
Effect of Post–Polyketide Synthase Modification Groups on Property and Activity of Polyene Macrolides

Abstract
:1. Introduction
2. Modification of the Carboxyl Group in the Side Chain of Lactone
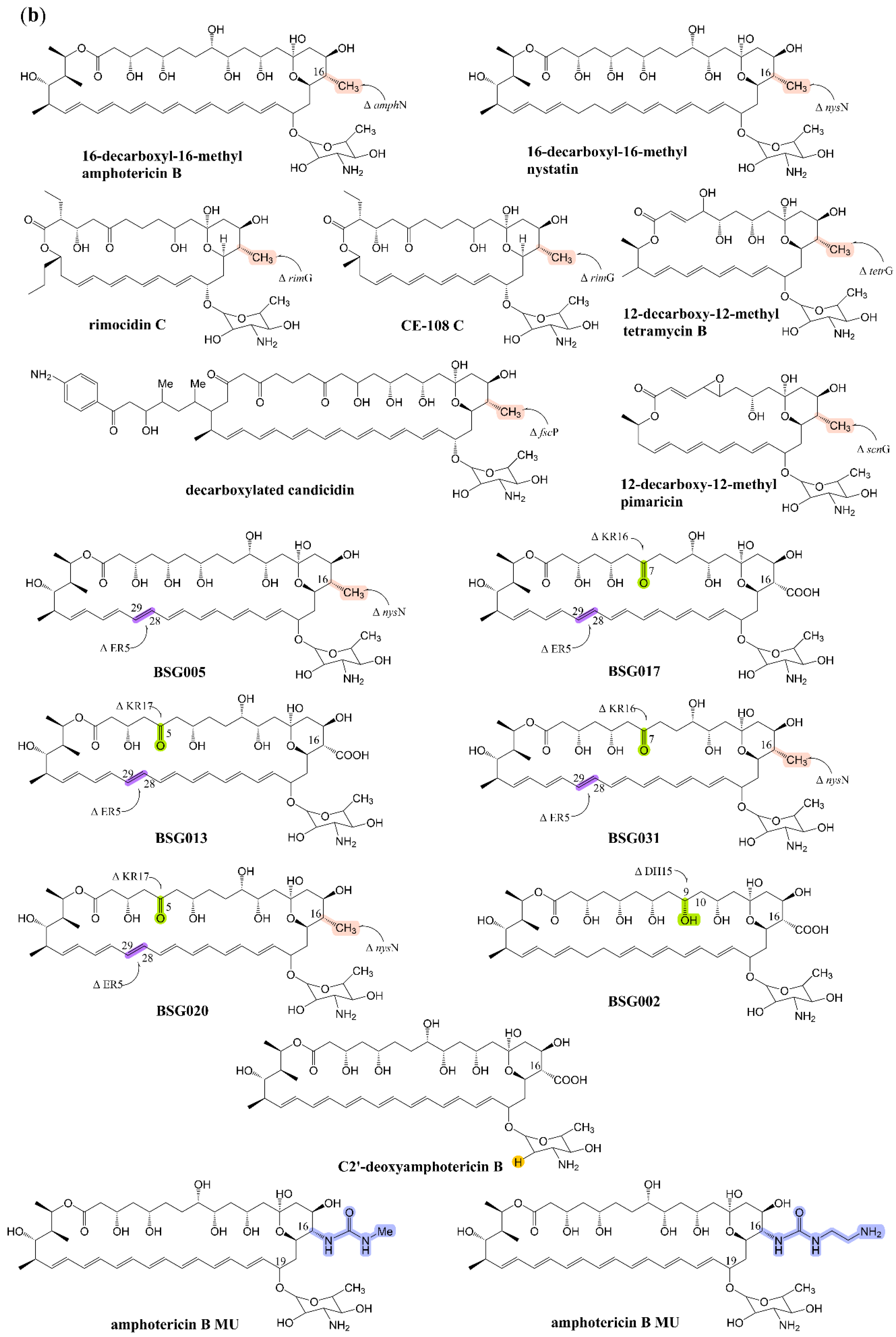
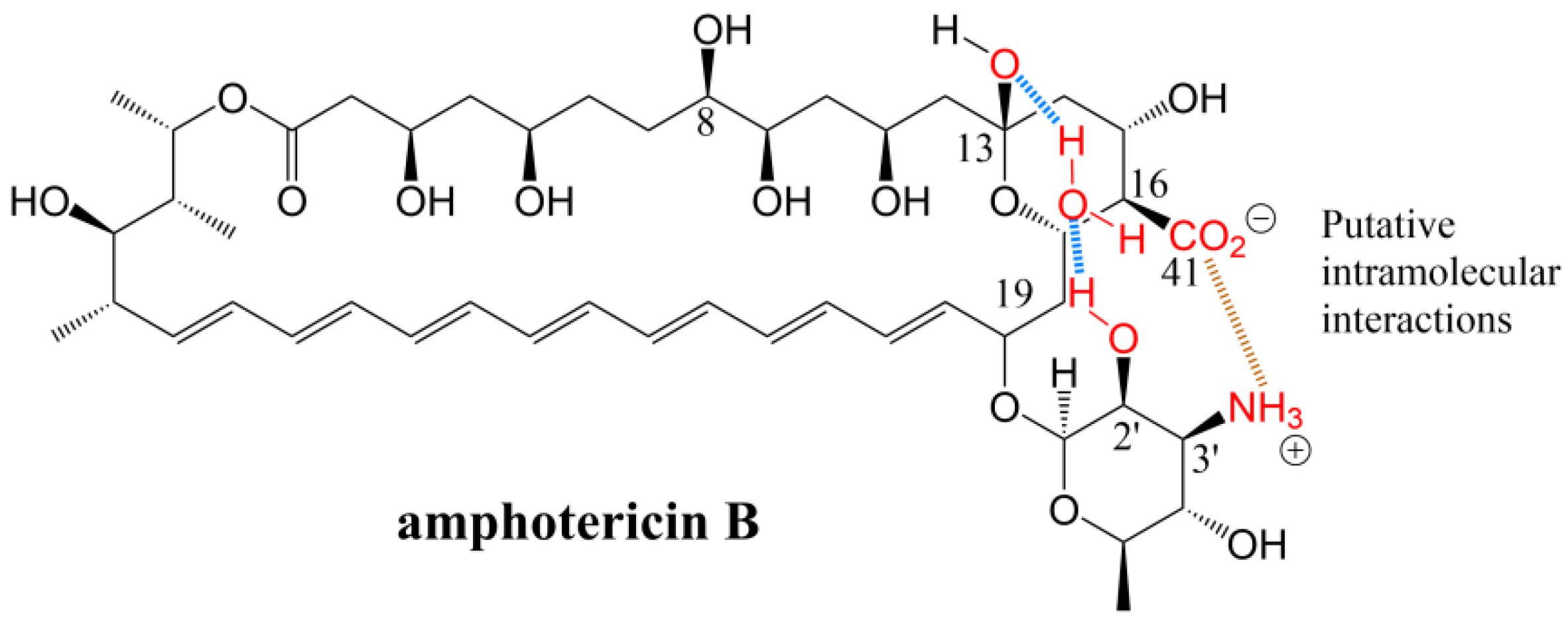
2.1. Effect of Side Chain Carboxyl on Antifungal Activity and Toxicity
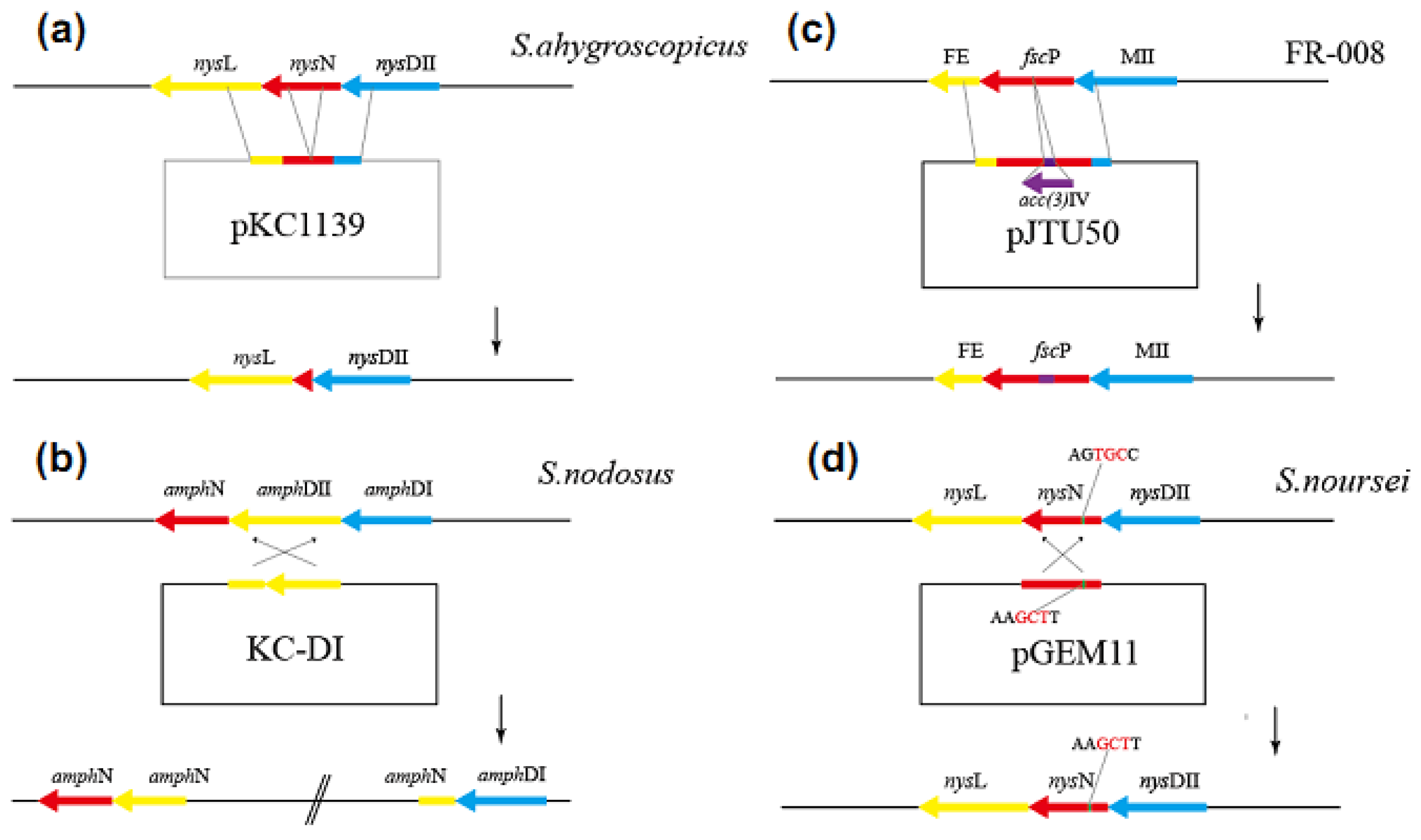
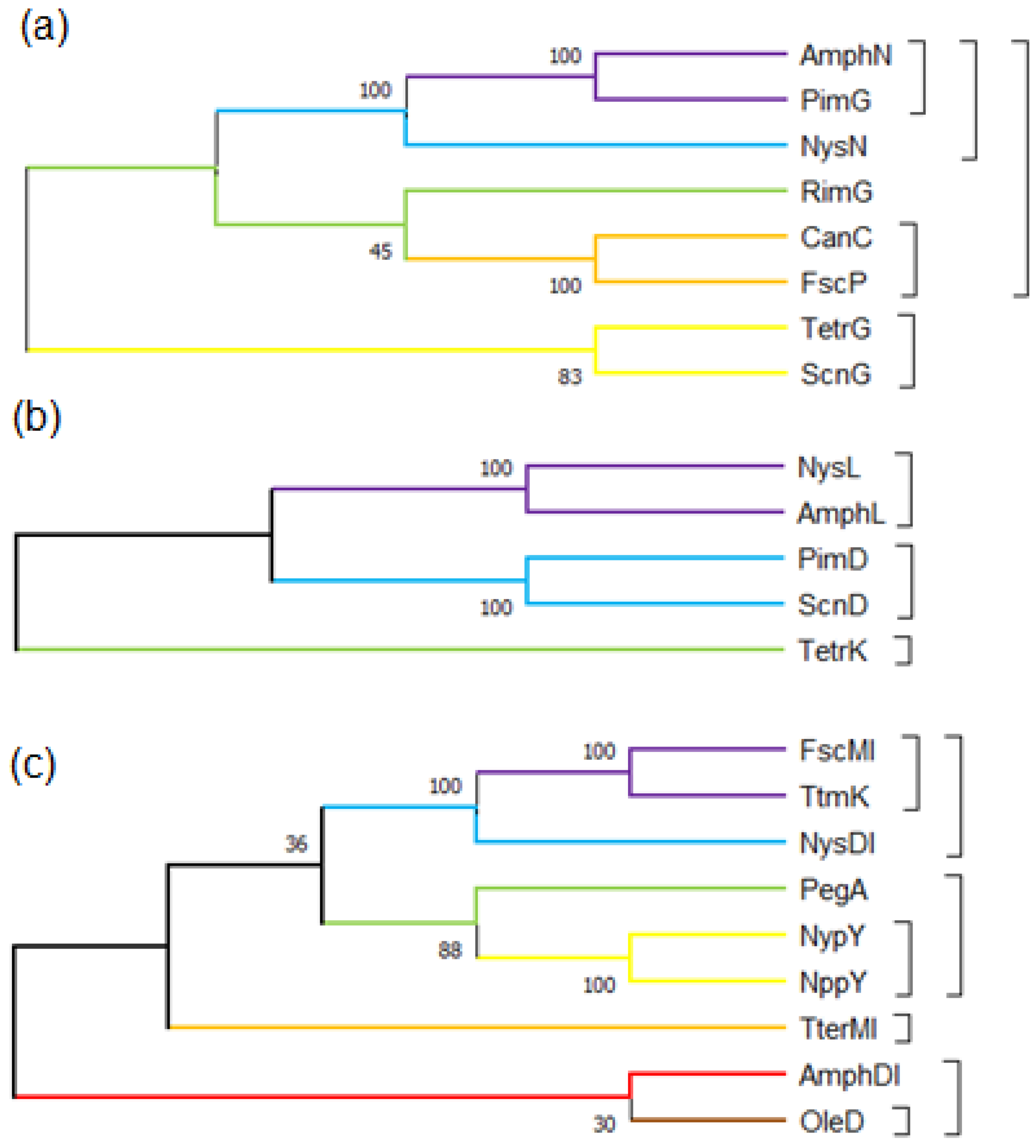
2.2. Genetic Engineering Methods for the Decarboxylation of Polyene Macrolides
3. Research Progress on Side Chain Glycosyl
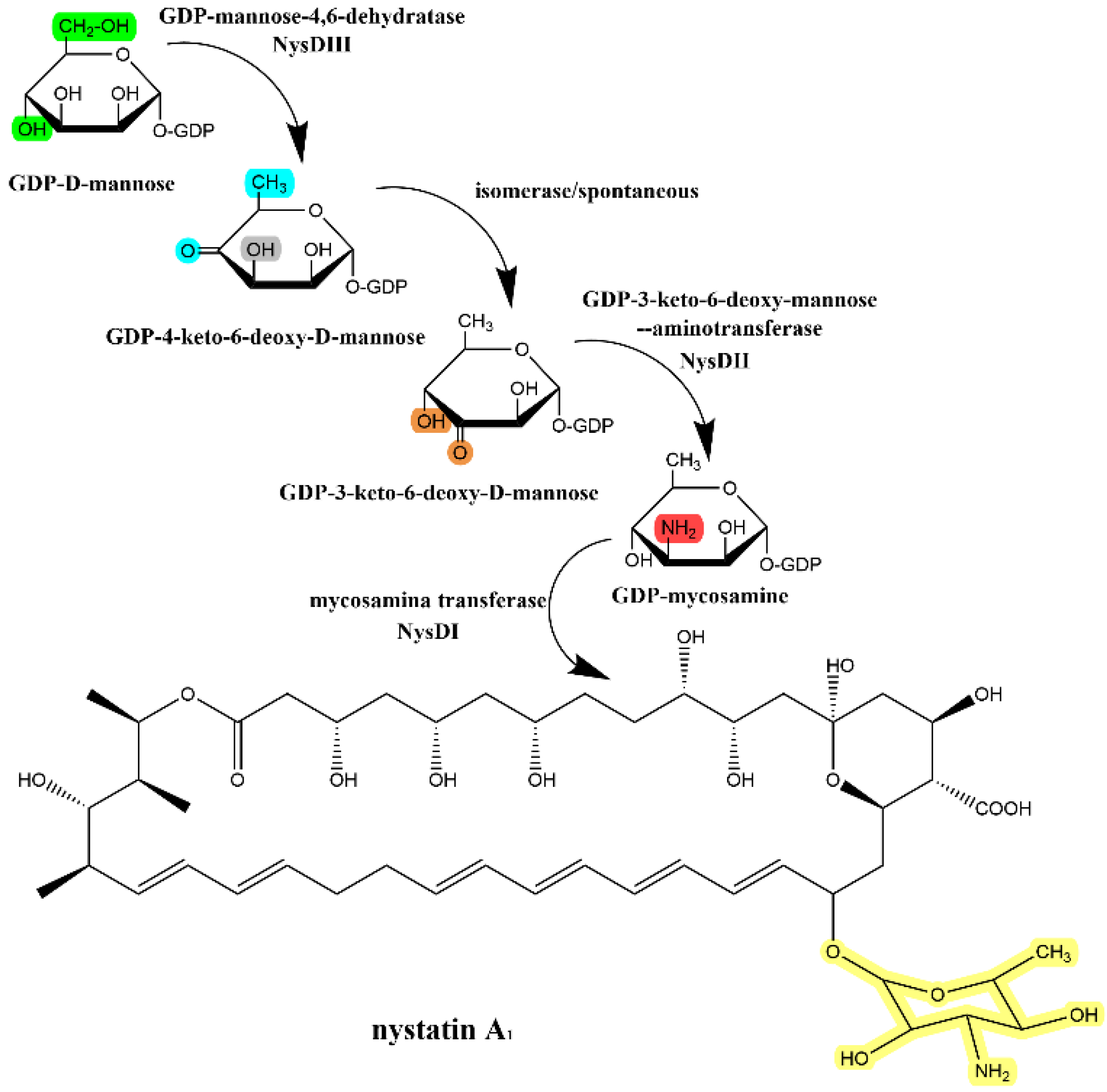
3.1. Glycosylation Mechanism of Polyene Macrolides Antibiotics
3.2. Structure–Activity Relationship between Glycosyl and Polyene Macrolides
3.3. Research Progress on the Second Glycosyl of Polyene Antibiotics
4. Research Progress on Epoxidation or Hydroxylation of Macrolactone Ring
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Omura, S. Production, Structure, and Antifungal Activity of Polyene Macrolides. In Macrolide Antibiotics; Academic Press: Cambridge, MA, USA, 1984; pp. 351–404. [Google Scholar]
- Abu-Salah, K.M. Amphotericin B: An update. Br. J. Biomed. Sci 1996, 53, 122–133. [Google Scholar] [PubMed]
- Spanakis, E.K.; Aperis, G.; Mylonakis, E. New agents for the treatment of fungal infections: Clinical efficacy and gaps in coverage. Clin. Infect. Dis. 2006, 43, 1060–1068. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sekurova, O.N.; Brautaset, T.; Sletta, H.; Borgos, S.E.; Jakobsen, M.O.; Ellingsen, T.E.; Strom, A.R.; Valla, S.; Zotchev, S.B. In vivo analysis of the regulatory genes in the nystatin biosynthetic gene cluster of Streptomyces noursei ATCC 11455 reveals their differential control over antibiotic biosynthesis. J. Bacteriol. 2004, 186, 1345–1354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gagos, M.; Gabrielska, J.; Dalla Serra, M.; Gruszecki, W.I. Binding of antibiotic amphotericin B to lipid membranes: Monomolecular layer technique and linear dichroism-FTIR studies. Mol. Membr. Biol. 2005, 22, 433–442. [Google Scholar] [CrossRef]
- Gagoś, M.; Kamiński, D.; Arczewska, M.; Krajnik, B.; Maćkowski, S. Spectroscopic evidence for self-organization of N-iodoacetylamphotericin B in crystalline and amorphous phases. J. Phys. Chem. B 2012, 116, 12706–12713. [Google Scholar] [CrossRef] [PubMed]
