmamouni, Y.; Essassi, E.M.; Ferreira, J.M.F.; Mabrouk, K.E. Surfactant-assisted hydrothermal synthesis of hydroxyapatite nanopowders. J. Nanosci. Nanotechnol. 2012, 12, 8042–8049. [Google Scholar] [CrossRef]
- Mann, S.; Archibald, D.D.; Didymus, J.M.; Douglas, T.; Heywood, B.R.; Meldrum, F.C.; Reeves, N.J. Crystallization at inorganic-organic interfaces: Biominerals and biomimetic synthesis. Science 1993, 261, 1286–1292. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Liu, L. Modern methods for delivery of drugs across the blood–brain barrier. Adv. Drug Del. Rev. 2012, 64, 640–665. [Google Scholar] [CrossRef]
- Yin, X.; Teradal, N.L.; Morag, A.; Jelinek, R. Catalytic Au wool-ball-shaped nanostructures. ChemCatChem 2017, 9, 2473–2479. [Google Scholar] [CrossRef]
- Mo, S.; Zhang, Q.; Li, J.; Sun, Y.; Ren, Q.; Zou, S.; Zhang, Q.; Lu, J.; Fu, M.; Mo, D.; et al. Highly efficient mesoporous MnO2 catalysts for the total toluene oxidation: Oxygen-Vacancy defect engineering and involved intermediates using in situ DRIFTS. Appl. Catal., B 2020, 264, 118464. [Google Scholar] [CrossRef]
- Shi, F.; Wang, F.; Dai, H.; Dai, J.; Deng, J.; Liu, Y.; Bai, G.; Ji, K.; Au, C.T. Rod-, flower-, and dumbbell-like MnO2: Highly active catalysts for the combustion of toluene. Appl. Catal., A 2012, 433-434, 206–213. [Google Scholar] [CrossRef]
- Miranda, B.; Díaz, E.; Ordóñez, S.; Vega, A.; Díez, F.V. Oxidation of trichloroethene over metal oxide catalysts: Kinetic studies and correlation with adsorption properties. Chemosphere 2007, 66, 1706–1715. [Google Scholar] [CrossRef]
- Sing, K.S.W.; Everett, D.H.; Haul, R.A.W.; Moscou, L.; Pierotti, R.A.; Rouquerol, J.; Siemieniewska, T. Reporting physisorption data for gas/solid systems. In Handbook of Heterogeneous Catalysis; Wiley-VCH: Weinheim, Germany, 2008; pp. 1217–1230. [Google Scholar]
- Saqer, S.M.; Kondarides, D.I.; Verykios, X.E. Catalytic oxidation of toluene over binary mixtures of copper, manganese and cerium oxides supported on γ-Al2O3. Appl. Catal. B 2011, 103, 275–286. [Google Scholar] [CrossRef]
- Kim, S.C.; Shim, W.G. Catalytic combustion of VOCs over a series of manganese oxide catalysts. Appl. Catal. B 2010, 98, 180–185. [Google Scholar] [CrossRef]
- Qu, Z.; Bu, Y.; Qin, Y.; Wang, Y.; Fu, Q. The effects of alkali metal on structure of manganese oxide supported on SBA-15 for application in the toluene catalytic oxidation. Chem. Eng. J. 2012, 209, 163–169. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Components | DE Raw (%) | DBM (HT-8h) (%) |
|---|---|---|
| Si | 87.50 | 47.51 |
| Fe | 4.94 | 19.73 |
| Ca | 1.98 | 9.80 |
| Al | 2.30 | 11.66 |
| K | 2.71 | 3.81 |
| P | 0.54 | 2.38 |
| Ti | - | 0.90 |
| Na | - | 4.17 |
| Sample | Specific Surface Area (m2/g) | Total Pore Volume (cm3/g) |
|---|---|---|
| DE | 3.1 | 0.001 |
| DBM-2h | 185.1 | 0.32 |
| DBM-6h | 133.5 | 0.23 |
| DBM-8h | 84.0 | 0.18 |
| Catalyst | Specific Surface Area (m2/g) | Toluene Concentration (ppm) | GHSV 1 | T90 2 (°C) | Ref. |
|---|---|---|---|---|---|
| Mn/DBM Ag/DBM | 75 64 | 160 400 400 | 40,000 h−1 (or 60,000 cm3⋅g−1⋅h−1) | 250 260 315 | This study |
| Mn/DE | 33 | 1000 | 30,000 cm3⋅g−1⋅h−1 | 279 | [5] |
| MnOx/Al2O3 CuO–MnOx/Al2O3 CeO2–MnOx/Al2O3 | 53 50 79 | 1000 | 200,000 cm3⋅g−1⋅h−1 | >300 270 | [49] |
| Mn3O4 Mn2O3 MnO2 0.5 wt. % K/Mn3O4 | 18 7 3 16 | 1000 | 15,000 cm3⋅g−1⋅h−1 | 270 280 340 250 | [50] |
| KMn/SBA-15 | 500–600 | 1600 | - | 300 | [51] |
| 5% MnOx/TiO2 MnCuOx/TiO2 | 50 | 500 | 5000 h−1 | 230 | [57] |
| 15 wt. % Cu/Al2O3 5 wt. % Cu/Al2O3 | 182 203 | 1000 160 | 4800 cm3⋅g−1⋅h−1 | 290 260 | [58] |
| FeMn oxide MnOx | 178 14 | 500 | 50,000 h−1 | 197 253 | [59] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Le, Q.-C.; Nguyen, C.C.; Le, T.T.N.; Lefèvre, T.; Dinh, M.T.N.; Hong, S.H.; Kim, S.Y.; Le, Q.V. Synthesis of Diatomite-Based Mesoporous Wool-Ball-Like Microspheres and Their Application for Toluene Total Oxidation Reaction. Nanomaterials 2020, 10, 339. https://doi.org/10.3390/nano10020339
Le Q-C, Nguyen CC, Le TTN, Lefèvre T, Dinh MTN, Hong SH, Kim SY, Le QV. Synthesis of Diatomite-Based Mesoporous Wool-Ball-Like Microspheres and Their Application for Toluene Total Oxidation Reaction. Nanomaterials. 2020; 10(2):339. https://doi.org/10.3390/nano10020339
Chicago/Turabian StyleLe, Quoc-Chon, Chinh Chien Nguyen, Thi Thanh Nhi Le, Thierry Lefèvre, Minh Tuan Nguyen Dinh, Sung Hyun Hong, Soo Young Kim, and Quyet Van Le. 2020. "Synthesis of Diatomite-Based Mesoporous Wool-Ball-Like Microspheres and Their Application for Toluene Total Oxidation Reaction" Nanomaterials 10, no. 2: 339. https://doi.org/10.3390/nano10020339