


A Comprehensive Overview of Antibacterial Agents for Combating Multidrug-Resistant Bacteria: The Current Landscape, Development, Future Opportunities, and Challenges
 , , , , , ,
, , , , , ,
Abstract
:1. Introduction
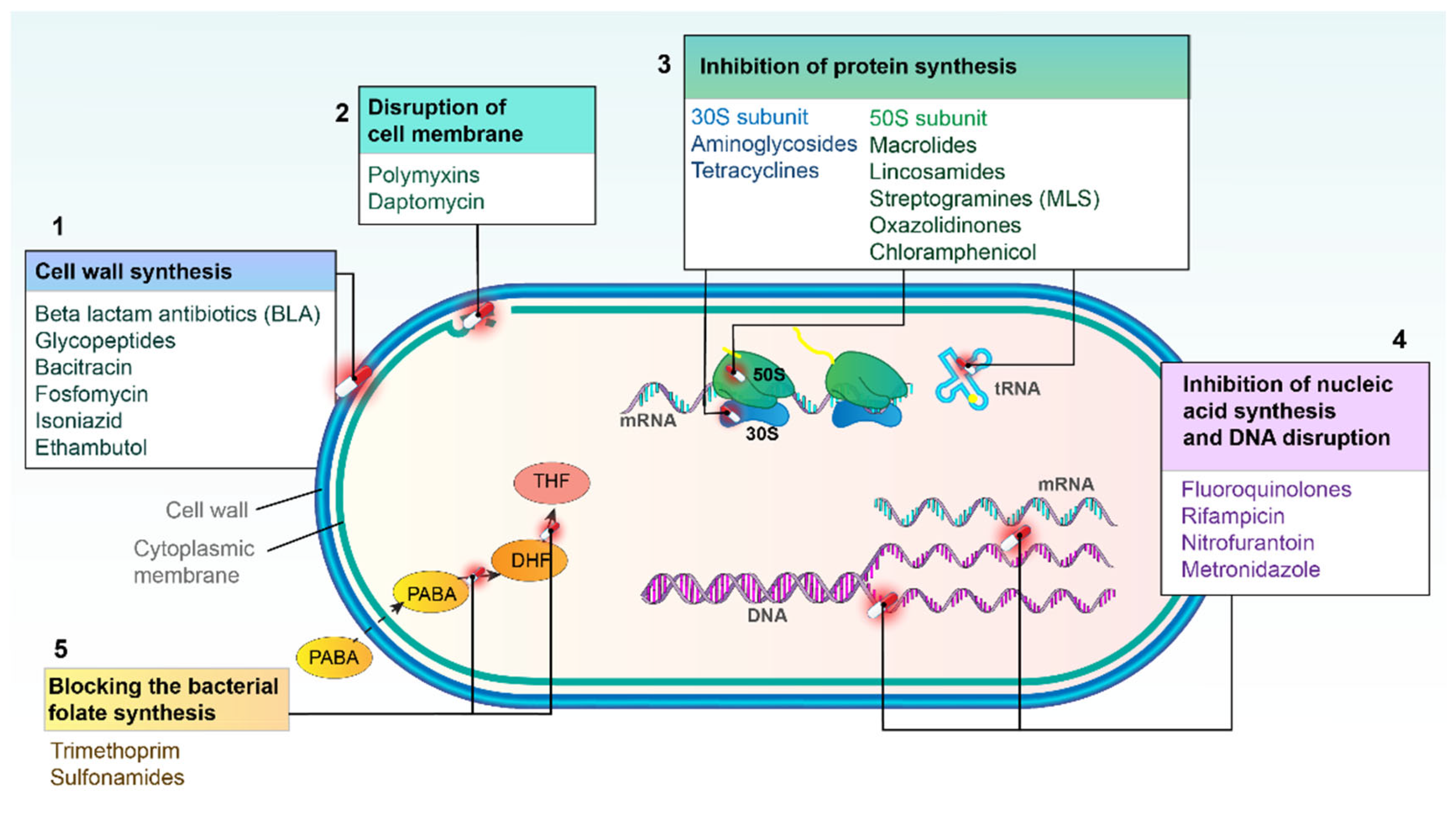
2. Major Mechanisms of Action of Antibacterial Agents
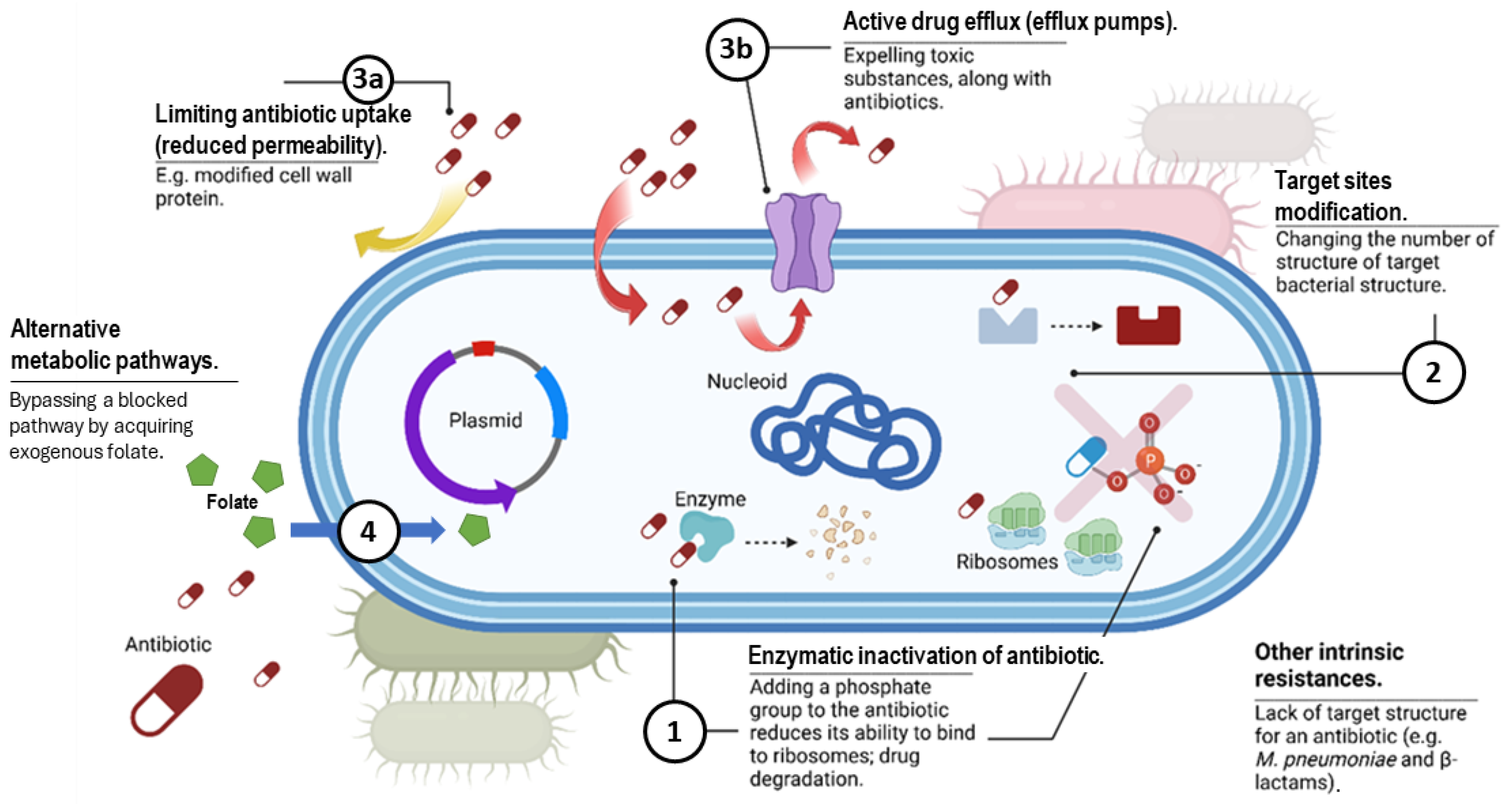
3. Overview of Antibiotic Resistance Mechanisms
3.1. Production of Enzymes That Deactivate the Drug
3.1.1. Beta-Lactamases
3.1.2. Enzymes That Alter Antibiotics
3.2. Modification of the Target Site of the Drug
3.3. Changes in Antibiotic Transport, Including Decreased Permeability and Increased Efflux
3.4. Alteration of Metabolic Pathways
4. Antibiotic Resistance in Key Gram-Negative Bacteria and Available Treatment Options
4.1. Carbapenem-Resistant Acinetobacter baumannii
4.2. Carbapenem-Resistant Pseudomonas aeruginosa
4.3. Third-Generation Cephalosporin-Resistant Enterobacterales
4.4. Carbapenem-Resistant Enterobacterales
4.5. Fluoroquinolone-Resistant Salmonella Typhi
4.6. Fluoroquinolone-Resistant Non-Typhoidal Salmonella
4.7. Fluoroquinolone-Resistant Shigella spp.
4.8. Resistance in Neisseria Gonorrhoeae
4.9. Resistance in Helicobacter pylori
5. Antibiotic Resistance in Important Gram-Positive Cocci and Available Treatment Options
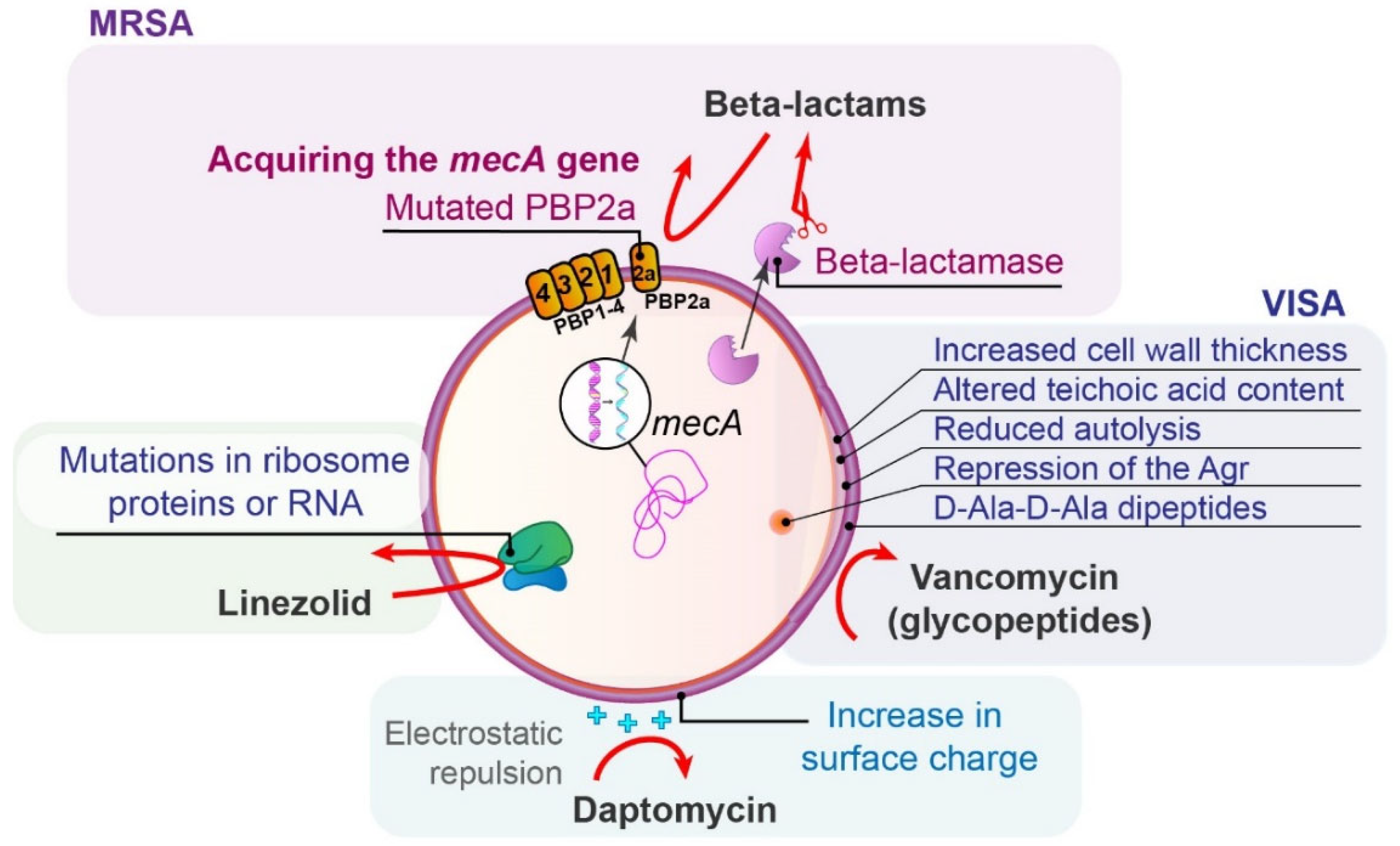
5.1. Methicillin-Resistant Staphylococcus aureus
5.2. Vancomycin-Resistant Enterococci
5.3. Macrolide-Resistant Streptococcus pneumoniae
5.4. Macrolide-Resistant Group A Streptococci
5.5. Penicillin-Resistant Group B Streptococci
5.6. Ampicillin-Resistant Haemophilus Influenzae
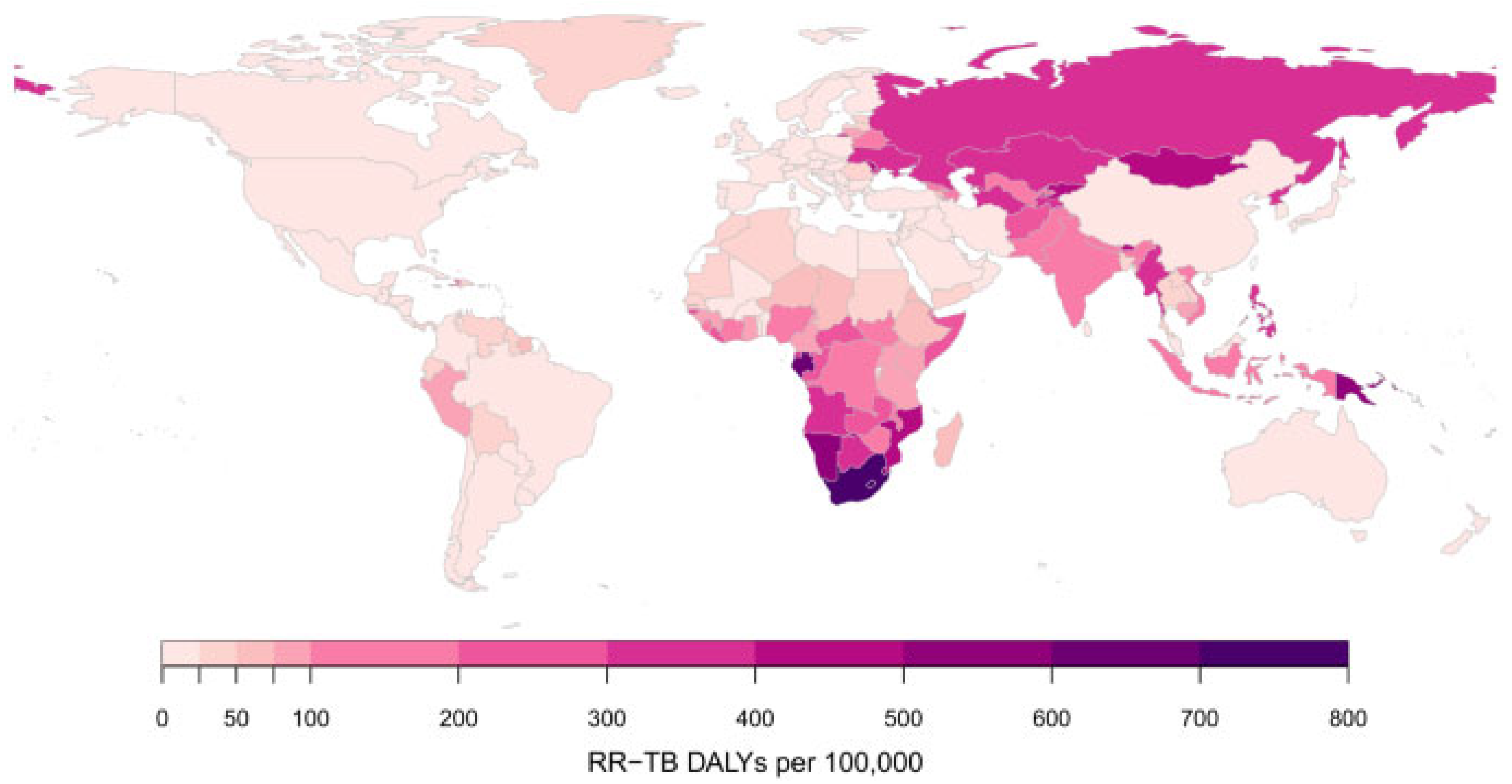
5.7. Rifampicin-Resistant Mycobacterium tuberculosis
5.8. Resistance in Clostridioides Difficile
6. Overview of Existing Antibiotics Effective Against Particular MDR Strains
6.1. Beta-Lactam Antibiotics/Inhibitors of Beta-Lactamases
Rare Genotypes of Gram-Negative Bacilli Resistant to New BLBLI