- Brautaset, T.; Sekurova, O.N.; Sletta, H.; Ellingsen, T.E.; Strøm, A.R.; Valla, S.; Zotchev, S.B. Biosynthesis of the polyene antifungal antibiotic nystatin in Streptomyces noursei ATCC 11455: Analysis of the gene cluster and deduction of the biosynthetic pathway. Chem. Biol. 2000, 7, 395–403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fjaervik, E.; Zotchev, S.B. Biosynthesis of the polyene macrolide antibiotic nystatin in Streptomyces noursei. Appl. Microbiol. Biotechnol. 2005, 67, 436–443. [Google Scholar] [CrossRef]
- Caffrey, P.; Lynch, S.; Flood, E.; Finnan, S.; Oliynyk, M. Amphotericin biosynthesis in Streptomyces nodosus. Chem. Biol. 2003, 10, 93–94. [Google Scholar] [CrossRef] [Green Version]
- Lee, M.Y.; Myeong, J.S.; Park, H.J.; Han, K.; Kim, E.S. Isolation and partial characterization of a cryptic polyene gene cluster in Pseudonocardia autotrophica. J. Ind. Microbiol. Biotechnol. 2006, 33, 84–87. [Google Scholar] [CrossRef]
- Cao, B.; Yao, F.; Zheng, X.; Cui, D.; Shao, Y.; Zhu, C.; Deng, Z.; You, D. Genome mining of the biosynthetic gene cluster of the polyene macrolide antibiotic tetramycin and characterization of a P450 monooxygenase involved in the hydroxylation of the tetramycin B polyol segment. ChemBioChem 2012, 13, 2234–2242. [Google Scholar] [CrossRef]
- Aparicio, J.F.; Colina, A.J.; Ceballos, E.; Martín, J.F. The biosynthetic gene cluster for the 26-membered ring polyene macrolide pimaricin: A new polyketide synthase organization encoded by two subclusters separated by functionalization genes. J. Biol. Chem. 1999, 274, 10133–10139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, Y.L.; Chen, S.F.; Cheng, L.Y.; Shen, X.L.; Tian, Y.; Li, Y.Q. Identification of a novel Streptomyces chattanoogensis L10 and enhancing its natamycin production by overexpressing positive regulator ScnRII. Microbiology 2009, 47, 506–513. [Google Scholar] [CrossRef] [PubMed]
- Campelo, A.B.; Gil, J.A. The candicidin gene cluster from Streptomyces griseus IMRU 3570. Microbiology 2002, 148, 51–59. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.; Huang, X.; Zhou, X.; Bai, L.; He, J.; Jeong, K.J.; Lee, S.Y.; Deng, Z. Organizational and mutational analysis of a complete FR-008/candicidin gene cluster encoding a structurally related polyene complex. Chem. Biol. 2003, 10, 1065–1076. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seco, E.M.; Perez-Zuniga, F.J.; Rolon, M.S.; Malpartida, F. Starter unit choice determines the production of two tetraene macrolides, rimocidin and CE-108, in Streptomyces diastaticus var. 108. Chem. Biol. 2004, 11, 357–366. [Google Scholar] [CrossRef] [Green Version]
