Simultaneous Determination of Uric Acid and Caffeine by Flow Injection Using Multiple-Pulse Amperometry


 ,
,  ,
, 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemical
2.2. Apparatus
2.3. Preparation of the BDD Electrode
2.4. Preparation of Urine and River Water Samples
2.5. Analytical Procedure
3. Results and Discussion
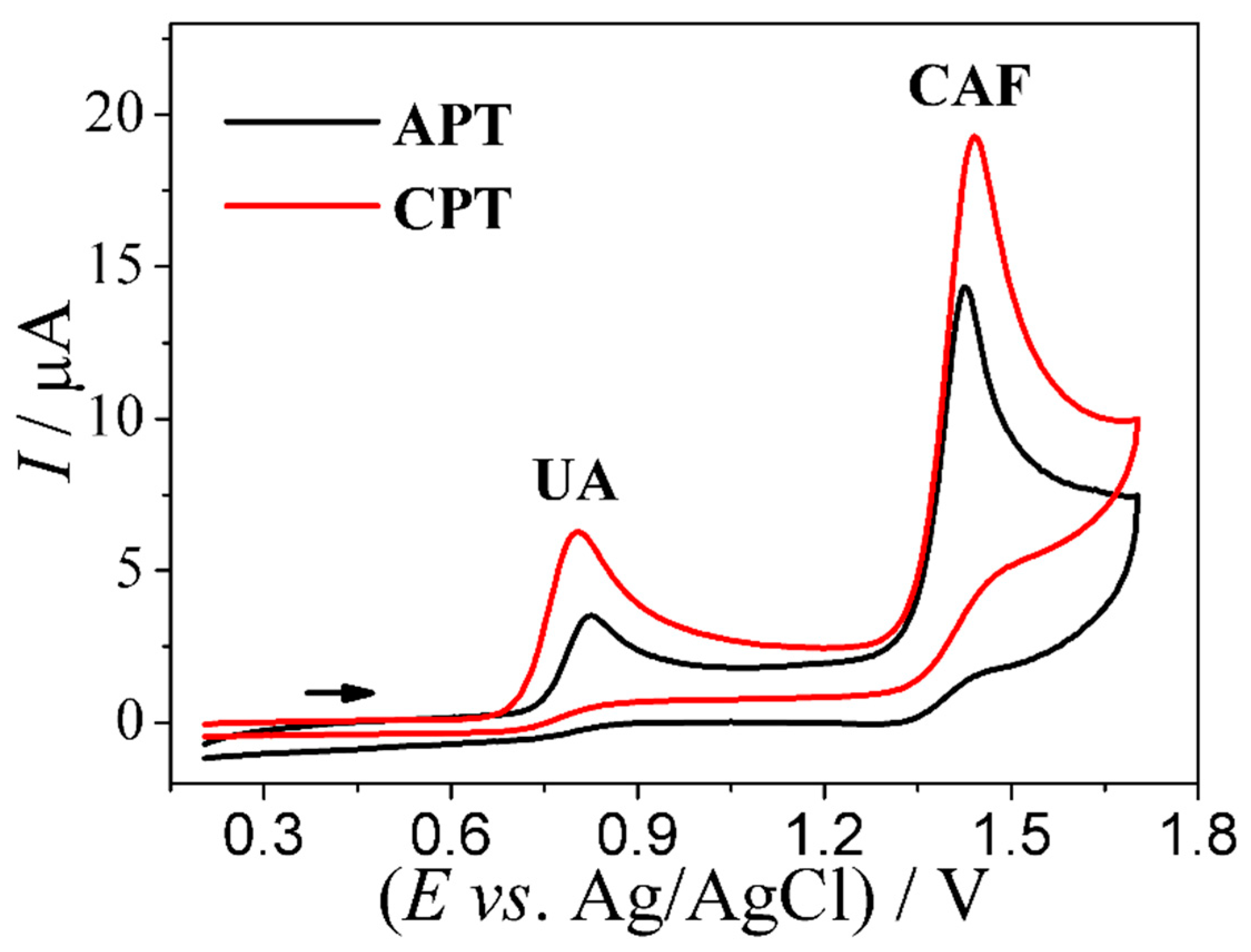
3.1. Voltammetric Profile of UA and CAF
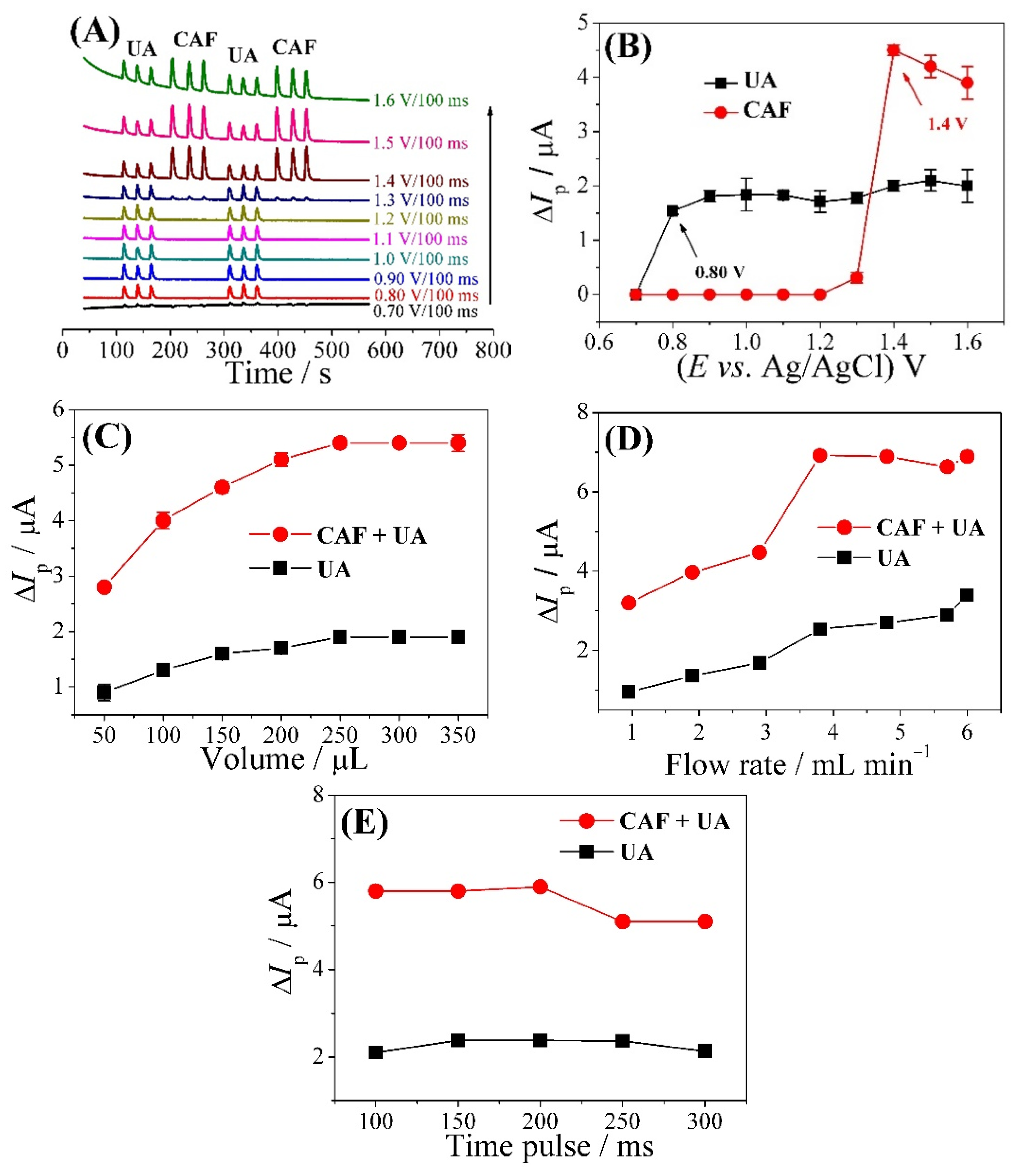
3.2. Optimization of the FIA-MPA System
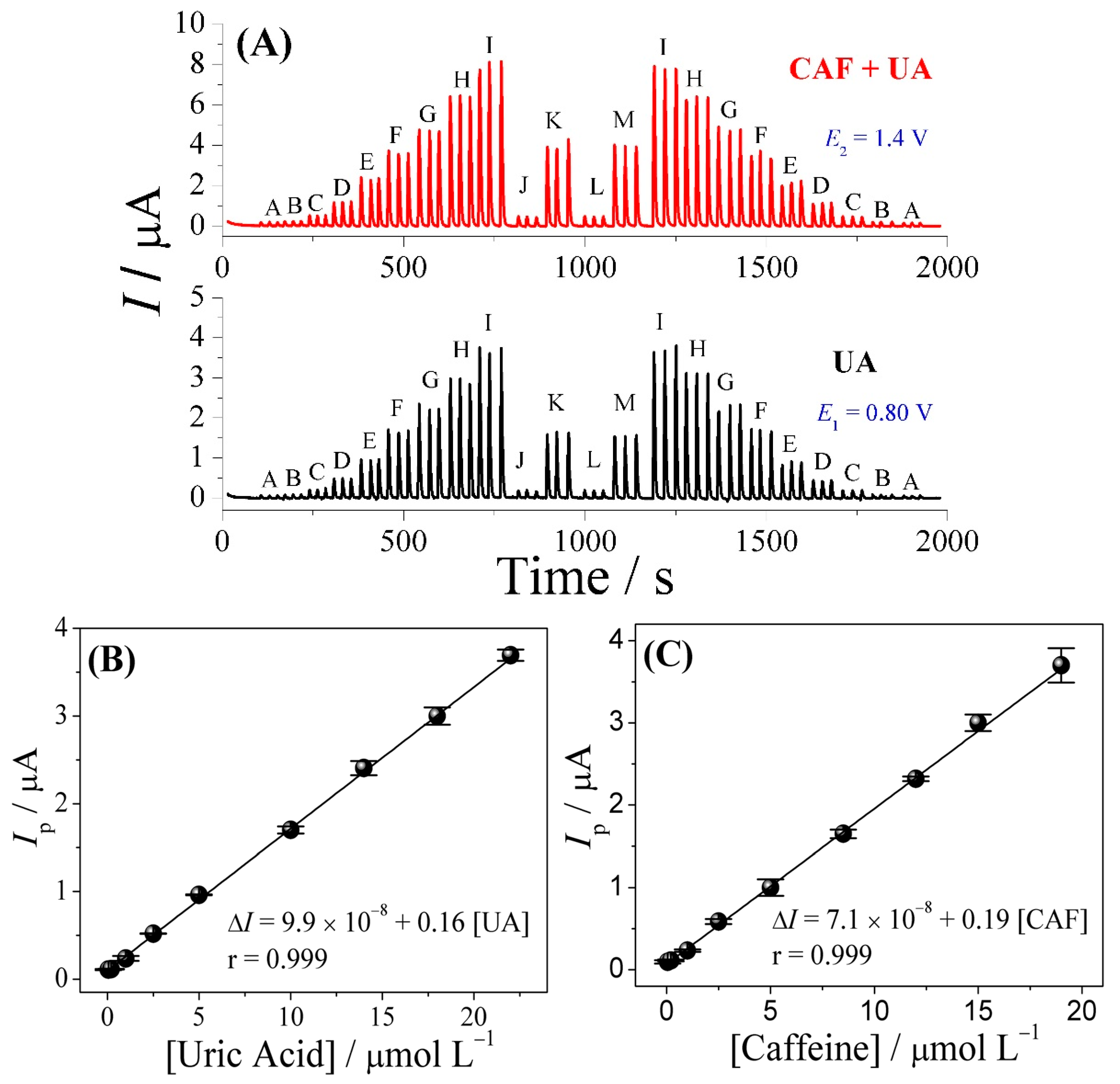
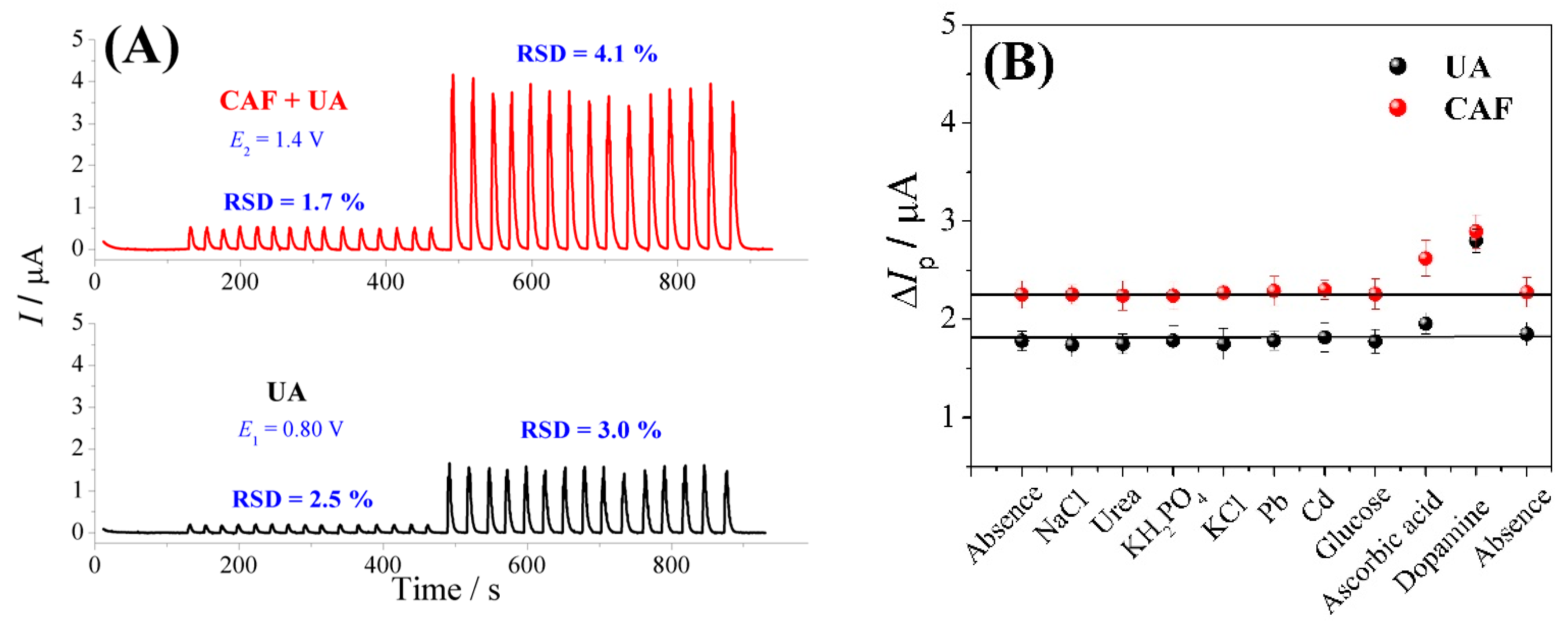
3.3. FIA-MPA of Uric Acid and Caffeine
3.4. Application of the FIA-MPA Method in River Water and Urine Samples
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ragab, G.; Elshahaly, M.; Bardin, T. Gout: An old disease in new perspective—A review. J. Adv. Res. 2017, 8, 495–511. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Chen, S.; Tang, X.; Lin, D.; Qiu, P. Ultrasensitive detection of uric acid in serum of patients with gout by a new assay based on Pt@Ag nanoflowers. RSC Adv. 2019, 9, 36578–36585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Islam, M.N.; Ahmed, I.; Anik, M.I.; Ferdous, M.S.; Khan, M.S. Developing paper based diagnostic technique to detect uric acid in urine. Front. Chem. 2018, 6, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weldegebreal, B.; Redi-Abshiro, M.; Chandravanshi, B.S. Development of new analytical methods for the determination of caffeine content in aqueous solution of green coffee beans. Chem. Cent. J. 2017, 11, 126. [Google Scholar] [CrossRef] [Green Version]