6.2. New Non-BLBLI Antibiotics Effective Against MDR Strains
7. Combination Antibiotic Therapy Against MDR Bacteria
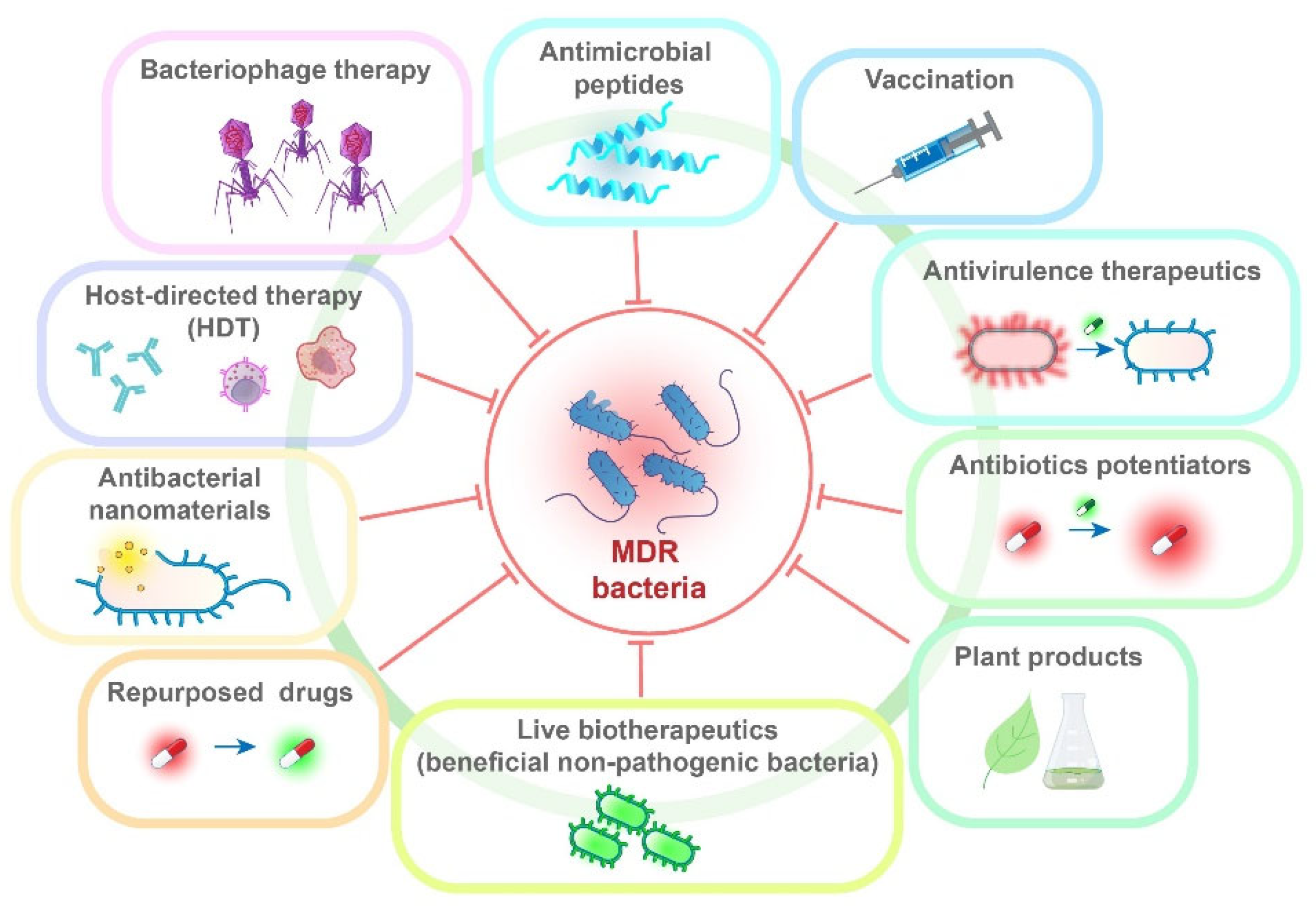
8. Alternative Therapeutic Approaches and Repurposed Drugs
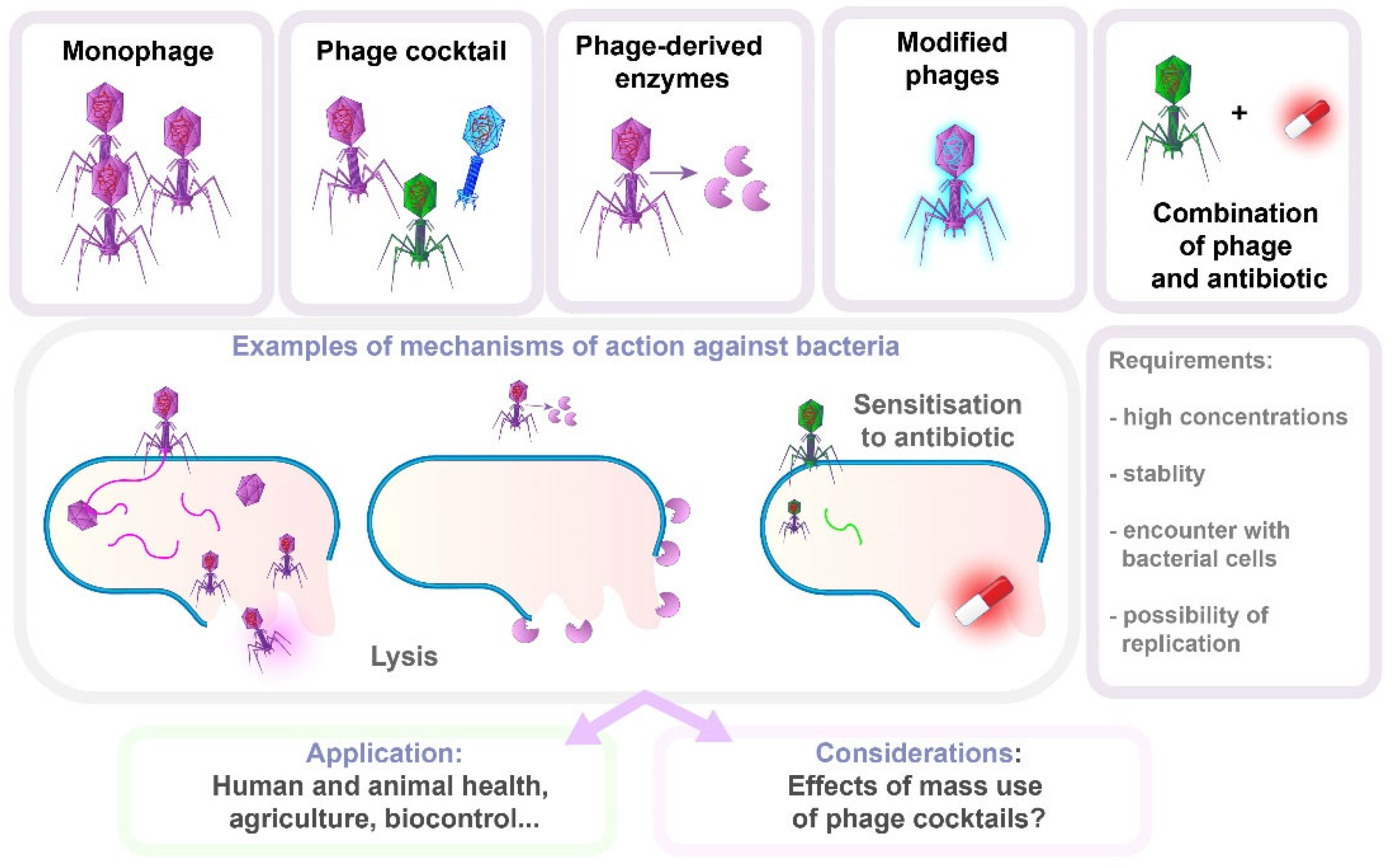
8.1. Bacteriophage Therapy
8.2. Antivirulence Therapeutics
8.3. Antimicrobial Peptides
8.4. Antibacterial Nanomaterials
8.5. Antibiotic Potentiators
8.6. Host-Directed Therapy
8.7. Repurposed Drugs
9. Antibiotic/Non-Antibiotic Combinations for Combating MDR Infections
10. Challenges in Developing New Antimicrobials: Science, Policy, and Economics
11. Biotechnology and Genomics: The Role of Biotechnology in Discovering New Antibacterial Agents, Including Genomic Approaches
12. Discussion
13. Conclusions and Future Directions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Rice, L.B. Federal Funding for the Study of Antimicrobial Resistance in Nosocomial Pathogens: No ESKAPE. J. Infect. Dis. 2008, 197, 1079–1081. [Google Scholar] [CrossRef]
- World Health Organization. WHO Bacterial Priority List, 2024: Bacterial Pathogens of Public Health Importance to Guide Research, Development and Strategies to Prevent and Control Antimicrobial Resistance; World Health Organization: Geneva, Switzerland, 2024. [Google Scholar]
- Naghavi, M.; Vollset, S.E.; Ikuta, K.S.; Swetschinski, L.R.; Gray, A.P.; Wool, E.E.; Robles Aguilar, G.; Mestrovic, T.; Smith, G.; Han, C.; et al. Global Burden of Bacterial Antimicrobial Resistance 1990–2021: A Systematic Analysis with Forecasts to 2050. Lancet 2024, 404, 1199–1226. [Google Scholar] [CrossRef]
- European Centre for Disease Prevention and Control Assessing the Health Burden of Infections with Antibiotic-Resistant Bacteria in the EU/EEA, 2016–2020. 2022. Available online: https://www.ecdc.europa.eu/sites/default/files/documents/Health-burden-infections-antibiotic-resistant-bacteria.pdf (accessed on 12 January 2025).