- Payero, T.D.; Vicente, C.M.; Rumbero, Á.; Barreales, E.G.; Santos-Aberturas, J.; de Pedro, A.; Aparicio, J.F. Functional analysis of filipin tailoring genes from Streptomyces filipinensis reveals alternative routes in filipin III biosynthesis and yields bioactive derivatives. Microb. Cell Factories 2015, 14, 114. [Google Scholar] [CrossRef] [Green Version]
- Wei, J.; Meng, X.; Wang, Q. Enhanced production of aureofuscin by over-expression of AURJ3M, positive regulator of aureofuscin biosynthesis in Streptomyces aureofuscus. Lett. Appl. Microbiol. 2011, 52, 322–329. [Google Scholar] [CrossRef]
- Martin, J.F.; McDaniel, L.E. Production of polyene macrolide antibiotics. Adv. Appl. Microbiol. 1977, 21, 1–52. [Google Scholar] [CrossRef]
- Birch, A.; Holzapfel, C.; Rickards, R.; Djerassi, C.; Suzuki, M.; Westley, J.; Dutcher, J.; Thomas, R. Nystatin. part V. biosynthetic definition of some structural features. Tetrahedron Lett. 1964, 5, 1485–1490. [Google Scholar] [CrossRef]
- Aparicio, J.F.; Caffrey, P.; Gil, J.A.; Zotchev, S.B. Polyene antibiotic biosynthesis gene clusters. Appl. Microbiol. Biotechnol. 2003, 61, 179–188. [Google Scholar] [CrossRef]
- Carmody, M.; Murphy, B.; Byrne, B.; Power, P.; Rai, D.; Rawlings, B.; Caffrey, P. Biosynthesis of amphotericin derivatives lacking exocyclic carboxyl groups. Biol. Chem. 2005, 280, 34420–34426. [Google Scholar] [CrossRef] [Green Version]
- Brautaset, T.; Sletta, H.; Nedal, A.; Borgos, S.E.; Degnes, K.F.; Bakke, I.; Volokhan, O.; Sekurova, O.N.; Treshalin, I.D.; Mirchink, E.P.; et al. Improved antifungal polyene macrolides via engineering of the nystatin biosynthetic genes in Streptomyces noursei. Chem. Biol. 2008, 15, 1198–1206. [Google Scholar] [CrossRef] [Green Version]
- Seco, E.M.; Fotso, S.; Laatsch, H.; Malpartida, F. A tailoring activity is responsible for generating polyene amide derivatives in Streptomyces diastaticus var. 108. Chem. Biol. 2005, 12, 1093–1101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, S.; Mao, X.; Shen, Y.; Zhou, Y.; Li, J.; Wang, L.; Tao, X.; Yang, L.; Wang, Y.; Zhou, X.; et al. Tailoring the P450 monooxygenase gene for FR-008/candicidin biosynthesis. Appl. Environ. Microbiol. 2009, 75, 1778–1781. [Google Scholar] [CrossRef] [Green Version]
