
Targeting N-Acetylglucosaminidase in Staphylococcus aureus with Iminosugar Inhibitors
 , ,
, ,  ,
,  and
and
Abstract
:1. Introduction
2. Results and Discussion
2.1. Iminosugar Dataset
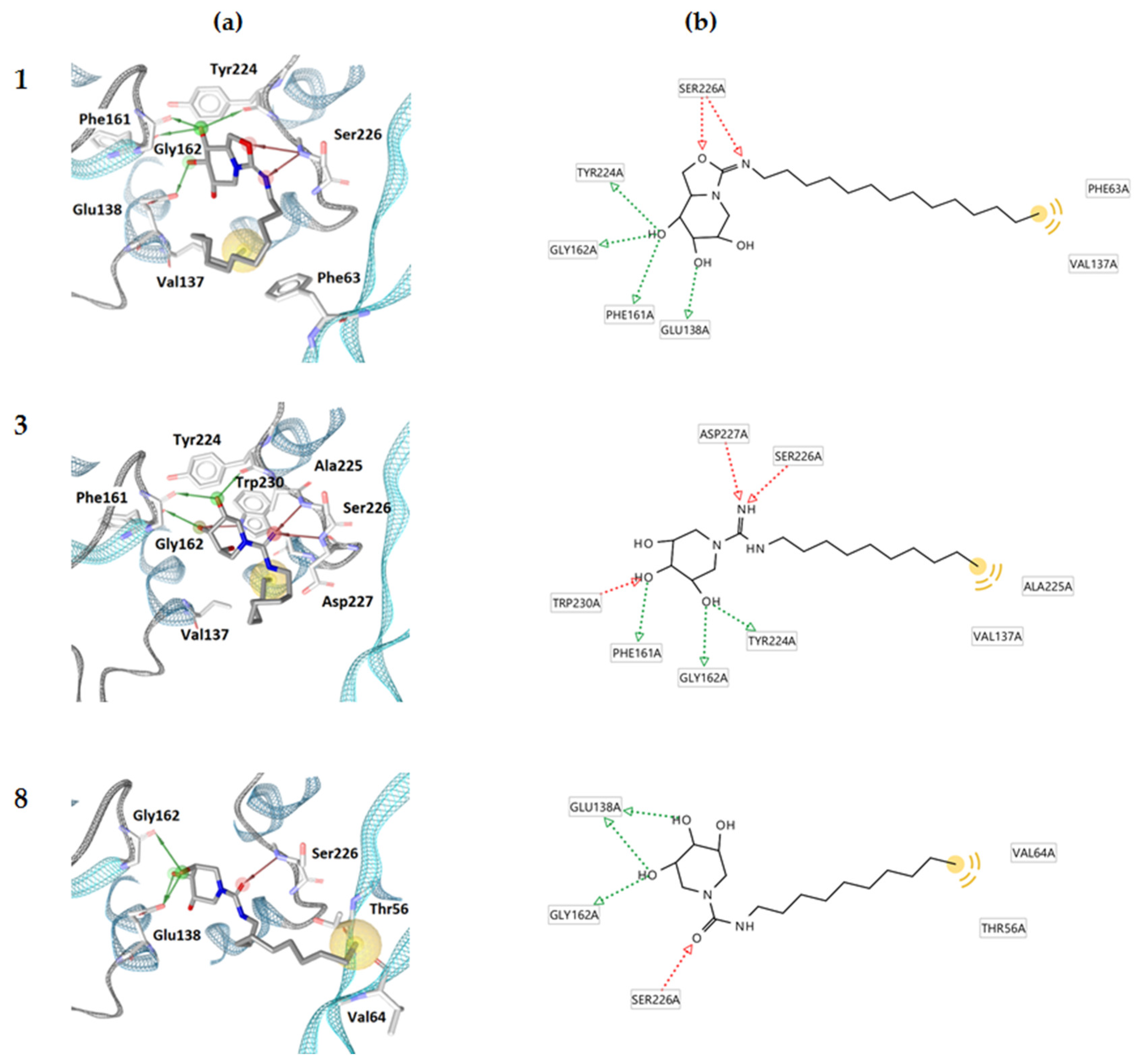
2.2. Molecular Docking Calculations
2.3. In Silico Physicochemical and Toxicity Assessment
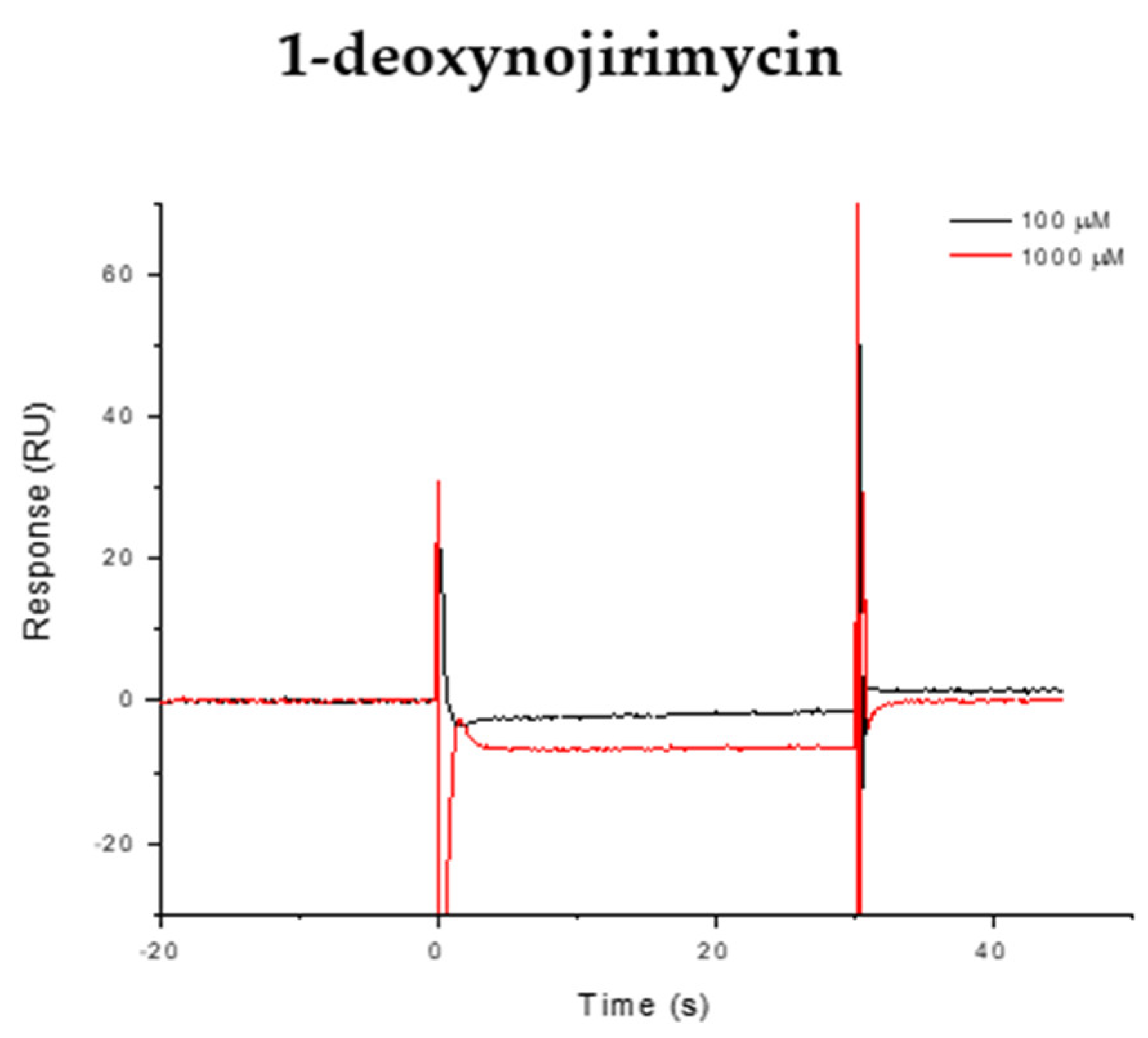
2.4. Surface Plasmon Resonance (SPR) Analysis
2.5. Minimal Inhibitory Concentration (MIC)
3. Materials and Methods
3.1. Dataset
3.2. Molecular Docking Calculations
3.3. In Silico Physicochemical and Toxicity Assessment
3.4. Surface Plasmon Resonance (SPR) Analysis
3.5. Minimal Inhibitory Concentration (MIC)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ikuta, K.S.; Swetschinski, L.R.; Aguilar, G.R.; Sharara, F.; Mestrovic, T.; Gray, A.P.; Weaver, N.D.; Wool, E.E.; Han, C.; Hayoon, A.G.; et al. Global Mortality Associated with 33 Bacterial Pathogens in 2019: A Systematic Analysis for the Global Burden of Disease Study 2019. Lancet 2022, 400, 2221–2248. [Google Scholar] [CrossRef] [PubMed]
- Mancuso, G.; Midiri, A.; Gerace, E.; Biondo, C. Bacterial Antibiotic Resistance: The Most Critical Pathogens. Pathogens 2021, 10, 1310. [Google Scholar] [CrossRef] [PubMed]
- Xia, J.; Gao, J.; Kokudo, N.; Hasegawa, K.; Tang, W. Methicillin-Resistant Staphylococcus aureus Antibiotic Resistance and Virulence. Biosci. Trends 2013, 7, 113–121. [Google Scholar] [PubMed]
- Archer, G.L. Staphylococcus aureus: A Well–Armed Pathogen. Clin. Infect. Dis. 1998, 26, 1179–1181. [Google Scholar] [CrossRef] [PubMed]
- Tong, S.Y.C.; Davis, J.S.; Eichenberger, E.; Holland, T.L.; Fowler, V.G. Staphylococcus aureus Infections: Epidemiology, Pathophysiology, Clinical Manifestations, and Management. Clin. Microbiol. Rev. 2015, 28, 603–661. [Google Scholar] [CrossRef] [PubMed]