- Cassini, A.; Högberg, L.D.; Plachouras, D.; Quattrocchi, A.; Hoxha, A.; Simonsen, G.S.; Colomb-Cotinat, M.; Kretzschmar, M.E.; Devleesschauwer, B.; Cecchini, M.; et al. Attributable Deaths and Disability-Adjusted Life-Years Caused by Infections with Antibiotic-Resistant Bacteria in the EU and the European Economic Area in 2015: A Population-Level Modelling Analysis. Lancet Infect. Dis. 2019, 19, 56–66. [Google Scholar] [CrossRef]
- Kakoullis, L.; Papachristodoulou, E.; Chra, P.; Panos, G. Mechanisms of Antibiotic Resistance in Important Gram-Positive and Gram-Negative Pathogens and Novel Antibiotic Solutions. Antibiotics 2021, 10, 415. [Google Scholar] [CrossRef] [PubMed]
- Centers for Disease Control and Prevention COVID-19 Impact on Healthcare-Associated Infections. 2024. Available online: https://www.cdc.gov/healthcare-associated-infections/php/data/covid-impact.html (accessed on 12 January 2025).
- Centers for Disease Control and Prevention Antimicrobial Resistance Threats in the United States, 2021–2022. 2024. Available online: https://www.cdc.gov/antimicrobial-resistance/data-research/threats/update-2022.html (accessed on 12 January 2025).
- Antibiotic Resistance Coordination and Strategy Unit within the Division of Healthcare Quality Promotion; National Center for Emerging and Zoonotic Infectious Diseases; Centers for Disease Control and Prevention. Antibiotic Resistance Threats in the United States. 2019. Available online: https://stacks.cdc.gov/view/cdc/82532 (accessed on 12 January 2025).
- Xiong, X.-S.; Zhang, X.-D.; Yan, J.-W.; Huang, T.-T.; Liu, Z.-Z.; Li, Z.-K.; Wang, L.; Li, F. Identification of Mycobacterium Tuberculosis Resistance to Common Antibiotics: An Overview of Current Methods and Techniques. Infect. Drug Resist. 2024, 17, 1491–1506. [Google Scholar] [CrossRef] [PubMed]
- Cox, G.; Wright, G.D. Intrinsic Antibiotic Resistance: Mechanisms, Origins, Challenges and Solutions. Int. J. Med. Microbiol. 2013, 303, 287–292. [Google Scholar] [CrossRef] [PubMed]
- Enault, F.; Briet, A.; Bouteille, L.; Roux, S.; Sullivan, M.B.; Petit, M.-A. Phages Rarely Encode Antibiotic Resistance Genes: A Cautionary Tale for Virome Analyses. ISME J. 2017, 11, 237–247. [Google Scholar] [CrossRef]
- Reygaert, W.C. An Overview of the Antimicrobial Resistance Mechanisms of Bacteria. AIMS Microbiol. 2018, 4, 482–501. [Google Scholar] [CrossRef]
- Lehtinen, S.; Blanquart, F.; Lipsitch, M.; Fraser, C. On the Evolutionary Ecology of Multidrug Resistance in Bacteria. PLoS Pathog. 2019, 15, e1007763. [Google Scholar] [CrossRef]
- Tooke, C.L.; Hinchliffe, P.; Bragginton, E.C.; Colenso, C.K.; Hirvonen, V.H.A.; Takebayashi, Y.; Spencer, J. β-Lactamases and β-Lactamase Inhibitors in the 21st Century. J. Mol. Biol. 2019, 431, 3472–3500. [Google Scholar] [CrossRef] [PubMed]
- Naas, T.; Oueslati, S.; Bonnin, R.A.; Dabos, M.L.; Zavala, A.; Dortet, L.; Retailleau, P.; Iorga, B.I. Beta-Lactamase Database (BLDB)–Structure and Function. J. Enzyme Inhib. Med. Chem. 2017, 32, 917–919. [Google Scholar] [CrossRef] [PubMed]
- Bush, K. Past and Present Perspectives on β-Lactamases. Antimicrob. Agents Chemother. 2018, 62, e01076-18. [Google Scholar] [CrossRef]
- Jain, P.; Bepari, A.K.; Sen, P.K.; Rafe, T.; Imtiaz, R.; Hossain, M.; Reza, H.M. High Prevalence of Multiple Antibiotic Resistance in Clinical E. Coli Isolates from Bangladesh and Prediction of Molecular Resistance Determinants Using WGS of an XDR Isolate. Sci. Rep. 2021, 11, 22859. [Google Scholar] [CrossRef] [PubMed]
- Castanheira, M.; Simner, P.J.; Bradford, P.A. Extended-Spectrum β-Lactamases: An Update on Their Characteristics, Epidemiology and Detection. JAC Antimicrob. Resist. 2021, 3, dlab092. [Google Scholar] [CrossRef]
- Bush, K.; Bradford, P.A. Interplay between β-Lactamases and New β-Lactamase Inhibitors. Nat. Rev. Microbiol. 2019, 17, 295–306. [Google Scholar] [CrossRef]
- Adam, M.A.; Elhag, W.I. Prevalence of Metallo-β-Lactamase Acquired Genes among Carbapenems Susceptible and Resistant Gram-Negative Clinical Isolates Using Multiplex PCR, Khartoum Hospitals, Khartoum Sudan. BMC Infect. Dis. 2018, 18, 668. [Google Scholar] [CrossRef]
- Lukovic, B.; Kabic, J.; Dragicevic, M.; Kuljanin, S.; Dimkić, I.; Jovcic, B.; Gajic, I. Genetic Basis of Antimicrobial Resistance, Virulence Features and Phylogenomics of Carbapenem-Resistant Acinetobacter baumannii Clinical Isolates. Infection 2024. ahead of print. [Google Scholar] [CrossRef] [PubMed]
- Koncz, M.; Stirling, T.; Hadj Mehdi, H.; Méhi, O.; Eszenyi, B.; Asbóth, A.; Apjok, G.; Tóth, Á.; Orosz, L.; Vásárhelyi, B.M.; et al. Genomic Surveillance as a Scalable Framework for Precision Phage Therapy against Antibiotic-Resistant Pathogens. Cell 2024, 187, 5901–5918. [Google Scholar] [CrossRef] [PubMed]
- Sandfort, M.; Hans, J.B.; Fischer, M.A.; Reichert, F.; Cremanns, M.; Eisfeld, J.; Pfeifer, Y.; Heck, A.; Eckmanns, T.; Werner, G.; et al. Increase in NDM-1 and NDM-1/OXA-48-Producing Klebsiella pneumoniae in Germany Associated with the War in Ukraine, 2022. Eurosurveillance 2022, 27, 2200926. [Google Scholar] [CrossRef]
- Saravanan, M.; Belete, M.A.; Arockiaraj, J. Carbapenem-Resistant Pseudomonas Aeruginosa in Intensive Care Units Increase Mortality as an Emerging Global Threat. Int. J. Surg. 2023, 109, 1034–1036. [Google Scholar] [CrossRef]
- Ivanov, I.; Sabtcheva, S.; Dobreva, E.; Asseva, G.; Padeshki, P.; Petrov, P. Prevalence of Carbapenemase Genes among 16S RRNA Methyltransferase-Producing Enterobacteriaceae Isolated from Cancer Patients. Probl. Infect. Parasit. Dis. 2014, 42, 10–13. [Google Scholar]
- Pradier, L.; Bedhomme, S. Ecology, More than Antibiotics Consumption, Is the Major Predictor for the Global Distribution of Aminoglycoside-Modifying Enzymes. Elife 2023, 12, e77015. [Google Scholar] [CrossRef]
- Peacock, S.J.; Paterson, G.K. Mechanisms of Methicillin Resistance in Staphylococcus aureus. Annu. Rev. Biochem. 2015, 84, 577–601. [Google Scholar] [CrossRef]
- Tenover, F.C. Mechanisms of Antimicrobial Resistance in Bacteria. Am. J. 2006, 119, S3–S10. [Google Scholar] [CrossRef]
- Brenciani, A.; Morroni, G.; Schwarz, S.; Giovanetti, E. Oxazolidinones: Mechanisms of Resistance and Mobile Genetic Elements Involved. J. Antimicrob. Chemother. 2022, 77, 2596–2621. [Google Scholar] [CrossRef]
- Fukuda, A.; Nakajima, C.; Suzuki, Y.; Usui, M. Transferable Linezolid Resistance Genes (OptrA and PoxtA) in Enterococci Derived from Livestock Compost at Japanese Farms. J. Glob. Antimicrob. Resist. 2024, 36, 336–344. [Google Scholar] [CrossRef]
- Fàbrega, A.; Madurga, S.; Giralt, E.; Vila, J. Mechanism of Action of and Resistance to Quinolones. Microb. Biotechnol. 2009, 2, 40–61. [Google Scholar] [CrossRef]
- Miller, W.R.; Bayer, A.S.; Arias, C.A. Mechanism of Action and Resistance to Daptomycin in Staphylococcus Aureus and Enterococci. Cold Spring Harb. Perspect. Med. 2016, 6, a026997. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.-H.; Bhuiyan, M.S.; Shen, H.-H.; Cameron, D.R.; Rupasinghe, T.W.T.; Wu, C.-M.; Le Brun, A.P.; Kostoulias, X.; Domene, C.; Fulcher, A.J.; et al. Antibiotic Resistance and Host Immune Evasion in Staphylococcus Aureus Mediated by a Metabolic Adaptation. Proc. Natl. Acad. Sci. USA 2019, 116, 3722–3727. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Hu, J.; Li, L.; Zhang, M.; Cui, Q.; Ma, Y.; Su, H.; Zhang, X.; Xu, H.; Wang, M. New Mutations in Cls Lead to Daptomycin Resistance in a Clinical Vancomycin- and Daptomycin-Resistant Enterococcus faecium Strain. Front. Microbiol. 2022, 13, 896916. [Google Scholar] [CrossRef] [PubMed]
- Miller, W.R.; Munita, J.M.; Arias, C.A. Mechanisms of Antibiotic Resistance in Enterococci. Expert Rev. Anti Infect. Ther. 2014, 12, 1221–1236. [Google Scholar] [CrossRef]
- Tran, T.T.; Panesso, D.; Mishra, N.N.; Mileykovskaya, E.; Guan, Z.; Munita, J.M.; Reyes, J.; Diaz, L.; Weinstock, G.M.; Murray, B.E.; et al. Daptomycin-Resistant Enterococcus faecalis Diverts the Antibiotic Molecule from the Division Septum and Remodels Cell Membrane Phospholipids. mBio 2013, 4, e00281-13. [Google Scholar] [CrossRef] [PubMed]
- Darby, E.M.; Trampari, E.; Siasat, P.; Solsona Gaya, M.; Alav, I.; Webber, M.A.; Blair, J.M.A. Molecular Mechanisms of Antibiotic Resistance Revisited. Nat. Rev. Microbiol. 2023, 21, 280–295. [Google Scholar] [CrossRef] [PubMed]
- Kumawat, M.; Nabi, B.; Daswani, M.; Viquar, I.; Pal, N.; Sharma, P.; Tiwari, S.; Sarma, D.K.; Shubham, S.; Kumar, M. Role of Bacterial Efflux Pump Proteins in Antibiotic Resistance across Microbial Species. Microb. Pathog. 2023, 181, 106182. [Google Scholar] [CrossRef] [PubMed]
- Estrada, A.; Wright, D.L.; Anderson, A.C. Antibacterial Antifolates: From Development through Resistance to the next Generation. Cold Spring Harb. Perspect. Med. 2016, 6, a028324. [Google Scholar] [CrossRef]
- Mugnier, P.D.; Poirel, L.; Naas, T.; Nordmann, P. Worldwide Dissemination of the BlaOXA-23 Carbapenemase Gene of Acinetobacter Baumannii1. Emerg. Infect. Dis. 2010, 16, 35–40. [Google Scholar] [CrossRef]
- Moubareck, C.A.; Halat, D.H. Insights into Acinetobacter baumannii: A Review of Microbiological, Virulence, and Resistance Traits in a Threatening Nosocomial Pathogen. Antibiotics 2020, 9, 119. [Google Scholar] [CrossRef] [PubMed]
- Kabic, J.; Novovic, K.; Kekic, D.; Trudic, A.; Opavski, N.; Dimkic, I.; Jovcic, B.; Gajic, I. Comparative Genomics and Molecular Epidemiology of Colistin-Resistant Acinetobacter baumannii. Comput. Struct. Biotechnol. J. 2023, 21, 574–585. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.; Yu, Y.; Hua, X. Resistance Mechanisms of Tigecycline in Acinetobacter baumannii. Front. Cell Infect. Microbiol. 2023, 13, 1141490. [Google Scholar] [CrossRef] [PubMed]
- Leite, G.C.; Oliveira, M.S.; Perdigão-Neto, L.V.; Rocha, C.K.D.; Guimarães, T.; Rizek, C.; Levin, A.S.; Costa, S.F. Antimicrobial Combinations against Pan- Resistant Acinetobacter Baumannii Isolates with Different Resistance Mechanisms. PLoS ONE 2016, 11, e0151270. [Google Scholar] [CrossRef] [PubMed]
- Tan, Y.C.; Lahiri, C. Promising Acinetobacter baumannii Vaccine Candidates and Drug Targets in Recent Years. Front. Immunol. 2022, 13, 900509. [Google Scholar] [CrossRef]
- Pahil, K.S.; Gilman, M.S.A.; Baidin, V.; Clairfeuille, T.; Mattei, P.; Bieniossek, C.; Dey, F.; Muri, D.; Baettig, R.; Lobritz, M.; et al. A New Antibiotic Traps Lipopolysaccharide in Its Intermembrane Transporter. Nature 2024, 625, 572–577. [Google Scholar] [CrossRef]
- Zampaloni, C.; Mattei, P.; Bleicher, K.; Winther, L.; Thäte, C.; Bucher, C.; Adam, J.M.; Alanine, A.; Amrein, K.E.; Baidin, V.; et al. A Novel Antibiotic Class Targeting the Lipopolysaccharide Transporter. Nature 2024, 625, 566–571. [Google Scholar] [CrossRef] [PubMed]
- Narimisa, N.; Keshtkar, A.; Dadgar-Zankbar, L.; Bostanghadiri, N.; Far, Y.R.; Shahroodian, S.; Zahedi Bialvaei, A.; Razavi, S. Prevalence of Colistin Resistance in Clinical Isolates of Pseudomonas aeruginosa: A Systematic Review and Meta-Analysis. Front. Microbiol. 2024, 15, 1477836. [Google Scholar] [CrossRef] [PubMed]
- Haidar, G.; Clancy, C.J.; Chen, L.; Samanta, P.; Shields, R.K.; Kreiswirth, B.N.; Nguyen, H. Identifying Spectra of Activity and Therapeutic Niches for Ceftazidime-Avibactam and Imipenem-Relebactam against Carbapenem-Resistant Enterobacteriaceae. Antimicrob. Agents Chemother. 2017, 61, e00642-17. [Google Scholar] [CrossRef] [PubMed]
- Goessens, W.H.F.; Van Der Bij, A.K.; Van Boxtel, R.; Pitout, J.D.D.; Van Ulsen, P.; Melles, D.C.; Tommassen, J. Antibiotic Trapping by Plasmid-Encoded Cmy-2-Lactamase Combined with Reduced Outer Membrane Permeability as a Mechanism of Carbapenem Resistance in Escherichia coli. Antimicrob. Agents Chemother. 2013, 57, 3941–3949. [Google Scholar] [CrossRef] [PubMed]
- Kherroubi, L.; Bacon, J.; Rahman, K.M. Navigating Fluoroquinolone Resistance in Gram-Negative Bacteria: A Comprehensive Evaluation. JAC Antimicrob. Resist. 2024, 6, dlae127. [Google Scholar] [CrossRef] [PubMed]
- Azargun, R.; Gholizadeh, P.; Sadeghi, V.; Hosainzadegan, H.; Tarhriz, V.; Memar, M.Y.; Pormohammad, A.; Eyvazi, S. Molecular Mechanisms Associated with Quinolone Resistance in Enterobacteriaceae: Review and Update. Trans. R Soc. Trop. Med. Hyg. 2020, 114, 770–781. [Google Scholar] [CrossRef] [PubMed]
- EUCAST European Committee on Antimicrobial Susceptibility Testing. Breakpoint Tables for Interpretation of MICs and Zone Diameters. Version 9.0. 2019. Available online: https://www.eucast.org/fileadmin/src/media/PDFs/EUCAST_files/Breakpoint_tables/v_9.0_Breakpoint_Tables.pdf (accessed on 12 January 2025).