- Qi, Z.; Kang, Q.; Jiang, C.; Han, M.; Bai, L. Engineered biosynthesis of pimaricin derivatives with improved antifungal activity and reduced cytotoxicity. Appl. Microbiol. Biotechnol. 2015, 99, 6745–6752. [Google Scholar] [CrossRef]
- Sheng, Y.; Ou, Y.; Hu, X.; Deng, Z.; Bai, L.; Kang, Q. Generation of tetramycin B derivative with improved pharmacological property based on pathway engineering. Appl. Microbiol. Biotechnol. 2020, 104, 2561–2573. [Google Scholar] [CrossRef]
- Walmsley, S.; De Poire, E.; Rawlings, B.; Caffrey, P. Engineered biosynthesis and characterisation of disaccharide-modified 8-deoxyamphoteronolides. Appl. Microbiol. Biotechnol. 2017, 101, 1899–1905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palacios, D.S.; Dailey, I.; Siebert, D.M.; Wilcock, B.C.; Burke, M.D. Synthesis-enabled functional group deletions reveal key underpinnings of amphotericin B ion channel and antifungal activities. Proc. Natl. Acad. Sci. USA 2011, 108, 6733–6738. [Google Scholar] [CrossRef] [Green Version]
- Wilcock, B.C.; Endo, M.M.; Uno, B.E.; Burke, M.D. C2′-OH of amphotericin B plays an important role in binding the primary sterol of human cells but not yeast cells. J. Am. Chem. Soc. 2013, 135, 8488–8491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davis, S.A.; Vincent, B.M.; Endo, M.M.; Whitesell, L.; Marchillo, K.; Andes, D.R.; Lindquist, S.; Burke, M.D. Nontoxic antimicrobials that evade drug resistance. Nat. Chem. Biol. 2015, 11, 481–487. [Google Scholar] [CrossRef]
- Caffrey, P.; Lynch, S.; Flood, E.; Finnan, S.; Oliynyk, M. Amphotericin biosynthesis in Streptomyces nodosus: Deductions from analysis of polyketide synthase and late genes. Chem. Biol. 2001, 8, 713–723. [Google Scholar] [CrossRef] [Green Version]
- Byrne, B.; Carmody, M.; Gibson, E.; Rawlings, B.; Caffrey, P. Biosynthesis of deoxyamphotericins and deoxyamphoteronolides by engineered strains of Streptomyces nodosus. Chem. Biol. 2003, 10, 1215–1224. [Google Scholar] [CrossRef] [Green Version]
- Nic Lochlainn, L.; Caffrey, P. Phosphomannose isomerase and phosphomannomutase gene disruptions in Streptomyces nodosus: Impact on amphotericin biosynthesis and implications for glycosylation engineering. Metab. Eng. 2009, 11, 40–47. [Google Scholar] [CrossRef] [Green Version]