- Sereshti, H.; Samadi, S. A rapid and simple determination of caffeine in teas, coffees and eight beverages. Food Chem. 2014, 158, 8–13. [Google Scholar] [CrossRef]
- Svorc, L. Determination of Caffeine: A Comprehensive Review on Electrochemical Methods. Int. J. Electrochem. Sci. 2013, 8, 5755–5773. [Google Scholar] [CrossRef]
- Fajara, B.E.P.; Susanti, H. HPLC determination of caffeine in coffee beverage. IOP Conf. Ser. Mater. Sci. Eng. 2017, 259, 012011. [Google Scholar] [CrossRef] [Green Version]
- Du, C.; Ma, C.; Gu, J.; Li, L.; Chen, G. Fluorescence sensing of caffeine in tea beverages with 3,5-diaminobenzoic acid. Sensors 2020, 20, 819. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Wang, J.; Chen, Z. Colorimetric determination of uric acid based on the suppression of oxidative etching of silver nanoparticles by chloroauric acid. Microchim. Acta 2020, 187, 18. [Google Scholar] [CrossRef]
- Dai, X.; Fang, X.; Zhang, C.; Xu, R.; Xu, B. Determination of serum uric acid using high-performance liquid chromatography (HPLC)/isotope dilution mass spectrometry (ID-MS) as a candidate reference method. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2007, 857, 287–295. [Google Scholar] [CrossRef]
- Yamaguchi, T.; Hasegawa, K.; Kamino, S.; Miyachi, K.; Tominaga, H.; Fujita, Y. Spectrophotometric determination of uric acid based on fading of o-hydroxyhydroquinonephthalein-palladium(II)-hexadecyltrimethylammonium complex. Anal. Sci. 2007, 23, 223–226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vuletić, N.; Bardić, L.; Odžak, R. Spectrophotometric determining of caffeine content in the selection of teas, soft and energy drinks available on the Croatian market. Food Res. 2021, 5, 325–330. [Google Scholar] [CrossRef] [PubMed]