- European Centre for Disease Prevention and Control. Gonococcal Antimicrobial Susceptibility Surveillance in the European Union/European Economic Area. 2022. Available online: https://www.ecdc.europa.eu/en/publications-data/gonococcal-antimicrobial-susceptibility-surveillance-eu-eea (accessed on 12 January 2025).
- Pleininger, S.; Indra, A.; Golparian, D.; Heger, F.; Schindler, S.; Jacobsson, S.; Heidler, S.; Unemo, M. Extensively Drug-Resistant (XDR) Neisseria Gonorrhoeae Causing Possible Gonorrhoea Treatment Failure with Ceftriaxone plus Azithromycin in Austria, April 2022. Eurosurveillance 2022, 27, 2200455. [Google Scholar] [CrossRef] [PubMed]
- Maubaret, C.; Caméléna, F.; Mrimèche, M.; Braille, A.; Liberge, M.; Mainardis, M.; Guillaume, C.; Noel, F.; Bébéar, C.; Molina, J.-M.; et al. Two Cases of Extensively Drug-Resistant (XDR) Neisseria gonorrhoeae Infection Combining Ceftriaxone-Resistance and High-Level Azithromycin Resistance. Eurosurveillance 2022, 28, 2300456. [Google Scholar] [CrossRef]
- Unemo, M.; Ross, J.D.C.; Serwin, A.B.; Gomberg, M.; Cusini, M.; Jensen, J.S. 2020 European Guideline for the Diagnosis and Treatment of Gonorrhoea in Adults. Int. J. STD AIDS 2020, 1–17. [Google Scholar] [CrossRef]
- Ouk, V.; Heng, L.S.; Virak, M.; Deng, S.; Lahra, M.M.; Frankson, R.; Kreisel, K.; McDonald, R.; Escher, M.; Unemo, M.; et al. High Prevalence of Ceftriaxone-Resistant and XDR Neisseria gonorrhoeae in Several Cities of Cambodia, 2022–23: WHO Enhanced Gonococcal Antimicrobial Surveillance Programme (EGASP). JAC Antimicrob. Resist. 2024, 6, dlae053. [Google Scholar] [CrossRef] [PubMed]
- Hook, E.W.; Newman, L.; Drusano, G.; Evans, S.; Handsfield, H.H.; Jerse, A.E.; Kong, F.Y.S.; Lee, J.Y.; Taylor, S.N.; Deal, C. Development of New Antimicrobials for Urogenital Gonorrhea Therapy: Clinical Trial Design Considerations. Clin. Infect. Dis. 2020, 70, 1495–1500. [Google Scholar] [CrossRef]
- The Global Antibiotic Research; Development Partnership. Positive Results Announced in Largest Pivotal Phase 3 Trial of a First-in-Class Oral Antibiotic to Treat Uncomplicated Gonorrhoea. 2023. Available online: https://gardp.org/positive-results-announced-in-largest-pivotal-phase-3-trial-of-a-first-in-class-oral-antibiotic-to-treat-uncomplicated-gonorrhoea/#:~:text=Positive%20results%20announced%20in%20largest,antibiotic%20to%20treat%20uncomplicated%20gonorrhoea&text=A%20phase%203%20study%20of,current%20international%20standard%20of%20care (accessed on 12 January 2025).
- Petousis-Harris, H.; Paynter, J.; Morgan, J.; Saxton, P.; McArdle, B.; Goodyear-Smith, F.; Black, S. Effectiveness of a Group B Outer Membrane Vesicle Meningococcal Vaccine against Gonorrhoea in New Zealand: A Retrospective Case-Control Study. Lancet 2017, 390, 1603–1610. [Google Scholar] [CrossRef] [PubMed]
- Semchenko, E.A.; Tan, A.; Borrow, R.; Seib, K.L. The Serogroup B Meningococcal Vaccine Bexsero Elicits Antibodies to Neisseria Gonorrhoeae. Clin. Infect. Dis. 2019, 69, 1101–1111. [Google Scholar] [CrossRef]
- Wu, J.-Y.; Liou, J.-M.; Graham, D.Y. Evidence-Based Recommendations for Successful Helicobacter Pylori Treatment. Expert. Rev. Gastroenterol. Hepatol. 2014, 8, 21–28. [Google Scholar] [CrossRef] [PubMed]
- Glupczynski, Y.; Mégraud, F.; Lopez-Brea, M.; Andersen, L. European Multicentre Survey of in Vitro Antimicrobial Resistance in Helicobacter Pylori. Eur. J. Clin. Microbiol. Infect. Dis. 2001, 20, 820–823. [Google Scholar] [CrossRef] [PubMed]
- Megraud, F. H Pylori Antibiotic Resistance: Prevalence, Importance, and Advances in Testing. Gut 2004, 53, 1374–1384. [Google Scholar] [CrossRef] [PubMed]
- Bujanda, L.; Nyssen, O.P.; Vaira, D.; Saracino, I.M.; Fiorini, G.; Lerang, F.; Georgopoulos, S.; Tepes, B.; Heluwaert, F.; Gasbarrini, A.; et al. Antibiotic Resistance Prevalence and Trends in Patients Infected with Helicobacter Pylori in the Period 2013–2020: Results of the European Registry on H. pylori Management (Hp-EuReg). Antibiotics 2021, 10, 1058. [Google Scholar] [CrossRef] [PubMed]
- Nyssen, O.P.; Martínez, B.; Mégraud, F.; Savarino, V.; Fallone, C.A.; Bazzoli, F.; Gisbert, J.P. Sequential versus Standard Triple Therapy for First-Line Helicobacter pylori Eradication: An Update. Antibiotics 2024, 13, 136. [Google Scholar] [CrossRef] [PubMed]
- Megraud, F.; Bruyndonckx, R.; Coenen, S.; Wittkop, L.; Huang, T.-D.; Hoebeke, M.; Bénéjat, L.; Lehours, P.; Goossens, H.; Glupczynski, Y. Helicobacter pylori Resistance to Antibiotics in Europe in 2018 and Its Relationship to Antibiotic Consumption in the Community. Gut 2021, 70, 1815–1822. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Liu, Y.; Wang, M.; Gao, L.; Liu, J.; Zhang, H.; Wu, M.; Chen, H.; Lou, J.; Wang, J.; et al. Safety, Pharmacokinetics, and Efficacy of Rifasutenizol, a Novel Dual-Targeted Antibacterial Agent in Healthy Participants and Patients in China with Helicobacter Pylori Infection: Four Randomised Clinical Trials. Lancet Infect. Dis. 2024, 24, 650–664. [Google Scholar] [CrossRef] [PubMed]
- Shim, H. Three Innovations of Next-Generation Antibiotics: Evolvability, Specificity, and Non-Immunogenicity. Antibiotics 2023, 12, 204. [Google Scholar] [CrossRef] [PubMed]
- Australian New Zealand Clinical Trials Registry. Testing Safety and Efficacy of a Live Bacterial Therapy for the Treatment of Helicobacter Pylori Infection; Australian New Zealand Clinical Trials Registry: Camperdown, Australia, 2020. [Google Scholar]
- Livermore, D.M. Beta-Lactamases in Laboratory and Clinical Resistance. Clin. Microbiol. Rev. 1995, 8, 557–584. [Google Scholar] [CrossRef]
- Malhotra-Kumar, S.; Haccuria, K.; Michiels, M.; Ieven, M.; Poyart, C.; Hryniewicz, W.; Goossens, H. Current Trends in Rapid Diagnostics for Methicillin-Resistant Staphylococcus Aureus and Glycopeptide-Resistant Enterococcus Species. J. Clin. Microbiol. 2008, 46, 1577–1587. [Google Scholar] [CrossRef]
- Howden, B.P.; Davies, J.K.; Johnson, P.D.R.; Stinear, T.P.; Grayson, M.L. Reduced Vancomycin Susceptibility in Staphylococcus aureus, Including Vancomycin-Intermediate and Heterogeneous Vancomycin-Intermediate Strains: Resistance Mechanisms, Laboratory Detection, and Clinical Implications. Clin. Microbiol. Rev. 2010, 23, 99–139. [Google Scholar] [CrossRef] [PubMed]
- Cong, Y.; Yang, S.; Rao, X. Vancomycin Resistant Staphylococcus aureus Infections: A Review of Case Updating and Clinical Features. J. Adv. Res. 2020, 21, 169–176. [Google Scholar] [CrossRef] [PubMed]
- Jubeh, B.; Breijyeh, Z.; Karaman, R. Resistance of Gram-Positive Bacteria to Current Antibacterial Agents and Overcoming Approaches. Molecules 2020, 25, 2888. [Google Scholar] [CrossRef]
- Hashemian, S.M.R.; Farhadi, T.; Ganjparvar, M. Linezolid: A Review of Its Properties, Function, and Use in Critical Care. Drug Des. Dev. Ther. 2018, 12, 1759–1767. [Google Scholar] [CrossRef] [PubMed]
- Carcione, D.; Intra, J.; Andriani, L.; Campanile, F.; Gona, F.; Carletti, S.; Mancini, N.; Brigante, G.; Cattaneo, D.; Baldelli, S.; et al. New Antimicrobials for Gram-Positive Sustained Infections: A Comprehensive Guide for Clinicians. Pharmaceuticals 2023, 16, 1304. [Google Scholar] [CrossRef] [PubMed]
- Bender, J.K.; Fleige, C.; Funk, F.; Moretó-Castellsagué, C.; Fischer, M.A.; Werner, G. Linezolid Resistance Genes and Mutations among Linezolid-Susceptible Enterococcus spp.—A Loose Cannon? Antibiotics 2024, 13, 101. [Google Scholar] [CrossRef]
- Schroeder, M.R.; Stephens, D.S. Macrolide Resistance in Streptococcus pneumoniae. Front. Cell Infect. Microbiol. 2016, 6, 98. [Google Scholar] [CrossRef] [PubMed]
- Calatayud, L.; Ardanuy, C.; Tubau, F.; Rolo, D.; Grau, I.; Pallareés, R.; Martiín, R.; Linñares, J. Serotype and Genotype Replacement among Macrolide-Resistant Invasive Pneumococci in Adults: Mechanisms of Resistance and Association with Different Transposons. J. Clin. Microbiol. 2010, 48, 1310–1316. [Google Scholar] [CrossRef] [PubMed]
- Opavski, N.; Jovićević, M.; Kabić, J.; Kekić, D.; Gajić, I. Effect of Childhood Pneumococcal Conjugate Vaccination on Invasive Disease Serotypes in Serbia. Vaccines 2024, 12, 940. [Google Scholar] [CrossRef]
- Lynch, J.; Zhanel, G. Streptococcus Pneumoniae: Does Antimicrobial Resistance Matter? Semin. Respir. Crit. Care Med. 2009, 30, 210–238. [Google Scholar] [CrossRef] [PubMed]
- Cherazard, R.; Epstein, M.; Doan, T.-L.; Salim, T.; Bharti, S.; Smith, M.A. Antimicrobial Resistant Streptococcus Pneumoniae: Prevalence, Mechanisms, and Clinical Implications. Am. J. Ther. 2017, 24, e361–e369. [Google Scholar] [CrossRef]
- Humphries, R.M.; Lu, J.; Martin, I.; Rauch, C.A.; Wojewoda, C.; McCarter, Y.; Long, T.; Simner, P.J. Detection of Penicillin Nonsusceptible Streptococcus agalactiae by Laboratories That Participate in the College of American Pathologist’s Proficiency Testing Program. J. Clin. Microbiol. 2023, 61, e00595-23. [Google Scholar] [CrossRef] [PubMed]
- Kimura, K.; Suzuki, S.; Wachino, J.; Kurokawa, H.; Yamane, K.; Shibata, N.; Nagano, N.; Kato, H.; Shibayama, K.; Arakawa, Y. First Molecular Characterization of Group B Streptococci with Reduced Penicillin Susceptibility. Antimicrob. Agents Chemother. 2008, 52, 2890–2897. [Google Scholar] [CrossRef] [PubMed]
- McGuire, E.; Ready, D.; Ellaby, N.; Potterill, I.; Pike, R.; Hopkins, K.L.; Guy, R.L.; Lamagni, T.; Mack, D.; Scobie, A.; et al. A Case of Penicillin-Resistant Group B Streptococcus Isolated from a Patient in the UK. J. Antimicrob. Chemother. 2024, 80, 399–404. [Google Scholar] [CrossRef] [PubMed]
- Seki, T.; Kimura, K.; Reid, M.E.; Miyazaki, A.; Banno, H.; Jin, W.; Wachino, J.; Yamada, K.; Arakawa, Y. High Isolation Rate of MDR Group B Streptococci with Reduced Penicillin Susceptibility in Japan. J. Antimicrob. Chemother. 2015, 70, 2725–2728. [Google Scholar] [CrossRef] [PubMed]
- Kekic, D.; Gajic, I.; Opavski, N.; Kojic, M.; Vukotic, G.; Smitran, A.; Boskovic, L.; Stojkovic, M.; Ranin, L. Trends in Molecular Characteristics and Antimicrobial Resistance of Group B Streptococci: A Multicenter Study in Serbia, 2015–2020. Sci. Rep. 2021, 11, 540. [Google Scholar] [CrossRef] [PubMed]
- Peltola, H. Worldwide Haemophilus Influenzae Type b Disease at the Beginning of the 21st Century: Global Analysis of the Disease Burden 25 Years after the Use of the Polysaccharide Vaccine and a Decade after the Advent of Conjugates. Clin. Microbiol. Rev. 2000, 13, 302–317. [Google Scholar] [CrossRef] [PubMed]
- Centers for Disease Control and Prevention. Active Bacterial Core Surveillance Report, Emerging Infections Program Network, Haemophilus influenzae. 2022. Available online: https://www.cdc.gov/abcs/downloads/HFLU_Surveillance_Report_2022.pdf (accessed on 12 January 2025).