- Nedal, A.; Sletta, H.; Brautaset, T.; Borgos, S.E.; Sekurova, O.N.; Ellingsen, T.E.; Zotchev, S.B. Analysis of the mycosamine biosynthesis and attachment genes in the nystatin biosynthetic gene cluster of Streptomyces noursei ATCC 11455. Appl. Environ. Microbiol. 2007, 73, 7400–7407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lei, X.; Kong, L.; Zhang, C.; Liu, Q.; Yao, F.; Zhang, W.; Deng, Z.; You, D. In vivo investigation of the substrate recognition capability and activity affecting amino acid residues of glycosyltransferase FscMI in the biosynthesis of candicidin. Mol. Biosyst. 2013, 9, 422–430. [Google Scholar] [CrossRef]
- Baginski, M.; Resat, H.; McCammon, J.A. Molecular properties of amphotericin B membrane channel: A molecular dynamics simulation. Mol. Pharmacol. 1997, 52, 560–570. [Google Scholar] [CrossRef] [Green Version]
- Matsumori, N.; Sawada, Y.; Murata, M. Mycosamine orientation of amphotericin B controlling interaction with ergosterol: Sterol-dependent activity of conformation-restricted derivatives with an amino-carbonyl bridge. J. Am. Chem. Soc. 2005, 127, 10667–10675. [Google Scholar] [CrossRef] [PubMed]
- Gray, K.C.; Palacios, D.S.; Dailey, I.; Endo, M.M.; Uno, B.E.; Wilcock, B.C.; Burke, M.D. Amphotericin primarily kills yeast by simply binding ergosterol. J. Am. Chem. Soc. 2012, 109, 2234–2239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Welscher, Y.M.; Jones, L.; van Leeuwen, M.R.; Dijksterhuis, J.; de Kruijff, B.; Eitzen, G.; Breukink, E. Natamycin inhibits vacuole fusion at the priming phase via a specific interaction with ergosterol. Antimicrob. Agents Chemother. 2010, 54, 2618–2625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anderson, T.M.; Clay, M.C.; Cioffi, A.G.; Diaz, K.A.; Hisao, G.S.; Tuttle, M.D.; Nieuwkoop, A.J.; Comellas, G.; Maryum, N.; Wang, S.; et al. Amphotericin forms an extramembranous and fungicidal sterol sponge. Nat. Chem. Biol. 2014, 10, 400–406. [Google Scholar] [CrossRef] [PubMed]
- Palacios, D.S.; Anderson, T.M.; Burke, M.D. A post-PKS oxidation of the amphotericin B skeleton predicted to be critical for channel formation is not required for potent antifungal activity. J. Am. Chem. Soc. 2007, 129, 13804–13805. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ganis, P.; Avitabile, G.; Mechlinski, W.; Schaffner, C.P. Polyene macrolide antibiotic amphotericin B. Crystal structure of the N-iodoacetyl derivative. J. Am. Chem. Soc. 1971, 93, 4560–4564. [Google Scholar] [CrossRef] [PubMed]