- Culp, E.J.; Waglechner, N.; Wang, W.; Fiebig-Comyn, A.A.; Hsu, Y.-P.; Koteva, K.; Sychantha, D.; Coombes, B.K.; Van Nieuwenhze, M.S.; Brun, Y.V.; et al. Evolution-Guided Discovery of Antibiotics That Inhibit Peptidoglycan Remodelling. Nature 2020, 578, 582–587. [Google Scholar] [CrossRef] [PubMed]
- Tinajero-Trejo, M.; Carnell, O.; Kabli, A.F.; Pasquina-Lemonche, L.; Lafage, L.; Han, A.; Hobbs, J.K.; Foster, S.J. The Staphylococcus aureus Cell Division Protein, DivIC, Interacts with the Cell Wall and Controls Its Biosynthesis. Commun. Biol. 2022, 5, 1228. [Google Scholar] [CrossRef] [PubMed]
- Saber, A.M.; Aghamollaei, H.; Esmaeili Gouvarchin Ghaleh, H.; Mohammadi, M.; Yaghoob Sehri, S.; Farnoosh, G. Design and Production of a Chimeric enzyme with Efficient Antibacterial Properties on Staphylococcus Aureus. Int. J. Pept. Res. Ther. 2024, 30, 7. [Google Scholar] [CrossRef]
- Krishnan, M.; Tham, H.Y.; Wan Nur Ismah, W.A.K.; Yusoff, K.; Song, A.A.L. Effect of Domain Manipulation in the Staphylococcal Phage Endolysin, Endo88, on Lytic Efficiency and Host Range. Mol. Biotechnol. 2024, 1–9. [Google Scholar] [CrossRef]
- Yuan, B.; Lu, X.; Yang, M.; He, Q.; Cha, Z.; Fang, Y.; Yang, Y.; Xu, L.; Yan, J.; Lai, R.; et al. A Designed Antimicrobial Peptide with Potential Ability against Methicillin Resistant Staphylococcus aureus. Front. Microbiol. 2022, 13, 1029366. [Google Scholar] [CrossRef] [PubMed]
- Tan, Z.; Yang, W.; O’Brien, N.A.; Pan, X.; Ramadan, S.; Marsh, T.; Hammer, N.; Cywes-Bentley, C.; Vinacur, M.; Pier, G.B.; et al. A Comprehensive Synthetic Library of Poly-N-Acetyl Glucosamines Enabled Vaccine against Lethal Challenges of Staphylococcus aureus. Nat. Commun. 2024, 15, 3420. [Google Scholar] [CrossRef]