- European Centre for Disease Prevention and Control (ECDC). Haemophilus influenzae. In ECDC. Annual Epidemiological Report for 2022; European Centre for Disease Prevention and Control: Stockholm, Sweden, 2024. [Google Scholar]
- Heinz, E. The Return of Pfeiffer’s Bacillus: Rising Incidence of Ampicillin Resistance in Haemophilus influenzae. Microb. Genom. 2018, 4, e000214. [Google Scholar] [CrossRef] [PubMed]
- Abavisani, M.; Keikha, M.; Karbalaei, M. First Global Report about the Prevalence of Multi-Drug Resistant Haemophilus Influenzae: A Systematic Review and Meta-Analysis. BMC Infect. Dis. 2024, 24, 90. [Google Scholar] [CrossRef]
- Menzies, N.A.; Allwood, B.W.; Dean, A.S.; Dodd, P.J.; Houben, R.M.G.J.; James, L.P.; Knight, G.M.; Meghji, J.; Nguyen, L.N.; Rachow, A.; et al. Global Burden of Disease Due to Rifampicin-Resistant Tuberculosis: A Mathematical Modeling Analysis. Nat. Commun. 2023, 14, 6182. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Rapid Communication: Key Changes to Treatment of Multidrug- and Rifampicin-Resistant Tuberculosis (MDR/RR-TB). 2018. Available online: https://www.who.int/publications/i/item/WHO-CDS-TB-2018.18 (accessed on 12 January 2025).
- World Health Organization. Global Tuberculosis Report 2023; World Health Organization: Geneva, Switzerland, 2023. [Google Scholar]
- World Health Organization. WHO Consolidated Guidelines on Tuberculosis. Module 4: Treatment—Drug-Resistant Tuberculosis Treatment, 2022 Update; World Health Organization: Geneva, Switzerland, 2022. [Google Scholar]
- U.S. Department of Health and Human Services; CDC. Antibiotic Resistance Threats in the United States, 2019; U.S. Department of Health and Human Services: Atlanta, GA, USA, 2019. [Google Scholar]
- Wickramage, I.; Spigaglia, P.; Sun, X. Mechanisms of Antibiotic Resistance of Clostridioides Difficile. J. Antimicrob. Chemother. 2021, 76, 3077–3090. [Google Scholar] [CrossRef] [PubMed]
- Boekhoud, I.M.; Hornung, B.V.H.; Sevilla, E.; Harmanus, C.; Bos-Sanders, I.M.J.G.; Terveer, E.M.; Bolea, R.; Corver, J.; Kuijper, E.J.; Smits, W.K. Plasmid-Mediated Metronidazole Resistance in Clostridioides difficile. Nat. Commun. 2020, 11, 598. [Google Scholar] [CrossRef]
- Mullane, K. Fidaxomicin in Clostridium Difficile Infection: Latest Evidence and Clinical Guidance. Ther. Adv. Chronic Dis. 2014, 5, 69–84. [Google Scholar] [CrossRef] [PubMed]
- Mason, C.S.; Avis, T.; Hu, C.; Nagalingam, N.; Mudaliar, M.; Coward, C.; Begum, K.; Gajewski, K.; Alam, M.J.; Bassères, E.; et al. The Novel DNA Binding Mechanism of Ridinilazole, a Precision Clostridiodes difficile Antibiotic. Antimicrob. Agents Chemother. 2023, 67, e01563-22. [Google Scholar] [CrossRef] [PubMed]
- Lomeli, B.K.; Galbraith, H.; Schettler, J.; Saviolakis, G.A.; El-Amin, W.; Osborn, B.; Ravel, J.; Hazleton, K.; Lozupone, C.A.; Evans, R.J.; et al. Multiple-Ascending-Dose Phase 1 Clinical Study of the Safety, Tolerability, and Pharmacokinetics of CRS3123, a Narrow-Spectrum Agent with Minimal Disruption of Normal Gut Microbiota. Antimicrob. Agents Chemother. 2019, 64, e01395-19. [Google Scholar] [CrossRef] [PubMed]
- Freeman, J.; Pilling, S.; Vernon, J.; Wilcox, M.H. In Vitro Activities of MCB3681 and Eight Comparators against Clostridium Difficile Isolates with Known Ribotypes and Diverse Geographical Spread. Antimicrob. Agents Chemother. 2017, 61, e02077-16. [Google Scholar] [CrossRef] [PubMed]
- Garey, K.W.; Begum, K.; Lancaster, C.; Gonzales-Luna, A.; Bui, D.; Mercier, J.; Seng Yue, C.; Ducharme, M.P.; Hu, M.; Vince, B.; et al. A Randomized, Double-Blind, Placebo-Controlled, Single and Multiple Ascending Dose Phase 1 Study to Determine the Safety, Pharmacokinetics and Food and Faecal Microbiome Effects of Ibezapolstat Administered Orally to Healthy Subjects. J. Antimicrob. Chemother. 2020, 75, 3635–3643. [Google Scholar] [CrossRef] [PubMed]
- Hind, C.; Clifford, M.; Woolley, C.; Harmer, J.; McGee, L.M.C.; Tyson-Hirst, I.; Tait, H.J.; Brooke, D.P.; Dancer, S.J.; Hunter, I.S.; et al. Insights into the Spectrum of Activity and Mechanism of Action of MGB-BP-3. ACS Infect. Dis. 2022, 8, 2552–2563. [Google Scholar] [CrossRef]
- Gaibani, P.; Giani, T.; Bovo, F.; Lombardo, D.; Amadesi, S.; Lazzarotto, T.; Coppi, M.; Rossolini, G.M.; Ambretti, S. Resistance to Ceftazidime/Avibactam, Meropenem/Vaborbactam and Imipenem/Relebactam in Gram-Negative MDR Bacilli: Molecular Mechanisms and Susceptibility Testing. Antibiotics 2022, 11, 628. [Google Scholar] [CrossRef]
- Fraile-Ribot, P.A.; Cabot, G.; Mulet, X.; Periañez, L.; Martín-Pena, M.L.; Juan, C.; Pérez, J.L.; Oliver, A. Mechanisms Leading to in Vivo Ceftolozane/Tazobactam Resistance Development during the Treatment of Infections Caused by MDR Pseudomonas Aeruginosa. J. Antimicrob. Chemother. 2018, 73, 658–663. [Google Scholar] [CrossRef] [PubMed]
- Contreras-Gómez, M.J.; Martinez, J.R.W.; Rivas, L.; Riquelme-Neira, R.; Ugalde, J.A.; Wozniak, A.; García, P.; Munita, J.M.; Olivares-Pacheco, J.; Alcalde-Rico, M. Role of the Multi-Drug Efflux Systems on the Baseline Susceptibility to Ceftazidime/Avibactam and Ceftolozane/Tazobactam in Clinical Isolates of Non-Carbapenemase-Producing Carbapenem-Resistant Pseudomonas aeruginosa. Front. Pharmacol. 2022, 13, 1007162. [Google Scholar] [CrossRef] [PubMed]
- McLeod, S.M.; O’Donnell, J.P.; Narayanan, N.; Mills, J.P.; Kaye, K.S. Sulbactam–Durlobactam: A β-Lactam/β-Lactamase Inhibitor Combination Targeting Acinetobacter baumannii. Future Microbiol. 2024, 19, 563–576. [Google Scholar] [CrossRef]
- Food and Drug Administration (FDA). XACDURO (Sulbactam for Injection; Durlobactam for Injection), Co-Packaged for Intravenous Use. Prescribing Information. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2023/216974s000lbl.pdf (accessed on 15 October 2024).