- Jiang, M.; Chen, J.S. A lable-free ECL biosensor for the detection of uric acid based on Au NRs at TiO2 nanocomposite. Int. J. Electrochem. Sci. 2019, 14, 2333–2344. [Google Scholar] [CrossRef]
- Wong, A.; Santos, A.M.; da Fonseca Alves, R.; Vicentini, F.C.; Fatibello-Filho, O.; Del Pilar Taboada Sotomayor, M. Simultaneous determination of direct yellow 50, tryptophan, carbendazim, and caffeine in environmental and biological fluid samples using graphite pencil electrode modified with palladium nanoparticles. Talanta 2021, 222, 121539. [Google Scholar] [CrossRef]
- Tasić, Ž.Z.; Petrović Mihajlović, M.B.; Simonović, A.T.; Radovanović, M.B.; Antonijević, M.M. Recent advances in electrochemical sensors for caffeine determination. Sensors 2022, 22, 9185. [Google Scholar] [CrossRef] [PubMed]
- Aafria, S.; Kumari, P.; Sharma, S.; Yadav, S.; Batra, B.; Rana, J.S.; Sharma, M. Electrochemical biosensing of uric acid: A review. Microchem. J. 2022, 182, 107945. [Google Scholar] [CrossRef]
- Lakshmi, D.; Whitcombe, M.J.; Davis, F.; Sharma, P.S.; Prasad, B.B. Electrochemical detection of uric acid in mixed and clinical samples: A review. Electroanalysis 2011, 23, 305–320. [Google Scholar] [CrossRef]
- Wong, A.; Santos, A.M.; Fatibello-Filho, O. Simultaneous determination of dopamine and cysteamine by flow injection with multiple pulse amperometric detection using a boron-doped diamond electrode. Diam. Relat. Mater. 2018, 85, 68–73. [Google Scholar] [CrossRef]