- Mihelič, M.; Vlahoviček-Kahlina, K.; Renko, M.; Mesnage, S.; Doberšek, A.; Taler-Verčič, A.; Jakas, A.; Turk, D. The Mechanism behind the Selection of Two Different Cleavage Sites in NAG-NAM Polymers. IUCrJ 2017, 4, 185–198. [Google Scholar] [CrossRef] [PubMed]
- Bose, J.L.; Lehman, M.K.; Fey, P.D.; Bayles, K.W. Contribution of the Staphylococcus aureus Atl AM and GL Murein Hydrolase Activities in Cell Division, Autolysis, and Biofilm Formation. PLoS ONE 2012, 7, e42244. [Google Scholar] [CrossRef] [PubMed]
- Foster, S.J. Molecular Characterization and Functional Analysis of the Major Autolysin of Staphylococcus aureus 8325/4. J. Bacteriol. 1995, 177, 5723–5725. [Google Scholar] [CrossRef] [PubMed]
- Wheeler, R.; Turner, R.D.; Bailey, R.G.; Salamaga, B.; Mesnage, S.; Mohamad, S.A.S.; Hayhurst, E.J.; Horsburgh, M.; Hobbs, J.K.; Foster, S.J. Bacterial Cell Enlargement Requires Control of Cell Wall Stiffness Mediated by Peptidoglycan Hydrolases. mBio 2015, 6, e00660-15. [Google Scholar] [CrossRef] [PubMed]
- van Heijenoort, J. Peptidoglycan Hydrolases of Escherichia coli. Microbiol. Mol. Biol. Rev. 2011, 75, 636–663. [Google Scholar] [CrossRef] [PubMed]
- Vollmer, W.; Blanot, D.; De Pedro, M.A. Peptidoglycan Structure and Architecture. FEMS Microbiol. Rev. 2008, 32, 149–167. [Google Scholar] [CrossRef] [PubMed]
- Reith, J.; Mayer, C. Peptidoglycan Turnover and Recycling in Gram-Positive Bacteria. Appl. Microbiol. Biotechnol. 2011, 92, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Davies, G.; Henrissat, B. Structures and mechanisms of glycosyl hydrolases. Structure 1995, 3, 853–859. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, H.; Gutelius, D.; Black, E.; Nadolny, C.; Basu, A.; Reid, C. Anti-Bacterial Glycosyl Triazoles-Identification of an N-Acetylglucosamine Derivative with Bacteriostatic Activity against Bacillus. Medchemcomm 2014, 5, 1213–1217. [Google Scholar] [CrossRef]