- Durand-Réville, T.F.; Guler, S.; Comita-Prevoir, J.; Chen, B.; Bifulco, N.; Huynh, H.; Lahiri, S.; Shapiro, A.B.; McLeod, S.M.; Carter, N.M.; et al. ETX2514 Is a Broad-Spectrum β-Lactamase Inhibitor for the Treatment of Drug-Resistant Gram-Negative Bacteria Including Acinetobacter baumannii. Nat. Microbiol. 2017, 2, 17104. [Google Scholar] [CrossRef]
- Iyer, R.; Moussa, S.H.; Durand-Réville, T.F.; Tommasi, R.; Miller, A. Acinetobacter Baumannii OmpA Is a Selective Antibiotic Permeant Porin. ACS Infect. Dis. 2018, 4, 373–381. [Google Scholar] [CrossRef] [PubMed]
- Barnes, M.D.; Kumar, V.; Bethel, C.R.; Moussa, S.H.; O’Donnell, J.; Rutter, J.D.; Good, C.E.; Hujer, K.M.; Hujer, A.M.; Marshall, S.H.; et al. Targeting Multidrug-Resistant Acinetobacter spp.: Sulbactam and the Diazabicyclooctenone β-Lactamase Inhibitor ETX2514 as a Novel Therapeutic Agent. mBio 2019, 10, 10–1128. [Google Scholar] [CrossRef] [PubMed]
- McLeod, S.M.; Shapiro, A.B.; Moussa, S.H.; Johnstone, M.; McLaughlin, R.E.; de Jonge, B.L.M.; Miller, A.A. Frequency and Mechanism of Spontaneous Resistance to Sulbactam Combined with the Novel β-Lactamase Inhibitor ETX2514 in Clinical Isolates of Acinetobacter baumannii. Antimicrob. Agents Chemother. 2018, 62, e01576-17. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Liao, X.; Ding, T.; Ahn, J. Role of β-Lactamase Inhibitors as Potentiators in Antimicrobial Chemotherapy Targeting Gram-Negative Bacteria. Antibiotics 2024, 13, 260. [Google Scholar] [CrossRef] [PubMed]
- Morrissey, I.; Magnet, S.; Hawser, S.; Shapiro, S.; Knechtle, P. In Vitro Activity of Cefepime-Enmetazobactam against Gram-Negative Isolates Collected from U.S. and European Hospitals during 2014–2015. Antimicrob. Agents Chemother. 2019, 63, e00514-19. [Google Scholar] [CrossRef]
- Tan, X.; Kim, H.S.; Baugh, K.; Huang, Y.; Kadiyala, N.; Wences, M.; Singh, N.; Wenzler, E.; Bulman, Z.P. Therapeutic Options for Metallo-β-Lactamase-Producing Enterobacterales. Infect. Drug Resist. 2021, 14, 125–142. [Google Scholar] [CrossRef] [PubMed]
- Falcone, M.; Daikos, G.L.; Tiseo, G.; Bassoulis, D.; Giordano, C.; Galfo, V.; Leonildi, A.; Tagliaferri, E.; Barnini, S.; Sani, S.; et al. Efficacy of Ceftazidime-Avibactam Plus Aztreonam in Patients with Bloodstream Infections Caused by Metallo-β-Lactamase–Producing Enterobacterales. Clin. Infect. Dis. 2021, 72, 1871–1878. [Google Scholar] [CrossRef] [PubMed]
- Sader, H.S.; Carvalhaes, C.G.; Kimbrough, J.H.; Mendes, R.E.; Castanheira, M. Activity of Aztreonam-Avibactam against Enterobacterales Resistant to Recently Approved Beta-Lactamase Inhibitor Combinations Collected in Europe, Latin America, and the Asia-Pacific Region (2020–2022). Int. J. Antimicrob. Agents 2024, 63, 107113. [Google Scholar] [CrossRef] [PubMed]
- McCreary, E.K.; Heil, E.L.; Tamma, P.D. New Perspectives on Antimicrobial Agents: Cefiderocol. Antimicrob. Agents Chemother. 2021, 65, e0217120. [Google Scholar] [CrossRef] [PubMed]
- Wunderink, R.G.; Matsunaga, Y.; Ariyasu, M.; Clevenbergh, P.; Echols, R.; Kaye, K.S.; Kollef, M.; Menon, A.; Pogue, J.M.; Shorr, A.F.; et al. Cefiderocol versus High-Dose, Extended-Infusion Meropenem for the Treatment of Gram-Negative Nosocomial Pneumonia (APEKS-NP): A Randomised, Double-Blind, Phase 3, Non-Inferiority Trial. Lancet Infect. Dis 2021, 21, 213–225. [Google Scholar] [CrossRef]
- Bassetti, M.; Echols, R.; Matsunaga, Y.; Ariyasu, M.; Doi, Y.; Ferrer, R.; Lodise, T.P.; Naas, T.; Niki, Y.; Paterson, D.L.; et al. Efficacy and Safety of Cefiderocol or Best Available Therapy for the Treatment of Serious Infections Caused by Carbapenem-Resistant Gram-Negative Bacteria (CREDIBLE-CR): A Randomised, Open-Label, Multicentre, Pathogen-Focused, Descriptive, Phase 3 Trial. Lancet Infect. Dis. 2021, 21, 226–240. [Google Scholar] [CrossRef] [PubMed]
- Choby, J.E.; Ozturk, T.; Satola, S.W.; Jacob, J.T.; Weiss, D.S. Widespread Cefiderocol Heteroresistance in Carbapenem-Resistant Gram-Negative Pathogens. Lancet Infect. Dis. 2021, 21, 597–598. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Yang, D.; Wang, Y.; Ni, W. Cefiderocol for the Treatment of Multidrug-Resistant Gram-Negative Bacteria: A Systematic Review of Currently Available Evidence. Front. Pharmacol. 2022, 13, 896971. [Google Scholar] [CrossRef] [PubMed]
- Macone, A.B.; Caruso, B.K.; Leahy, R.G.; Donatelli, J.; Weir, S.; Draper, M.P.; Tanaka, S.K.; Levy, S.B. In Vitro and In Vivo Antibacterial Activities of Omadacycline, a Novel Aminomethylcycline. Antimicrob. Agents Chemother. 2014, 58, 1127–1135. [Google Scholar] [CrossRef]
- Clark, J.A.; Burgess, D.S. Plazomicin: A New Aminoglycoside in the Fight against Antimicrobial Resistance. Ther. Adv. Infect. Dis. 2020, 7, 204993612095260. [Google Scholar] [CrossRef]
- Wetzel, C.; Lonneman, M.; Wu, C. Polypharmacological Drug Actions of Recently FDA Approved Antibiotics. Eur. J. Med. Chem. 2021, 209, 112931. [Google Scholar] [CrossRef] [PubMed]
- Woods, R.J.; Read, A.F. Combination Antimicrobial Therapy to Manage Resistance. Evol. Med. Public Health 2023, 11, 185–186. [Google Scholar] [CrossRef]
- Falcone, M.; Giordano, C.; Leonildi, A.; Galfo, V.; Lepore, A.; Suardi, L.R.; Riccardi, N.; Barnini, S.; Tiseo, G. Clinical Features and Outcomes of Infections Caused by Metallo-β-Lactamase–Producing Enterobacterales: A 3-Year Prospective Study from an Endemic Area. Clin. Infect. Dis. 2024, 78, 1111–1119. [Google Scholar] [CrossRef] [PubMed]
- Tamma, P.D.; Heil, E.L.; Justo, J.A.; Mathers, A.J.; Satlin, M.J.; Bonomo, R.A. Infectious Diseases Society of America 2024 Guidance on the Treatment of Antimicrobial-Resistant Gram-Negative Infections. Clin. Infect. Dis. 2024, ciae403. [Google Scholar] [CrossRef] [PubMed]
- Makris, D.; Petinaki, E.; Tsolaki, V.; Manoulakas, E.; Mantzarlis, K.; Apostolopoulou, O.; Sfyras, D.; Zakynthinos, E. Colistin versus Colistin Combined with Ampicillin. Sulbactam for Multiresistant Acinetobacter baumannii Ventilator. Associated Pneumonia Treatment: An Open. Label Prospective Study. Indian J. Crit. Care Med. 2018, 22, 67–77. [Google Scholar] [CrossRef] [PubMed]
- Bassetti, M.; Righi, E. Eravacycline for the Treatment of Intra-Abdominal Infections. Expert Opin. Investig. Drugs 2014, 23, 1575–1584. [Google Scholar] [CrossRef] [PubMed]
- Tsilibaris, V.; Maenhaut-Michel, G.; Van Melderen, L. Biological Roles of the Lon ATP-Dependent Protease. Res. Microbiol. 2006, 157, 701–713. [Google Scholar] [CrossRef]
- Xu, C.; Wei, X.; Jin, Y.; Bai, F.; Cheng, Z.; Chen, S.; Pan, X.; Wu, W. Development of Resistance to Eravacycline by Klebsiella pneumoniae and Collateral Sensitivity-Guided Design of Combination Therapies. Microbiol. Spectr. 2022, 10, e01390-22. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Hua, X.; Xu, Q.; Yang, Y.; Zhang, L.; He, J.; Mu, X.; Hu, L.; Leptihn, S.; Yu, Y. Mechanism of Eravacycline Resistance in Acinetobacter Baumannii Mediated by a Deletion Mutation in the Sensor Kinase AdeS, Leading to Elevated Expression of the Efflux Pump AdeABC. Infect. Genet. Evol. 2020, 80, 104185. [Google Scholar] [CrossRef] [PubMed]
- Shi, Z.; Zhang, J.; Tian, L.; Xin, L.; Liang, C.; Ren, X.; Li, M. A Comprehensive Overview of the Antibiotics Approved in the Last Two Decades: Retrospects and Prospects. Molecules 2023, 28, 1762. [Google Scholar] [CrossRef] [PubMed]
- Xiao, X.-Y.; Hunt, D.K.; Zhou, J.; Clark, R.B.; Dunwoody, N.; Fyfe, C.; Grossman, T.H.; O’Brien, W.J.; Plamondon, L.; Rönn, M.; et al. Fluorocyclines. 1. 7-Fluoro-9-Pyrrolidinoacetamido-6-Demethyl-6-Deoxytetracycline: A Potent, Broad Spectrum Antibacterial Agent. J. Med. Chem. 2012, 55, 597–605. [Google Scholar] [CrossRef] [PubMed]
- Eljaaly, K.; Alharbi, A.; Alshehri, S.; Ortwine, J.K.; Pogue, J.M. Plazomicin: A Novel Aminoglycoside for the Treatment of Resistant Gram-Negative Bacterial Infections. Drugs 2019, 79, 243–269. [Google Scholar] [CrossRef]
- Tang, H.-J.; Lai, C.-C. Plazomicin-Associated Nephrotoxicity. Clin. Infect. Dis. 2020, 71, 1130–1131. [Google Scholar] [CrossRef]
- Jospe-Kaufman, M.; Siomin, L.; Fridman, M. The Relationship between the Structure and Toxicity of Aminoglycoside Antibiotics. Bioorg. Med. Chem. Lett. 2020, 30, 127218. [Google Scholar] [CrossRef] [PubMed]
- Adhikary, S.; Duggal, M.K.; Nagendran, S.; Chintamaneni, M.; Tuli, H.S.; Kaur, G. Lefamulin: A New Hope in the Field of Community-Acquired Bacterial Pneumonia. Curr. Pharmacol. Rep. 2022, 8, 418–426. [Google Scholar] [CrossRef]
- Kocsis, B.; Gulyás, D.; Szabó, D. Delafloxacin, Finafloxacin, and Zabofloxacin: Novel Fluoroquinolones in the Antibiotic Pipeline. Antibiotics 2021, 10, 1506. [Google Scholar] [CrossRef]
- Baym, M.; Stone, L.K.; Kishony, R. Multidrug Evolutionary Strategies to Reverse Antibiotic Resistance. Science 2016, 351, aad3292. [Google Scholar] [CrossRef]
- Rybak, M.J.; McGrath, B.J. Combination Antimicrobial Therapy for Bacterial Infections. Drugs 1996, 52, 390–405. [Google Scholar] [CrossRef] [PubMed]
- Bulik, C.C.; Nicolau, D.P. Double-Carbapenem Therapy for Carbapenemase-Producing Klebsiella pneumoniae. Antimicrob. Agents Chemother. 2011, 55, 3002–3004. [Google Scholar] [CrossRef] [PubMed]
- Giamarellou, H.; Galani, L.; Baziaka, F.; Karaiskos, I. Effectiveness of a Double-Carbapenem Regimen for Infections in Humans Due to Carbapenemase-Producing Pandrug-Resistant Klebsiella pneumoniae. Antimicrob. Agents Chemother. 2013, 57, 2388–2390. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wang, J.; Wang, R.; Cai, Y. Double-Carbapenem Therapy in the Treatment of Multidrug Resistant Gram-Negative Bacterial Infections: A Systematic Review and Meta-Analysis. BMC Infect. Dis. 2020, 20, 408. [Google Scholar] [CrossRef] [PubMed]
- Lodise, T.P.; Smith, N.M.; O’Donnell, N.; Eakin, A.E.; Holden, P.N.; Boissonneault, K.R.; Zhou, J.; Tao, X.; Bulitta, J.B.; Fowler, V.G.; et al. Determining the Optimal Dosing of a Novel Combination Regimen of Ceftazidime/Avibactam with Aztreonam against NDM-1-Producing Enterobacteriaceae Using a Hollow-Fibre Infection Model. J. Antimicrob. Chemother. 2020, 75, 2622–2632. [Google Scholar] [CrossRef] [PubMed]
- Park, H.J.; Cho, J.H.; Kim, H.J.; Han, S.H.; Jeong, S.H.; Byun, M.K. Colistin Monotherapy versus Colistin/Rifampicin Combination Therapy in Pneumonia Caused by Colistin-Resistant Acinetobacter Baumannii: A Randomised Controlled Trial. J. Glob. Antimicrob. Resist. 2019, 17, 66–71. [Google Scholar] [CrossRef] [PubMed]
- Shariati, A.; Arshadi, M.; Khosrojerdi, M.A.; Abedinzadeh, M.; Ganjalishahi, M.; Maleki, A.; Heidary, M.; Khoshnood, S. The Resistance Mechanisms of Bacteria against Ciprofloxacin and New Approaches for Enhancing the Efficacy of This Antibiotic. Front. Public Health 2022, 10, 1025633. [Google Scholar] [CrossRef] [PubMed]
- D’Costa, V.M.; McGrann, K.M.; Hughes, D.W.; Wright, G.D. Sampling the Antibiotic Resistome. Science 2006, 311, 374–377. [Google Scholar] [CrossRef]
- Dhand, A.; Sakoulas, G. Daptomycin in Combination with Other Antibiotics for the Treatment of Complicated Methicillin-Resistant Staphylococcus Aureus Bacteremia. Clin. Ther. 2014, 36, 1303–1316. [Google Scholar] [CrossRef] [PubMed]
- Rand, K.H.; Houck, H.J. Synergy of Daptomycin with Oxacillin and Other β-Lactams against Methicillin-Resistant Staphylococcus aureus. Antimicrob. Agents Chemother. 2004, 48, 2871–2875. [Google Scholar] [CrossRef]