- Sotomayor, P.T.; Raimundo, I.M.; De Oliveira Neto, G.; De Oliveiira, W.A. Evaluation of fibre optical chemical sensors for flow analysis systems. Sens. Actuators B Chem. 1998, 51, 382–390. [Google Scholar] [CrossRef]
- Ghalkhani, M.; Bakirhan, N.K.; Ozkan, S.A. Combination of Efficiency with Easiness, Speed, and Cheapness in Development of Sensitive Electrochemical Sensors. Crit. Rev. Anal. Chem. 2020, 50, 538–553. [Google Scholar] [CrossRef]
- Hayashi, T.; Sakurada, I.; Honda, K.; Motohashi, S.; Uchikura, K. Electrochemical detection of sugar-related compounds using boron-doped diamond electrodes. Anal. Sci. 2012, 28, 127–133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lourenção, B.C.; Brocenschi, R.F.; Medeiros, R.A.; Fatibello-Filho, O.; Rocha-Filho, R.C. Analytical applications of electrochemically pretreated boron-doped diamond electrodes. ChemElectroChem 2020, 7, 1291–1311. [Google Scholar] [CrossRef]
- Pleskacova, A.; Brejcha, S.; Pacal, L.; Kankova, K.; Tomandl, J. Simultaneous determination of uric acid, xanthine and hypoxanthine in human plasma and serum by hplc–uv: Uric acid metabolism tracking. Chromatographia 2017, 80, 529–536. [Google Scholar] [CrossRef]
- Lopez-Sanchez, R.D.C.; Lara-Diaz, V.J.; Aranda-Gutierrez, A.; Martinez-Cardona, J.A.; Hernandez, J.A. HPLC method for quantification of caffeine and its three major metabolites in human plasma using fetal bovine serum matrix to evaluate prenatal drug exposure. J. Anal. Methods Chem. 2018, 2018, 2085059. [Google Scholar] [CrossRef] [Green Version]
- Girard, H.; Simon, N.; Ballutaud, D.; Herlem, M.; Etcheberry, A. Effect of anodic and cathodic treatments on the charge transfer of boron doped diamond electrodes. Diam. Relat. Mater. 2007, 16, 316–325. [Google Scholar] [CrossRef]