- Szweda, P.; Schielmann, M.; Kotlowski, R.; Gorczyca, G.; Zalewska, M.; Milewski, S. Peptidoglycan Hydrolases-Potential Weapons against Staphylococcus aureus. Appl. Microbiol. Biotechnol. 2012, 96, 1157–1174. [Google Scholar] [CrossRef] [PubMed]
- Borišek, J.; Pintar, S.; Ogrizek, M.; Turk, D.; Perdih, A.; Novič, M. A Water-Assisted Catalytic Mechanism in Glycoside Hydrolases Demonstrated on the Staphylococcus aureus Autolysin E. ACS Catal. 2018, 8, 4334–4345. [Google Scholar] [CrossRef]
- Pintar, S.; Borišek, J.; Usenik, A.; Perdih, A.; Turk, D. Domain Sliding of Two Staphylococcus aureus N-Acetylglucosaminidases Enables Their Substrate-Binding Prior to Its Catalysis. Commun. Biol. 2020, 3, 178. [Google Scholar] [CrossRef] [PubMed]
- Leonard, A.C.; Goncheva, M.I.; Gilbert, S.E.; Shareefdeen, H.; Petrie, L.E.; Thompson, L.K.; Khursigara, C.M.; Heinrichs, D.E.; Cox, G. Autolysin-Mediated Peptidoglycan Hydrolysis Is Required for the Surface Display of Staphylococcus aureus Cell Wall-Anchored Proteins. Proc. Natl. Acad. Sci. USA 2023, 120, e2301414120. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Ma, S.X.; John, A.S.; Torres, V.J. The Major Autolysin Atl Regulates the Virulence of Staphylococcus aureus by Controlling the Sorting of LukAB. Infect. Immun. 2022, 90, e0005622. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, S.J.; Verma, D.; Griswold, K.E.; Bailey-Kellogg, C. Building Blocks and Blueprints for Bacterial Autolysins. PLoS Comput. Biol. 2021, 17, e1008889. [Google Scholar] [CrossRef] [PubMed]
- Tibaut, T.; Drgan, V.; Novič, M. Application of SAR Methods toward Inhibition of Bacterial Peptidoglycan Metabolizing enzymes. J. Chemom. 2018, 32, e3007. [Google Scholar] [CrossRef]