- Duggan, K.C.; Hermanson, D.J.; Musee, J.; Prusakiewicz, J.J.; Scheib, J.L.; Carter, B.D.; Banerjee, S.; Oates, J.; Marnett, L.J. (R)-Profens are substrate-selective inhibitors of endocannabinoid oxygenation by COX-2. Nat. Chem. Biol. 2011, 7, 803–809. [Google Scholar] [CrossRef] [Green Version]
- Kim, B.G.; Lee, M.J.; Seo, J.; Hwang, Y.B.; Lee, M.Y.; Han, K.; Sherman, D.H.; Kim, E.S. Identification of functionally clustered nystatin-like biosynthetic genes in a rare actinomycetes, Pseudonocardia autotrophica. J. Ind. Microbiol. Biotechnol. 2009, 36, 1425–1434. [Google Scholar] [CrossRef]
- Lee, M.J.; Kong, D.; Han, K.; Sherman, D.H.; Bai, L.; Deng, Z.; Lin, S.; Kim, E.S. Structural analysis and biosynthetic engineering of a solubility-improved and less-hemolytic nystatin-like polyene in Pseudonocardia autotrophica. Appl. Microbiol. Biotechnol. 2012, 95, 157–168. [Google Scholar] [CrossRef]
- Kim, H.J.; Kim, M.K.; Lee, M.J.; Won, H.J.; Choi, S.S.; Kim, E.S. Post-PKS tailoring steps of a disaccharide-containing polyene NPP in Pseudonocardia autotrophica. PLoS ONE 2015, 10, e0123270. [Google Scholar] [CrossRef] [PubMed]
- Stephens, N.; Rawlings, B.; Caffrey, P. Versatility of enzymes catalyzing late steps in polyene 67-121C biosynthesis. Biosci. Biotechnol. Biochem. 2013, 77, 880–883. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barke, J.; Seipke, R.F.; Grüschow, S.; Heavens, D.; Drou, N.; Bibb, M.J.; Goss, R.J.; Yu, D.W.; Hutchings, M.I. A mixed community of actinomycetes produce multiple antibiotics for the fungus farming ant Acromyrmex octospinosus. BMC Biol. 2010, 8, 109. [Google Scholar] [CrossRef] [Green Version]
- De Poire, E.; Stephens, N.; Rawlings, B.; Caffrey, P. Engineered biosynthesis of disaccharide-modified polyene macrolides. Appl. Environ. Microbiol. 2013, 79, 6156–6159. [Google Scholar] [CrossRef] [Green Version]
- Aparicio, J.F.; Mendes, M.V.; Antón, N.; Recio, E.; Martín, J. Polyene macrolide antiobiotic biosynthesis. Curr. Med. Chem. 2004, 11, 1643–1656. [Google Scholar] [CrossRef]