- Suffredini, H.B.; Pedrosa, V.A.; Codognoto, L.; Machado, S.A.S.; Rocha-Filho, R.C.; Avaca, L.A. Enhanced electrochemical response of boron-doped diamond electrodes brought on by a cathodic surface pre-treatment. Electrochim. Acta 2004, 49, 4021–4026. [Google Scholar] [CrossRef]
- Iyyappan, E.; Samuel Justin, S.J.; Wilson, P.; Palaniappan, A. Nanoscale hydroxyapatite for electrochemical sensing of uric acid: Roles of mesopore volume and surface acidity. ACS Appl. Nano Mater. 2020, 3, 7761–7773. [Google Scholar] [CrossRef]
- Du, J.; Yue, R.; Yao, Z.; Jiang, F.; Du, Y.; Yang, P.; Wang, C. Nonenzymatic uric acid electrochemical sensor based on graphene-modified carbon fiber electrode. Colloids Surf. A Physicochem. Eng. Asp. 2013, 419, 94–99. [Google Scholar] [CrossRef]
- Wang, Y.; Tong, L.-L. Electrochemical sensor for simultaneous determination of uric acid, xanthine and hypoxanthine based on poly (bromocresol purple) modified glassy carbon electrode. Sens. Actuators B 2010, 150, 43–49. [Google Scholar] [CrossRef]
- Zhang, X.; Zhang, Y.-C.; Ma, L.-X. One-pot facile fabrication of graphene-zinc oxide composite and its enhanced sensitivity for simultaneous electrochemical detection of ascorbic acid, dopamine and uric acid. Sens. Actuators B 2016, 227, 488–496. [Google Scholar] [CrossRef]
- Geto, A.; Brett, C.M.A. Electrochemical sensor for caffeine in coffee and beverages using a naphthalene sulfonic acid polymer film–based modified electrode. Food Anal. Methods 2021, 14, 2386–2394. [Google Scholar] [CrossRef]