- Tibaut, T.; Tomašič, T.; Hodnik, V.; Anderluh, M.; Pintar, S.; Novič, M.; Turk, D. Application of Fragment Based Virtual Screening towards Inhibition of Bacterial N-Acetyglucosaminidase$. SAR QSAR Environ. Res. 2018, 29, 647–660. [Google Scholar] [CrossRef] [PubMed]
- Borišek, J.; Pintar, S.; Ogrizek, M.; Grdadolnik, S.G.; Hodnik, V.; Turk, D.; Perdih, A.; Novič, M. Discovery of (Phenylureido)Piperidinyl Benzamides as Prospective Inhibitors of Bacterial Autolysin E. from Staphylococcus aureus. J. Enzym. Inhib. Med. Chem. 2018, 33, 1239–1247. [Google Scholar] [CrossRef]
- Sevšek, A.; Čelan, M.; Erjavec, B.; Quarles Van Ufford, L.; Sastre Toraño, J.; Moret, E.E.; Pieters, R.J.; Martin, N.I. Bicyclic Isoureas Derived from 1-Deoxynojirimycin Are Potent Inhibitors of β-Glucocerebrosidase. Org. Biomol. Chem. 2016, 14, 8670–8673. [Google Scholar] [CrossRef] [PubMed]
- Sevšek, A.; Šrot, L.; Rihter, J.; Čelan, M.; van Ufford, L.Q.; Moret, E.E.; Martin, N.I.; Pieters, R.J. N-Guanidino Derivatives of 1,5-Dideoxy-1,5-Imino-d-Xylitol Are Potent, Selective, and Stable Inhibitors of β-Glucocerebrosidase. ChemMedChem 2017, 12, 483–486. [Google Scholar] [CrossRef] [PubMed]
- Sevšek, A.; Sastre Toraño, J.; Quarles Van Ufford, L.; Moret, E.E.; Pieters, R.J.; Martin, N.I. Orthoester Functionalized: N-Guanidino Derivatives of 1,5-Dideoxy-1,5-Imino-d-Xylitol as PH-Responsive Inhibitors of β-Glucocerebrosidase. Medchemcomm 2017, 8, 2050–2054. [Google Scholar] [CrossRef] [PubMed]
- Arévalo, N.B.; Lamaizon, C.M.; Cavieres, V.A.; Burgos, P.V.; Álvarez, A.R.; Yañez, M.J.; Zanlungo, S. Neuronopathic Gaucher Disease: Beyond Lysosomal Dysfunction. Front. Mol. Neurosci. 2022, 15, 934820. [Google Scholar] [CrossRef] [PubMed]
- Compain, P.; Martin, O.R. Iminosugars: From Synthesis to Therapeutic Applications; John Wiley & Sons, Ltd.: Chichester, UK, 2007; pp. 1–486. ISBN 9780470517437. [Google Scholar]