- Lefebvre, M.; Jacqueline, C.; Amador, G.; Le Mabecque, V.; Miegeville, A.; Potel, G.; Caillon, J.; Asseray, N. Efficacy of Daptomycin Combined with Rifampicin for the Treatment of Experimental Meticillin-Resistant Staphylococcus Aureus (MRSA) Acute Osteomyelitis. Int. J. Antimicrob. Agents 2010, 36, 542–544. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Peng, Y. Synergistic Effect of Clinically Used Antibiotics and Peptide Antibiotics against Gram-Positive and Gram-Negative Bacteria. Exp. Ther. Med. 2013, 6, 1000–1004. [Google Scholar] [CrossRef] [PubMed]
- Konwar, A.N.; Hazarika, S.N.; Bharadwaj, P.; Thakur, D. Emerging Non-Traditional Approaches to Combat Antibiotic Resistance. Curr. Microbiol. 2022, 79, 330. [Google Scholar] [CrossRef]
- Stoitsova, S.; Paunova-Krasteva, T.; Dimitrova, P.D.; Damyanova, T. The Concept for the Antivirulence Therapeutics Approach as Alternative to Antibiotics: Hope or Still a Fiction? Biotechnol. Biotechnol. Equip. 2022, 36, 697–705. [Google Scholar] [CrossRef]
- Đukanović, S.; Ganić, T.; Lončarević, B.; Cvetković, S.; Nikolić, B.; Tenji, D.; Randjelović, D.; Mitić-Ćulafić, D. Elucidating the Antibiofilm Activity of Frangula Emodin against Staphylococcus Aureus Biofilms. J. Appl. Microbiol. 2022, 132, 1840–1855. [Google Scholar] [CrossRef] [PubMed]
- Sulakvelidze, A.; Alavidze, Z.; Morris, J.G. Bacteriophage Therapy. Antimicrob. Agents Chemother. 2001, 45, 649–659. [Google Scholar] [CrossRef] [PubMed]
- Hibstu, Z.; Belew, H.; Akelew, Y.; Mengist, H.M. Phage Therapy: A Different Approach to Fight Bacterial Infections. Biologics 2022, 16, 173–186. [Google Scholar] [CrossRef]
- Brives, C.; Pourraz, J. Phage Therapy as a Potential Solution in the Fight against AMR: Obstacles and Possible Futures. Palgrave Commun. 2020, 6, 100. [Google Scholar] [CrossRef]
- Sawa, T.; Moriyama, K.; Kinoshita, M. Current Status of Bacteriophage Therapy for Severe Bacterial Infections. J. Intensive Care 2024, 12, 44. [Google Scholar] [CrossRef]
- Levin, B.R. The accessory genetic elements of bacteria: Existence conditions and (co)evolution. Curr. Opin. Genet. Dev. 1993, 3, 849–854. [Google Scholar] [CrossRef] [PubMed]
- Newsom, S.; Parameshwaran, H.P.; Martin, L.; Rajan, R. The CRISPR-Cas Mechanism for Adaptive Immunity and Alternate Bacterial Functions Fuels Diverse Biotechnologies. Front. Cell Infect. Microbiol. 2021, 10, 619763. [Google Scholar] [CrossRef] [PubMed]
- Shabbir, M.A.B.; Hao, H.; Shabbir, M.Z.; Hussain, H.I.; Iqbal, Z.; Ahmed, S.; Sattar, A.; Iqbal, M.; Li, J.; Yuan, Z. Survival and Evolution of CRISPR–Cas System in Prokaryotes and Its Applications. Front. Immunol. 2016, 7, 375. [Google Scholar] [CrossRef] [PubMed]
- Barrangou, R.; Marraffini, L.A. CRISPR-Cas Systems: Prokaryotes Upgrade to Adaptive Immunity. Mol. Cell 2014, 54, 234–244. [Google Scholar] [CrossRef]
- Yang, Q.; Le, S.; Zhu, T.; Wu, N. Regulations of Phage Therapy across the World. Front. Microbiol. 2023, 14, 1250848. [Google Scholar] [CrossRef] [PubMed]
- Hotinger, J.A.; Morris, S.T.; May, A.E. The Case against Antibiotics and for Anti-Virulence Therapeutics. Microorganisms 2021, 9, 2049. [Google Scholar] [CrossRef] [PubMed]
- Ganić, T.; Vuletić, S.; Nikolić, B.; Stevanović, M.; Kuzmanović, M.; Kekić, D.; Đurović, S.; Cvetković, S.; Mitić-Ćulafić, D. Cinnamon Essential Oil and Its Emulsion as Efficient Antibiofilm Agents to Combat Acinetobacter Baumannii. Front. Microbiol. 2022, 13, 989667. [Google Scholar] [CrossRef]
- Đukanović, S.; Cvetković, S.; Lončarević, B.; Lješević, M.; Nikolić, B.; Simin, N.; Bekvalac, K.; Kekić, D.; Mitić-Ćulafić, D. Antistaphylococcal and Biofilm Inhibitory Activities of Frangula Alnus Bark Ethyl-Acetate Extract. Ind. Crops Prod. 2020, 158, 113013. [Google Scholar] [CrossRef]
- Tomić, N.; Stevanović, M.M.; Filipović, N.; Ganić, T.; Nikolić, B.; Gajić, I.; Ćulafić, D.M. Resveratrol/Selenium Nanocomposite with Antioxidative and Antibacterial Properties. Nanomaterials 2024, 14, 368. [Google Scholar] [CrossRef]
- Mazurkiewicz-Pisarek, A.; Baran, J.; Ciach, T. Antimicrobial Peptides: Challenging Journey to the Pharmaceutical, Biomedical, and Cosmeceutical Use. Int. J. Mol. Sci. 2023, 24, 9031. [Google Scholar] [CrossRef]
- Xuan, J.; Feng, W.; Wang, J.; Wang, R.; Zhang, B.; Bo, L.; Chen, Z.-S.; Yang, H.; Sun, L. Antimicrobial Peptides for Combating Drug-Resistant Bacterial Infections. Drug Resist. Updates 2023, 68, 100954. [Google Scholar] [CrossRef]
- Boparai, J.K.; Sharma, P.K. Mini Review on Antimicrobial Peptides, Sources, Mechanism and Recent Applications. Protein Pept. Lett. 2019, 27, 4–16. [Google Scholar] [CrossRef] [PubMed]
- McManus, A.M.; Dawson, N.F.; Wade, J.D.; Carrington, L.E.; Winzor, D.J.; Craik, D.J. Three-Dimensional Structure of RK-1: A Novel α-Defensin Peptide. Biochemistry 2000, 39, 15757–15764. [Google Scholar] [CrossRef] [PubMed]
- Rozek, A.; Friedrich, C.L.; Hancock, R.E.W. Structure of the Bovine Antimicrobial Peptide Indolicidin Bound to Dodecylphosphocholine and Sodium Dodecyl Sulfate Micelles, Biochemistry 2000, 39, 15765–15774. [Google Scholar] [CrossRef]
- Peschel, A.; Sahl, H.-G. The Co-Evolution of Host Cationic Antimicrobial Peptides and Microbial Resistance. Nat. Rev. Microbiol. 2006, 4, 529–536. [Google Scholar] [CrossRef] [PubMed]
- Benfield, A.H.; Henriques, S.T. Mode-of-Action of Antimicrobial Peptides: Membrane Disruption vs. Intracellular Mechanisms. Front. Med. Technol. 2020, 2, 610997. [Google Scholar] [CrossRef] [PubMed]
- Moretta, A.; Scieuzo, C.; Petrone, A.M.; Salvia, R.; Manniello, M.D.; Franco, A.; Lucchetti, D.; Vassallo, A.; Vogel, H.; Sgambato, A.; et al. Antimicrobial Peptides: A New Hope in Biomedical and Pharmaceutical Fields. Front. Cell Infect. Microbiol. 2021, 11, 668632. [Google Scholar] [CrossRef] [PubMed]
- Okuda, K.; Zendo, T.; Sugimoto, S.; Iwase, T.; Tajima, A.; Yamada, S.; Sonomoto, K.; Mizunoe, Y. Effects of Bacteriocins on Methicillin-Resistant Staphylococcus aureus Biofilm. Antimicrob. Agents Chemother. 2013, 57, 5572–5579. [Google Scholar] [CrossRef]
- Luca, V.; Stringaro, A.; Colone, M.; Pini, A.; Mangoni, M.L. Esculentin(1-21), an Amphibian Skin Membrane-Active Peptide with Potent Activity on Both Planktonic and Biofilm Cells of the Bacterial Pathogen Pseudomonas Aeruginosa. Cell. Mol. Life Sci. 2013, 70, 2773–2786. [Google Scholar] [CrossRef] [PubMed]
- Luong, H.X.; Thanh, T.T.; Tran, T.H. Antimicrobial Peptides—Advances in Development of Therapeutic Applications. Life Sci. 2020, 260, 118407. [Google Scholar] [CrossRef] [PubMed]
- Baptista, P.V.; McCusker, M.P.; Carvalho, A.; Ferreira, D.A.; Mohan, N.M.; Martins, M.; Fernandes, A.R. Nano-Strategies to Fight Multidrug Resistant Bacteria—“A Battle of the Titans”. Front Microbiol. 2018, 9, 1441. [Google Scholar] [CrossRef]
- Mondal, S.K.; Chakraborty, S.; Manna, S.; Mandal, S.M. Antimicrobial Nanoparticles: Current Landscape and Future Challenges. RSC Pharm. 2024, 1, 388–402. [Google Scholar] [CrossRef]
- Ioannou, P.; Baliou, S.; Samonis, G. Nanotechnology in the Diagnosis and Treatment of Antibiotic-Resistant Infections. Antibiotics 2024, 13, 121. [Google Scholar] [CrossRef] [PubMed]
- Aflakian, F.; Mirzavi, F.; Aiyelabegan, H.T.; Soleimani, A.; Gholizadeh Navashenaq, J.; Karimi-Sani, I.; Rafati Zomorodi, A.; Vakili-Ghartavol, R. Nanoparticles-Based Therapeutics for the Management of Bacterial Infections: A Special Emphasis on FDA Approved Products and Clinical Trials. Eur. J. Pharm. Sci. 2023, 188, 106515. [Google Scholar] [CrossRef]
- Hetta, H.F.; Ramadan, Y.N.; Al-Harbi, A.I.; Ahmed, A.E.; Battah, B.; Abd Ellah, N.H.; Zanetti, S.; Donadu, M.G. Nanotechnology as a Promising Approach to Combat Multidrug Resistant Bacteria: A Comprehensive Review and Future Perspectives. Biomedicines 2023, 11, 413. [Google Scholar] [CrossRef]
- Abed, N.; Couvreur, P. Nanocarriers for Antibiotics: A Promising Solution to Treat Intracellular Bacterial Infections. Int. J. Antimicrob. Agents 2014, 43, 485–496. [Google Scholar] [CrossRef]
- Shukla, R.K.; Badiye, A.; Vajpayee, K.; Kapoor, N. Genotoxic Potential of Nanoparticles: Structural and Functional Modifications in DNA. Front. Genet. 2021, 12, 728250. [Google Scholar] [CrossRef]
- Li, H.; Xu, H. Mechanisms of Bacterial Resistance to Environmental Silver and Antimicrobial Strategies for Silver: A Review. Environ. Res. 2024, 248, 118313. [Google Scholar] [CrossRef] [PubMed]
- McCusker, M.P.; Alves Ferreira, D.; Cooney, D.; Martins Alves, B.; Fanning, S.; Pagès, J.-M.; Martins, M.; Davin-Regli, A. Modulation of Antimicrobial Resistance in Clinical Isolates of Enterobacter Aerogenes: A Strategy Combining Antibiotics and Chemosensitisers. J. Glob. Antimicrob. Resist. 2019, 16, 187–198. [Google Scholar] [CrossRef]
- Laws, M.; Shaaban, A.; Rahman, K.M. Antibiotic Resistance Breakers: Current Approaches and Future Directions. FEMS Microbiol. Rev. 2019, 43, 490–516. [Google Scholar] [CrossRef] [PubMed]
- Gill, E.E.; Franco, O.L.; Hancock, R.E.W. Antibiotic Adjuvants: Diverse Strategies for Controlling Drug-Resistant Pathogens. Chem. Biol. Drug Des. 2015, 85, 56–78. [Google Scholar] [CrossRef] [PubMed]
- Allen, R.C.; Brown, S.P. Modified Antibiotic Adjuvant Ratios Can Slow and Steer the Evolution of Resistance: Co-Amoxiclav as a Case Study. mBio 2019, 10, e01831-19. [Google Scholar] [CrossRef] [PubMed]
- Melander, R.J.; Melander, C. The Challenge of Overcoming Antibiotic Resistance: An Adjuvant Approach? ACS Infect. Dis. 2017, 3, 559–563. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.; Yasmeen, N.; Pandey, A.; Ahmad Chaudhary, A.; Alawam, A.S.; Ahmad Rudayni, H.; Islam, A.; Lakhawat, S.S.; Sharma, P.K.; Shahid, M. Antibiotic Adjuvants: Synergistic Tool to Combat Multi-Drug Resistant Pathogens. Front. Cell Infect. Microbiol. 2023, 13, 1293633. [Google Scholar] [CrossRef] [PubMed]
- Varma, D.M.; Zahid, M.S.H.; Bachelder, E.M.; Ainslie, K.M. Formulation of Host-Targeted Therapeutics against Bacterial Infections. Transl. Res. 2020, 220, 98–113. [Google Scholar] [CrossRef] [PubMed]
- Zumla, A.; Rao, M.; Wallis, R.S.; Kaufmann, S.H.E.; Rustomjee, R.; Mwaba, P.; Vilaplana, C.; Yeboah-Manu, D.; Chakaya, J.; Ippolito, G.; et al. Host-Directed Therapies for Infectious Diseases: Current Status, Recent Progress, and Future Prospects. Lancet Infect. Dis. 2016, 16, e47–e63. [Google Scholar] [CrossRef] [PubMed]
- Jeong, E.K.; Lee, H.J.; Jung, Y.J. Host-Directed Therapies for Tuberculosis. Pathogens 2022, 11, 1291. [Google Scholar] [CrossRef]
- Natsheh, I.Y.; Alsaleh, M.M.; Alkhawaldeh, A.K.; Albadawi, D.K.; Darwish, M.M.; Shammout, M.J.A. The Dark Side of Drug Repurposing. From Clinical Trial Challenges to Antimicrobial Resistance: Analysis Based on Three Major Fields. Drug Target Insights 2024, 18, 8–19. [Google Scholar] [CrossRef] [PubMed]
- Glajzner, P.; Bernat, A.; Jasińska-Stroschein, M. Improving the Treatment of Bacterial Infections Caused by Multidrug-Resistant Bacteria through Drug Repositioning. Front. Pharmacol. 2024, 15, 1397602. [Google Scholar] [CrossRef] [PubMed]
- Taheri-Araghi, S. Synergistic Action of Antimicrobial Peptides and Antibiotics: Current Understanding and Future Directions. Front. Microbiol. 2024, 15, 1390765. [Google Scholar] [CrossRef] [PubMed]
- Almaaytah, A.; Abualhaijaa, A.; Alqudah, O. The Evaluation of the Synergistic Antimicrobial and Antibiofilm Activity of AamAP1-Lysine with Conventional Antibiotics against Representative Resistant Strains of Both Gram-Positive and Gram-Negative Bacteria. Infect. Drug Resist. 2019, 12, 1371–1380. [Google Scholar] [CrossRef]