- Brunetti, B.; Desimoni, E.; Casati, P. Determination of caffeine at a nafion-covered glassy carbon electrode. Electroanalysis 2007, 19, 385–388. [Google Scholar] [CrossRef]
- Muzvidziwa, T.; Moyo, M.; Okonkwo, J.O.; Shumba, M.; Nharingo, T.; Guyo, U. Electrodeposition of zinc oxide nanoparticles on multiwalled carbon nanotube-modified electrode for determination of caffeine in wastewater effluent. Int. J. Environ. Anal. Chem. 2017, 97, 623–636. [Google Scholar] [CrossRef]
- Tyszczuk-Rotkoz, K.; Szwagierek, A. Green electrochemical sensor for caffeine determination in environmental water samples: The bismuth film screen-printed carbon electrode. J. Electrochem. Soc. 2017, 164, B342–B348. [Google Scholar] [CrossRef]
- Raj, M.A.; John, S.A.; John, S.A. Simultaneous determination of uric acid, xanthine, hypoxanthine and caffeine in human blood serum and urine samples using electrochemically reduced graphene oxide modified electrode. Anal. Chim. Acta 2013, 771, 14–20. [Google Scholar] [CrossRef] [PubMed]
- Sarakhman, O.; Benkova, A.; Svorc, Ľ. A modern and powerful electrochemical sensing platform for purines determination: Voltammetric determination of uric acid and caffeine in biological samples on miniaturized thick-film boron-doped diamond electrode. Microchem. J. 2022, 175, 107132. [Google Scholar] [CrossRef]
- Silva, W.C.; Pereira, P.F.; Marra, M.C.; Gimenes, D.T.; Cunha, R.R.; da Silva, R.A.B.; Munoz, R.A.A.; Richter, E.M. A simple strategy for simultaneous determination of paracetamol and caffeine using flow injection analysis with multiple pulse amperometric detection. Electroanalysis 2011, 23, 2764–2770. [Google Scholar] [CrossRef]