- Aparicio, J.F.; Fouces, R.; Mendes, M.V.; Olivera, N.; Martín, J.F. A complex multienzyme system encoded by five polyketide synthase genes is involved in the biosynthesis of the 26-membered polyene macrolide pimaricin in Streptomyces natalensis. Chem. Biol. 2000, 7, 895–905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mendes, M.V.; Recio, E.; Fouces, R.; Luiten, R.; Martín, J.F.; Aparicio, J.F. Engineered biosynthesis of novel polyenes: A pimaricin derivative produced by targeted gene disruption in Streptomyces natalensis. J. Chem. Biol. 2001, 8, 635–644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Volokhan, O.; Sletta, H.; Ellingsen, T.E.; Zotchev, S.B. Characterization of the P450 monooxygenase NysL, responsible for C-10 hydroxylation during biosynthesis of the polyene macrolide antibiotic nystatin in Streptomyces noursei. Appl. Environ. Microbiol. 2006, 72, 2514–2519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santos-Aberturas, J.; Engel, J.; Dickerhoff, J.; Dörr, M.; Rudroff, F.; Weisz, K.; Bornscheuer, U.T. Exploration of the substrate promiscuity of biosynthetic tailoring enzymes as a new source of structural diversity for polyene macrolide antifungals. ChemCatChem 2015, 7, 490–500. [Google Scholar] [CrossRef]
- Yamamoto, T.; Umegawa, Y.; Tsuchikawa, H.; Matsumori, N.; Hanashima, S.; Murata, M.; Haser, R.; Rawlings, B.J.; Caffrey, P. Role of polyol moiety of amphotericin B in ion channel formation and sterol selectivity in bilayer membrane. Bioorg. Med. Chem. 2015, 23, 5782–5788. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Antifungal Activity | Test Strain | Hemolytic Activity | ||
|---|---|---|---|---|---|
| amphotericin B | MIC | 1.25 μg/mL | Candida. albicans | MHC | 5 μg/mL |
| 16-descarboxyl-16-methyl amphotericin B | MIC | 1 μg/mL | C. albicans | MHC | 50 μg/mL |
| nystatin | MIC50 MIC90 | 1.2 ± 0.2 μg/mL 2.0 ± 0.3 μg/mL | C. albicans ATCC 14053 | HC50 | 85 μg/mL |
| 16-descarboxyl-16-methyl nystatin | MIC50 MIC90 | 1.3 ± 0.4 μg/mL 1.8 ± 0.5 μg/mL | C. albicans ATCC 14053 | HC50 | 175 μg/mL |
| S44HP | MIC50 MIC90 | 0.12 ± 0.03 μg/mL 0.20 ± 0.03 μg/mL | C. albicans ATCC 14053 | HC50 | 2.5 μg/mL |
| BSG005 | MIC50 MIC90 | 0.07 ± 0.02 μg/mL 0.20 ± 0.03 μg/mL | C. albicans ATCC 14053 | HC50 | 4.0 μg/mL |
| BSG013 | MIC50 MIC90 | 0.25 ± 0.07 μg/mL 0.43 ± 0.07 μg/mL | C. albicans ATCC 14053 | HC50 | 3.0 μg/mL |
| BSG020 | MIC50 MIC90 | 0.15 ± 0.03 μg/mL 0.19 ± 0.03 μg/mL | C. albicans ATCC 14053 | HC50 | 9.0 μg/mL |
| BSG017 | MIC50 MIC90 | 0.47 ± 0.15 μg/mL 0.92 ± 0.03 μg/mL | C. albicans ATCC 14053 | HC50 | 3.3 μg/mL |
| BSG031 | MIC50 MIC90 | 0.18 ± 0.06 μg/mL 0.37 ± 0.07 μg/mL | C. albicans ATCC 14053 | HC50 | 3.8 μg/mL |