- Wolber, G.; Langer, T. LigandScout: 3-D Pharmacophores Derived from Protein-Bound Ligands and Their Use as Virtual Screening Filters. J. Chem. Inf. Model. 2005, 45, 160–169. [Google Scholar] [CrossRef] [PubMed]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A Free Web Tool to Evaluate Pharmacokinetics, Drug-Likeness and Medicinal Chemistry Friendliness of Small Molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [PubMed]
- Sluga, J.; Venko, K.; Drgan, V.; Novič, M. QSPR Models for Prediction of Aqueous Solubility: Exploring the Potency of Randic-Type Indices. Croat. Chem. Acta 2020, 93, 311–319. [Google Scholar] [CrossRef]
- Jones, G.; Willett, P.; Glen, R.C.; Leach, A.R.; Taylor, R. Development and Validation of a Genetic Algorithm for Flexible Docking. J. Mol. Biol. 1997, 267, 727–748. [Google Scholar] [CrossRef] [PubMed]
- Verdonk, M.L.; Cole, J.C.; Hartshorn, M.J.; Murray, C.W.; Taylor, R.D. Improved Protein-Ligand Docking Using GOLD. Proteins Struct. Funct. Genet. 2003, 52, 609–623. [Google Scholar] [CrossRef] [PubMed]
- Kirchmair, J.; Markt, P.; Distinto, S.; Wolber, G.; Langer, T. Evaluation of the Performance of 3D Virtual Screening Protocols: RMSD Comparisons, Enrichment Assessments, and Decoy Selection—What Can We Learn from Earlier Mistakes? J. Comput. Aided Mol. Des. 2008, 22, 213–228. [Google Scholar] [CrossRef] [PubMed]
- Benfenati, E.; Manganaro, A.; Gini, G. VEGA-QSAR: AI inside a Platform for Predictive Toxicology. PAI@AI*IA 2013, 1107, 21–28. [Google Scholar]
- Martin, T. User’s Guide for T. E. S. T. (Toxicity Estimation Software Tool) Version 5.1 A Java Application to Estimate Toxicities and Physical Properties from Molecular Structure, U.S. Environmental Protection Agency. 2020. Available online: https://www.epa.gov/chemical-research/toxicity-estimation-software-tool-test (accessed on 15 January 2024).