- Ramos, D.L.O.; Ribeiro, M.M.A.C.; Munoz, R.A.A.; Richter, E.M. Portable amperometric method for selective determination of caffeine in samples with the presence of interfering electroactive chemical species. J. Electroanal. Chem. 2022, 906, 116006. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Electrode | Analyte | Technique | Linear Range (µmol L−1) | LOD (µmol L−1) | Reference |
|---|---|---|---|---|---|
| n-HA/CPE a | UA | DPV | 0.068–50 | 0.05 | [27] |
| Graphene-modified carbon fiber | UA | Amperometry | 0.194–49.68 | 0.132 | [28] |
| Poly(bromocresol purple)/GCE | UA | DPV | 0.5–120 | 0.2 | [29] |
| RGO−ZnO/GCE b | UA | DPV | 1.0–70.0 | 0.33 | [30] |
| Poly(7A4HN2SA)/GCE c | CAF | DPV | 10–500 | 0.23 | [31] |
| Nafion/GCE | CAF | DPV | 0.99–10.6 | 0.79 | [32] |
| ZnO/MWCNT/GCE d | CAF | DPV | 0.02–24.9 | 0.01 | [33] |
| BiF-SPCE/CNFs e | CAF | DPAdSV | 0.20–1.0 | 0.05 | [34] |
| FN/rGO-200/GCE f | UA and CAF | DPV | 4.0–21.5 | 1.3 and 1.6 | [35] |
| BDD | UA and CAF | SWV | 9.2–95.0 and 4.6–95.7 | 6.0 and 3.9 | [36] |
| BDD | PCT and CAF | FIA-MPA | (0.050–1.3) × 103 and (0.051–1.3) × 102 | 0.66 and 0.87 | [37] |
| BDD | CAF | BIA-MPA | 10–1445 | 0.26 | [38] |
| CPT-BDD | UA and CAF | FIA-MPA | 0.050–22 and 0.050–19 | 0.011 and 0.013 | This work |
| Samples | Analytes | Added/mol L−1 | Comparative Method/mol L−1 | Proposed Method/mol L−1 | Recovery ** (Sensor, %) | Error *** % |
|---|---|---|---|---|---|---|
| Found * | Found * | |||||
| River water | UA | 8.0 × 10−7 | (8.2 ± 0.1) × 10−7 | (8.3 ± 0.1) × 10−7 | 104 | +1.2 |
| 8.0 × 10−6 | (8.0 ± 0.1) × 10−6 | (7.8 ± 0.1) × 10−6 | 98 | −2.5 | ||