- Jorge, P.; Lourenço, A.; Pereira, M.O. New Trends in Peptide-Based Anti-Biofilm Strategies: A Review of Recent Achievements and Bioinformatic Approaches. Biofouling 2012, 28, 1033–1061. [Google Scholar] [CrossRef]
- Yu, G.; Baeder, D.Y.; Regoes, R.R.; Rolff, J. Combination Effects of Antimicrobial Peptides. Antimicrob. Agents Chemother. 2016, 60, 1717–1724. [Google Scholar] [CrossRef] [PubMed]
- Mhlongo, J.T.; Waddad, A.Y.; Albericio, F.; de la Torre, B.G. Antimicrobial Peptide Synergies for Fighting Infectious Diseases. Adv. Sci. 2023, 10, 2300472. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Starr, C.G.; Wimley, W.C. A Lack of Synergy between Membrane-Permeabilizing Cationic Antimicrobial Peptides and Conventional Antibiotics. Biochim. Biophys. Acta Biomembranes 2015, 1848, 8–15. [Google Scholar] [CrossRef] [PubMed]
- Shi, G.; Kang, X.; Dong, F.; Liu, Y.; Zhu, N.; Hu, Y.; Xu, H.; Lao, X.; Zheng, H. DRAMP 3.0: An Enhanced Comprehensive Data Repository of Antimicrobial Peptides. Nucleic Acids Res. 2022, 50, D488–D496. [Google Scholar] [CrossRef] [PubMed]
- Thapa, R.K.; Diep, D.B.; Tønnesen, H.H. Topical Antimicrobial Peptide Formulations for Wound Healing: Current Developments and Future Prospects. Acta Biomater. 2020, 103, 52–67. [Google Scholar] [CrossRef] [PubMed]
- Huan, Y.; Kong, Q.; Mou, H.; Yi, H. Antimicrobial Peptides: Classification, Design, Application and Research Progress in Multiple Fields. Front. Microbiol. 2020, 11, 582779. [Google Scholar] [CrossRef] [PubMed]
- Theuretzbacher, U.; Baraldi, E.; Ciabuschi, F.; Callegari, S. Challenges and Shortcomings of Antibacterial Discovery Projects. Clin. Microbiol. Infect. 2023, 29, 610–615. [Google Scholar] [CrossRef]
- Luepke, K.H.; Mohr, J.F. The Antibiotic Pipeline: Reviving Research and Development and Speeding Drugs to Market. Expert Rev. Anti Infect. Ther. 2017, 15, 425–433. [Google Scholar] [CrossRef] [PubMed]
- Butler, M.S.; Gigante, V.; Sati, H.; Paulin, S.; Al-Sulaiman, L.; Rex, J.H.; Fernandes, P.; Arias, C.A.; Paul, M.; Thwaites, G.E.; et al. Analysis of the Clinical Pipeline of Treatments for Drug-Resistant Bacterial Infections: Despite Progress, More Action Is Needed. Antimicrob. Agents Chemother. 2022, 66, e0199121. [Google Scholar] [CrossRef] [PubMed]
- Boyd, N.K.; Teng, C.; Frei, C.R. Brief Overview of Approaches and Challenges in New Antibiotic Development: A Focus on Drug Repurposing. Front. Cell Infect. Microbiol. 2021, 11, 684515. [Google Scholar] [CrossRef] [PubMed]
- Gigante, V.; Sati, H.; Beyer, P. Recent Advances and Challenges in Antibacterial Drug Development. ADMET DMPK 2022, 10, 147–151. [Google Scholar] [CrossRef] [PubMed]
- Committee on New Directions in the Study of Antimicrobial Therapeutics. New Classes of Antimicrobials. In Challenges for the Development of New Antimicrobials—Rethinking the Approaches; National Academies Press: Washington, DC, USA, 2006. [Google Scholar]
- Keck, J.M.; Viteri, A.; Schultz, J.; Fong, R.; Whitman, C.; Poush, M.; Martin, M. New Agents Are Coming, and So Is the Resistance. Antibiotics 2024, 13, 648. [Google Scholar] [CrossRef] [PubMed]
- Todd, M.H.; Klug, D.M.; Idiris, F.I.M.; Blaskovich, M.A.T.; von Delft, F.; Dowson, C.G.; Kirchhelle, C.; Roberts, A.P.; Singer, A.C. There Is No Market for New Antibiotics: This Allows an Open Approach to Research and Development. Welcome Open Res. 2021, 6, 146. [Google Scholar] [CrossRef]
- Schurer, M.; Patel, R.; van Keep, M.; Horgan, J.; Matthijsse, S.; Madin-Warburton, M. Recent Advances in Addressing the Market Failure of New Antimicrobials: Learnings from NICE’s Subscription-Style Payment Model. Front. Med. Technol. 2023, 5, 1010247. [Google Scholar] [CrossRef]
- Wells, N.; Nguyen, V.K.; Harbarth, S. Novel Insights from Financial Analysis of the Failure to Commercialise Plazomicin: Implications for the Antibiotic Investment Ecosystem. Humanit. Soc. Sci. Commun. 2024, 11, 941. [Google Scholar] [CrossRef]
- Dutescu, I.A.; Hillie, S.A. Encouraging the Development of New Antibiotics: Are Financial Incentives the Right Way Forward? A Systematic Review and Case Study. Infect. Drug Resist. 2021, 14, 415–434. [Google Scholar] [CrossRef]
- Ren, M.; So, A.D.; Chandy, S.J.; Mpundu, M.; Peralta, A.Q.; Åkerfeldt, K.; Sjöblom, A.K.; Cars, O. Equitable Access to Antibiotics: A Core Element and Shared Global Responsibility for Pandemic Preparedness and Response. J. Law Med. Ethics 2022, 50, 34–39. [Google Scholar] [CrossRef] [PubMed]
- Papapetropoulos, A.; Topouzis, S.; Alexander, S.P.H.; Cortese-Krott, M.; Kendall, D.A.; Martemyanov, K.A.; Mauro, C.; Nagercoil, N.; Panettieri, R.A.; Patel, H.H.; et al. Novel Drugs Approved by the EMA, the FDA, and the MHRA in 2023: A Year in Review. Br. J. Pharmacol. 2024, 181, 1553–1575. [Google Scholar] [CrossRef]
- Årdal, C.; Lacotte, Y.; Ploy, M.C. Financing Pull Mechanisms for Antibiotic-Related Innovation: Opportunities for Europe. Clin. Infect. Dis. 2020, 71, 1994–1999. [Google Scholar] [CrossRef]
- Global AMR R&D Hub; WHO. Incentivising the Development of New Antibacterial Treatments. 2023. Available online: https://globalamrhub.org/news/incentivising-the-development-of-new-antibacterial-treatments-progress-report-by-the-global-amr-rd-hub-and-who-2023/ (accessed on 12 January 2025).
- Anderson, M.; Panteli, D.; van Kessel, R.; Ljungqvist, G.; Colombo, F.; Mossialos, E. Challenges and Opportunities for Incentivising Antibiotic Research and Development in Europe. Lancet Reg. Health Eur. 2023, 33, 100705. [Google Scholar] [CrossRef]
- Allsop, A.E. Bacterial Genome Sequencing and Drug Discovery. Curr. Opin. Biotechnol. 1998, 9, 637–642. [Google Scholar] [CrossRef] [PubMed]
- Alkatheri, A.H.; Yap, P.S.-X.; Abushelaibi, A.; Lai, K.-S.; Cheng, W.-H.; Erin Lim, S.-H. Microbial Genomics: Innovative Targets and Mechanisms. Antibiotics 2023, 12, 190. [Google Scholar] [CrossRef] [PubMed]
- Read, T.D.; Massey, R.C. Characterizing the Genetic Basis of Bacterial Phenotypes Using Genome-Wide Association Studies: A New Direction for Bacteriology. Genome Med. 2014, 6, 109. [Google Scholar] [CrossRef] [PubMed]
- Medema, M.H.; Fischbach, M.A. Computational Approaches to Natural Product Discovery. Nat. Chem. Biol. 2015, 11, 639–648. [Google Scholar] [CrossRef] [PubMed]
- Bikard, D.; Barrangou, R. Using CRISPR-Cas Systems as Antimicrobials. Curr. Opin. Microbiol. 2017, 37, 155–160. [Google Scholar] [CrossRef]
- Ebrahimi, S.; Khosravi, M.A.; Raz, A.; Karimipoor, M.; Parvizi, P. CRISPR-Cas Technology as a Revolutionary Genome Editing Tool: Mechanisms and Biomedical Applications. Iran Biomed. J. 2023, 27, 219–246. [Google Scholar] [CrossRef]
- Handelsman, J. Metagenomics: Application of Genomics to Uncultured Microorganisms. Microbiol. Mol. Biol. Rev. 2004, 68, 669–685. [Google Scholar] [CrossRef]
- Schreiber, S.L. Target-Oriented and Diversity-Oriented Organic Synthesis in Drug Discovery. Science 2000, 287, 1964–1969. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. 2023 Antibacterial Agents in Clinical and Preclinical Development: An Overview and Analysis; World Health Organization: Geneva, Switzerland, 2023. [Google Scholar]
- Mühlberg, E.; Umstätter, F.; Kleist, C.; Domhan, C.; Mier, W.; Uhl, P. Renaissance of Vancomycin: Approaches for Breaking Antibiotic Resistance in Multidrug-Resistant Bacteria. Can. J. Microbiol. 2020, 66, 11–16. [Google Scholar] [CrossRef] [PubMed]
- Guan, D.; Chen, F.; Qiu, Y.; Jiang, B.; Gong, L.; Lan, L.; Huang, W. Sulfonium, an Underestimated Moiety for Structural Modification, Alters the Antibacterial Profile of Vancomycin Against Multidrug-Resistant Bacteria. Angew. Chem. Int. Ed. 2019, 58, 6678–6682. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Beta-Lactamases | Enzyme | Active Site Residue | Host Bacteria | Target | Beta-Lactamase Inhibitor | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 2nd Generation | 1st Generation | ||||||||||
| Ambler Class | Avibactam | Enmetazobactam | Vaborbactam | Relebactam | Tazobactam | Clavulanic acid | Sulbactam | ||||
| A | Narrow spectrum (SHV-1, TEM-1) | Serine-containing active site | G+ and G− | Penicillins, cephalosporins, and monobactams | |||||||
| ESBL (CTX-M, TEM-type, SHV-type) | G− | Extended-spectrum cephalosporins and monobactams (exception—carbapenems, cephamycins) [17] | |||||||||
| Carbapenemase (KPC) | G− | BLAs including carbapenems | |||||||||
| B | Carbapenemase (NDM, IMP, VIM) | Zn-containing active site (MBLs) | G− | BLAs including carbapenems (exception—monobactams) | |||||||
| C | AmpC | Serine-containing active site | Primarily G− | Penicillins, monobactams, and cephalosporins, including ceftaroline and ceftobiprole (exception—cefepime) | |||||||
| D | Carbapenemase (OXA-48, other OXAs) | Serine-containing active site | Primarily G− | BLAs, including cephalosporins, cephems, and/or monobactams; some OXA-type enzymes are also capable of hydrolyzing carbapenems | |||||||
| Antibacterial Agent | Antibiotic Class | Approved by FDA | Clinical Indication | Spectrum of Activity | Reference |
|---|---|---|---|---|---|
| Eravacycline | Tetracyclines | 2018 | CIAI, cUTIs | G+, G−, anaerobes (except Pseudomonas aeruginosa) | [128,129] |
| Omadacycline | Tetracyclines | 2018 | SSTI, CABP | G+, G−, anaerobes (except P. aeruginosa) | [130] |
| Plazomicin | Aminoglycosides | 2018 | cUTI, BSI, VAP | ESBL and CRE | [131] |
| Lefamulin | Pleuromutilin | 2019 | CABP | G+, G−, Chlamydia trachomatis, Neisseria gonorrhoeae, Mycoplasma genitalium | [132] |
| Delafloxacin | Fluoroquinolones | 2017 | SSTI, BC, AOE | P. aeruginosa, Staphylococcus aureus | [133] |
| Lascufloxacin | Fluoroquinolones | 2019 (Japan) | RTI, SSTI | G+, G− | [134] |
| Levonadifloxacin | Fluoroquinolones | 2020 (India) | RTI, SSTI | G+, G− | [134] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gajic, I.; Tomic, N.; Lukovic, B.; Jovicevic, M.; Kekic, D.; Petrovic, M.; Jankovic, M.; Trudic, A.; Mitic Culafic, D.; Milenkovic, M.; et al. A Comprehensive Overview of Antibacterial Agents for Combating Multidrug-Resistant Bacteria: The Current Landscape, Development, Future Opportunities, and Challenges. Antibiotics 2025, 14, 221. https://doi.org/10.3390/antibiotics14030221
Gajic I, Tomic N, Lukovic B, Jovicevic M, Kekic D, Petrovic M, Jankovic M, Trudic A, Mitic Culafic D, Milenkovic M, et al. A Comprehensive Overview of Antibacterial Agents for Combating Multidrug-Resistant Bacteria: The Current Landscape, Development, Future Opportunities, and Challenges. Antibiotics. 2025; 14(3):221. https://doi.org/10.3390/antibiotics14030221
Chicago/Turabian StyleGajic, Ina, Nina Tomic, Bojana Lukovic, Milos Jovicevic, Dusan Kekic, Milos Petrovic, Marko Jankovic, Anika Trudic, Dragana Mitic Culafic, Marina Milenkovic, and et al. 2025. "A Comprehensive Overview of Antibacterial Agents for Combating Multidrug-Resistant Bacteria: The Current Landscape, Development, Future Opportunities, and Challenges" Antibiotics 14, no. 3: 221. https://doi.org/10.3390/antibiotics14030221
APA StyleGajic, I., Tomic, N., Lukovic, B., Jovicevic, M., Kekic, D., Petrovic, M., Jankovic, M., Trudic, A., Mitic Culafic, D., Milenkovic, M., & Opavski, N. (2025). A Comprehensive Overview of Antibacterial Agents for Combating Multidrug-Resistant Bacteria: The Current Landscape, Development, Future Opportunities, and Challenges. Antibiotics, 14(3), 221. https://doi.org/10.3390/antibiotics14030221