| rimocidin A | Same antifungal activity, data not shown | ||||
| rimocidin C | Same antifungal activity, data not shown | 2.5–5 times that of rimocidin | |||
| CE-108 | Same antifungal activity, data not shown | ||||
| CE-108 C | Same antifungal activity, data not shown | ||||
| candicidin | MIC | 0.00039–0.00078 μg/mL | Saccharomyces cerevisiae Y029 | ||
| decarboxylated candicidin | MIC | 0.00312–0.00625 μg/mL | S. cerevisiae Y029 | 50 times that of candicidin | |
| pimaricin | MIC50 MIC90 | 0.51 ± 0.01 μg/mL 0.77 ± 0.02 μg/mL | C. albicans ATCC 14053 | HC50 | 114.0 ± 1.68 μg/mL |
| 12-decarboxy-12-methyl pimaricin | MIC50 MIC90 | 1.09 ± 0.02 μg/mL 1.61 ± 0.04 μg/mL | C. albicans ATCC 14053 | HC50 | 478.4 ± 8.58 μg/mL |
| tetramycin B | MIC50 MIC90 | 2.74 ± 0.19 μg/mL 6.01 ± 0.04 μg/mL | S. cerevisiae S288C | ||
| 12-decarboxy-12-methyl tetramycin B | MIC50 MIC90 | 1.89 ± 0.11 μg/mL 3.11 ± 0.02 μg/mL | S. cerevisiae S288C | ||
| tetramycin B | MIC50 MIC90 | 3.95 ± 0.21 μg/mL 8.38 ± 0.03 μg/mL | Rhodotorula glutinis CGMCC2.4238 | HC50 | 98.4 ± 6.97 μg/mL |
| 12-decarboxy-12-methyl tetramycin B | MIC50 MIC90 | 2.26 ± 0.14 μg/mL 5.51 ± 0.02 μg/mL | R. glutinis CGMCC2.4238 | HC50 | 167.0 ± 4.20 μg/mL |
| amphotericin B | MIC50 | 1.44 μg/mL | C. albicans ATCC10231 | MHC0 MHC50 MHC100 | 0.58 μg/mL 1.46 μg/mL 2.92 μg/mL |
| mannosyl-8-deoxyamphotericin B | MIC50 | 1.46 μg/mL | C. albicans ATCC10231 | MHC0 MHC50 MHC100 | 0.94 μg/mL 2.35 μg/mL 7.43 μg/mL |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Qiao, L.; Dong, Y.; Zhou, H.; Cui, H. Effect of Post–Polyketide Synthase Modification Groups on Property and Activity of Polyene Macrolides. Antibiotics 2023, 12, 119. https://doi.org/10.3390/antibiotics12010119
Qiao L, Dong Y, Zhou H, Cui H. Effect of Post–Polyketide Synthase Modification Groups on Property and Activity of Polyene Macrolides. Antibiotics. 2023; 12(1):119. https://doi.org/10.3390/antibiotics12010119
Chicago/Turabian StyleQiao, Liqin, Yao Dong, Hongli Zhou, and Hao Cui. 2023. "Effect of Post–Polyketide Synthase Modification Groups on Property and Activity of Polyene Macrolides" Antibiotics 12, no. 1: 119. https://doi.org/10.3390/antibiotics12010119
APA StyleQiao, L., Dong, Y., Zhou, H., & Cui, H. (2023). Effect of Post–Polyketide Synthase Modification Groups on Property and Activity of Polyene Macrolides. Antibiotics, 12(1), 119. https://doi.org/10.3390/antibiotics12010119