| CAF | 8.0 × 10−7 | (8.1 ± 0.1) × 10−7 | (8.2 ± 0.1) × 10−7 | 102 | +1.2 | |
| 8.0 × 10−6 | (8.0 ± 0.2) × 10−6 | (8.0 ± 0.1) × 10−6 | 100 | 0 | ||
| Synthetic Urine | UA | 8.0 × 10−7 | (8.1 ± 0.1) × 10−7 | (8.0 ± 0.1) × 10−7 | 100 | −1.2 |
| 8.0 × 10−6 | (8.1 ± 0.2) × 10−6 | (8.2 ± 0.1) × 10−6 | 102 | +1.2 | ||
| CAF | 8.0 × 10−7 | (8.2 ± 0.2) × 10−7 | (8.3 ± 0.4) × 10−7 | 104 | +1.2 | |
| 8.0 × 10−6 | (7.9 ± 0.2) × 10−6 | (7.9 ± 0.3) × 10−6 | 99 | 0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wong, A.; Santos, A.M.; Feitosa, M.H.A.; Fatibello-Filho, O.; Moraes, F.C.; Sotomayor, M.D.P.T. Simultaneous Determination of Uric Acid and Caffeine by Flow Injection Using Multiple-Pulse Amperometry. Biosensors 2023, 13, 690. https://doi.org/10.3390/bios13070690
Wong A, Santos AM, Feitosa MHA, Fatibello-Filho O, Moraes FC, Sotomayor MDPT. Simultaneous Determination of Uric Acid and Caffeine by Flow Injection Using Multiple-Pulse Amperometry. Biosensors. 2023; 13(7):690. https://doi.org/10.3390/bios13070690
Chicago/Turabian StyleWong, Ademar, Anderson M. Santos, Maria H. A. Feitosa, Orlando Fatibello-Filho, Fernando C. Moraes, and Maria D. P. T. Sotomayor. 2023. "Simultaneous Determination of Uric Acid and Caffeine by Flow Injection Using Multiple-Pulse Amperometry" Biosensors 13, no. 7: 690. https://doi.org/10.3390/bios13070690
APA StyleWong, A., Santos, A. M., Feitosa, M. H. A., Fatibello-Filho, O., Moraes, F. C., & Sotomayor, M. D. P. T. (2023). Simultaneous Determination of Uric Acid and Caffeine by Flow Injection Using Multiple-Pulse Amperometry. Biosensors, 13(7), 690. https://doi.org/10.3390/bios13070690