- Pires, D.E.V.; Blundell, T.L.; Ascher, D.B. PkCSM: Predicting Small-Molecule Pharmacokinetic and Toxicity Properties Using Graph-Based Signatures. J. Med. Chem. 2015, 58, 4066–4072. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Lou, C.; Sun, L.; Li, J.; Cai, Y.; Wang, Z.; Li, W.; Liu, G.; Tang, Y. AdmetSAR 2.0: Web-Service for Prediction and Optimization of Chemical ADMET Properties. Bioinformatics 2019, 35, 1067–1069. [Google Scholar] [CrossRef] [PubMed]
- Cheng, F.; Li, W.; Zhou, Y.; Shen, J.; Wu, Z.; Liu, G.; Lee, P.W.; Tang, Y. AdmetSAR: A Comprehensive Source and Free Tool for Assessment of Chemical ADMET Properties. J. Chem. Inf. Model. 2012, 52, 3099–3105. [Google Scholar] [CrossRef] [PubMed]
- Montanari, F.; Knasmüller, B.; Kohlbacher, S.; Hillisch, C.; Baierová, C.; Grandits, M.; Ecker, G.F. Vienna LiverTox Workspace—A Set of Machine Learning Models for Prediction of Interactions Profiles of Small Molecules With Transporters Relevant for Regulatory Agencies. Front. Chem. 2020, 7, 899. [Google Scholar] [CrossRef] [PubMed]
- Kolšek, K.; Mavri, J.; Sollner Dolenc, M.; Gobec, S.; Turk, S. Endocrine Disruptome—An Open Source Prediction Tool for Assessing Endocrine Disruption Potential through Nuclear Receptor Binding. J. Chem. Inf. Model. 2014, 54, 1254–1267. [Google Scholar] [CrossRef] [PubMed]
- CLSI Document M07-A9; Clinical Laboratory Standards Institute Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria that Grow Aerobically, Approved Standard—Eleven Edition. Clinical and Laboratory Standards Institute: Wayne, PA, USA, 2018.
- EUCAST Testing Breakpoint Tables for Interpretation of MICs and Zone Diameters. Available online: https://www.eucast.org/mic_and_zone_distributions_and_ecoffs/new_and_revised_ecoffs (accessed on 1 March 2024).



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ID | Structure | X | Goldscore Fitness | KD (µM) |
|---|---|---|---|---|
| 1 (14 d [30]) | 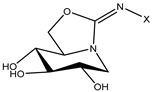 | C14 | 52.1 | 19 |
| 2 (14 e [30]) | CH2CON(C10)2 | 51.1 | LR | |
| 3 (14 b [31]) | 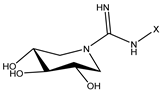 | C10 | 44.5 | 410 |
| 4 (14 c [31]) | C12 | 51.3 | LR | |
| 5 (14 d [31]) | C14 | 42.4 | LR | |
| 6 (14 e [31]) | CH2CON(C10)2 | 52.8 | LR | |
| 7 (17 a [31]) | 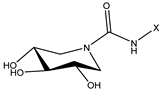 | C8 | 38.7 | >1000 |
| 8 (17 b [31]) | C10 | 46.7 | 88 | |
| 9 (17 c [31]) | C12 | 46.7 | >1000 | |
| 10 (17 d [31]) | C14 | 47.5 | >1000 | |
| 11 (10 [32]) | 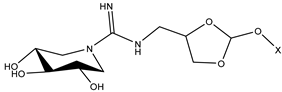 | C4 | 44.1 | >1000 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sluga, J.; Tomašič, T.; Anderluh, M.; Rambaher, M.H.; Bajc, G.; Sevšek, A.; Martin, N.I.; Pieters, R.J.; Novič, M.; Venko, K. Targeting N-Acetylglucosaminidase in Staphylococcus aureus with Iminosugar Inhibitors. Antibiotics 2024, 13, 751. https://doi.org/10.3390/antibiotics13080751
Sluga J, Tomašič T, Anderluh M, Rambaher MH, Bajc G, Sevšek A, Martin NI, Pieters RJ, Novič M, Venko K. Targeting N-Acetylglucosaminidase in Staphylococcus aureus with Iminosugar Inhibitors. Antibiotics. 2024; 13(8):751. https://doi.org/10.3390/antibiotics13080751
Chicago/Turabian StyleSluga, Janja, Tihomir Tomašič, Marko Anderluh, Martina Hrast Rambaher, Gregor Bajc, Alen Sevšek, Nathaniel I. Martin, Roland J. Pieters, Marjana Novič, and Katja Venko. 2024. "Targeting N-Acetylglucosaminidase in Staphylococcus aureus with Iminosugar Inhibitors" Antibiotics 13, no. 8: 751. https://doi.org/10.3390/antibiotics13080751