
Structure–Activity Study of the Antimicrobial Lipopeptide Humimycin A and Screening Against Multidrug-Resistant Staphylococcus aureus

 ,
,  ,
, 

Abstract

1. Introduction

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| # | Peptide | Lipid Chain | Peptide Sequence | C-Term | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | ||||
| 1 | Hum A [19] | 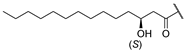 | Tyr | Ser | d-Tyr | Phe | d-Thr | Val | Val | COOH |
| 2 | Hum B [19] | Tyr | Ile | |||||||
| 3 | Hum W [24] | Trp | ||||||||
| 4 | Hum 17S [22] | Trp | d-Trp | Trp | ||||||
| 5 | Hum A_isoC15 | 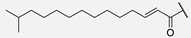 | ||||||||
| 6 | Hum A_isoC13 | 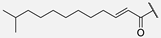 | ||||||||
| 7 | Hum W_geranoyl | 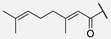 | Trp | |||||||
| 8 | Hum W_octanoyl |  | Trp | |||||||
| 9 | Hum W_mandelic | 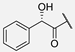 | Trp | |||||||
| 10 | Hum W_maleic | 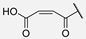 | Trp | |||||||
| 11 | Hum W_fumaric | 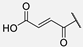 | Trp | |||||||
| 12 | Hum W1,3 |  | Trp | d-Trp | ||||||
| 13 | Hum W1,3-A6 | Trp | d-Trp | Ala | ||||||
| 14 | Hum W1,3,5 | Trp | d-Trp | d-Trp | ||||||
| 15 | Hum W amide | Trp | CONH2 | |||||||
| 16 | Hum W1,3 amide | Trp | d-Trp | CONH2 | ||||||
| 17 | Hum W1,3-A6 amide | Trp | d-Trp | Ala | CONH2 | |||||
2. Results
2.1. Synthesis of Humimycin Analogs
2.2. Antimicrobial Activity of the Humimycin Analogs Against Gram-Positive and Gram-Negative Bacteria
2.3. Antimicrobial Activity of the Humimycin Analogs Against S. aureus Isolates
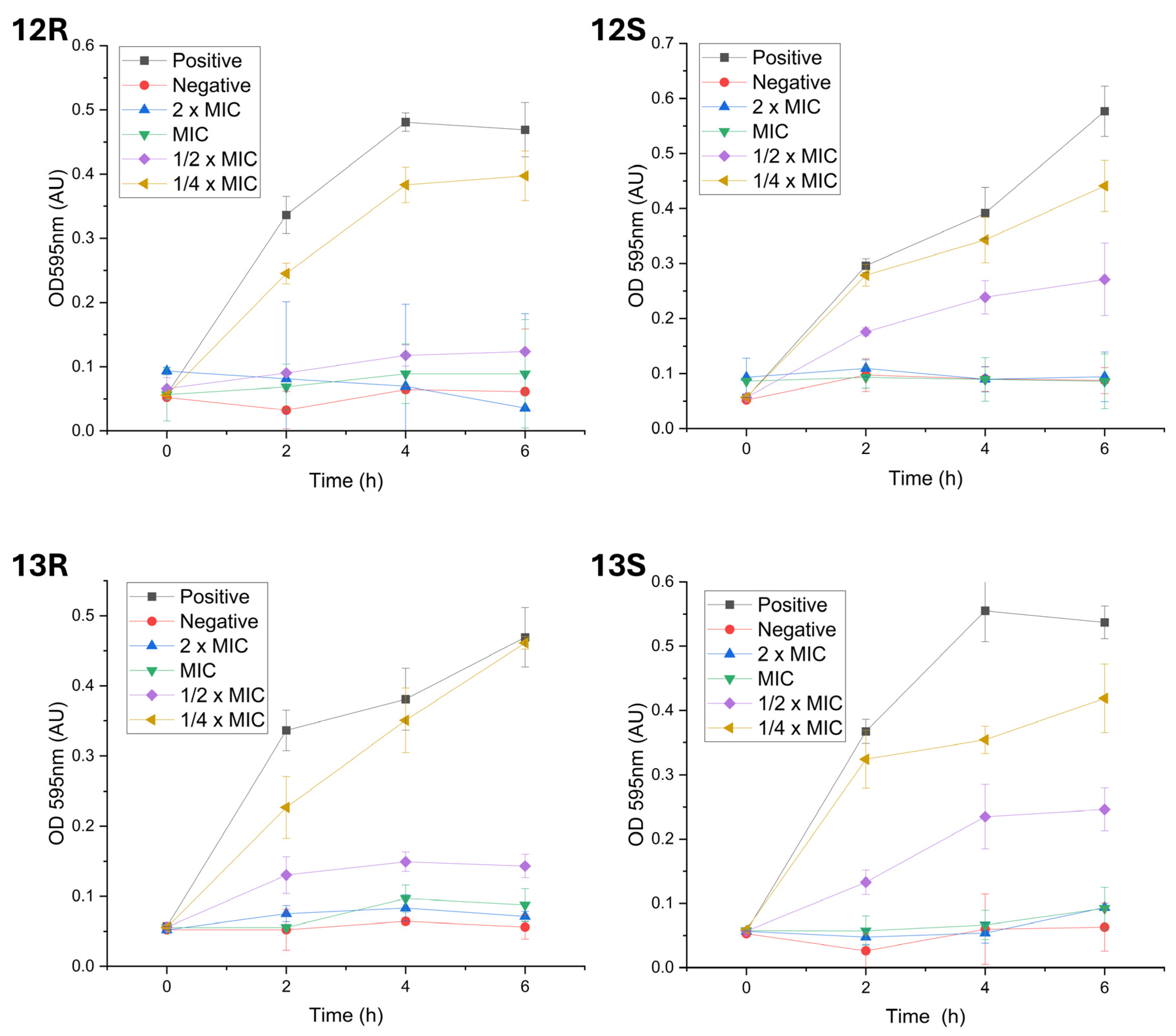
2.4. Impact of Humimycin Analogs on Bacterial Growth
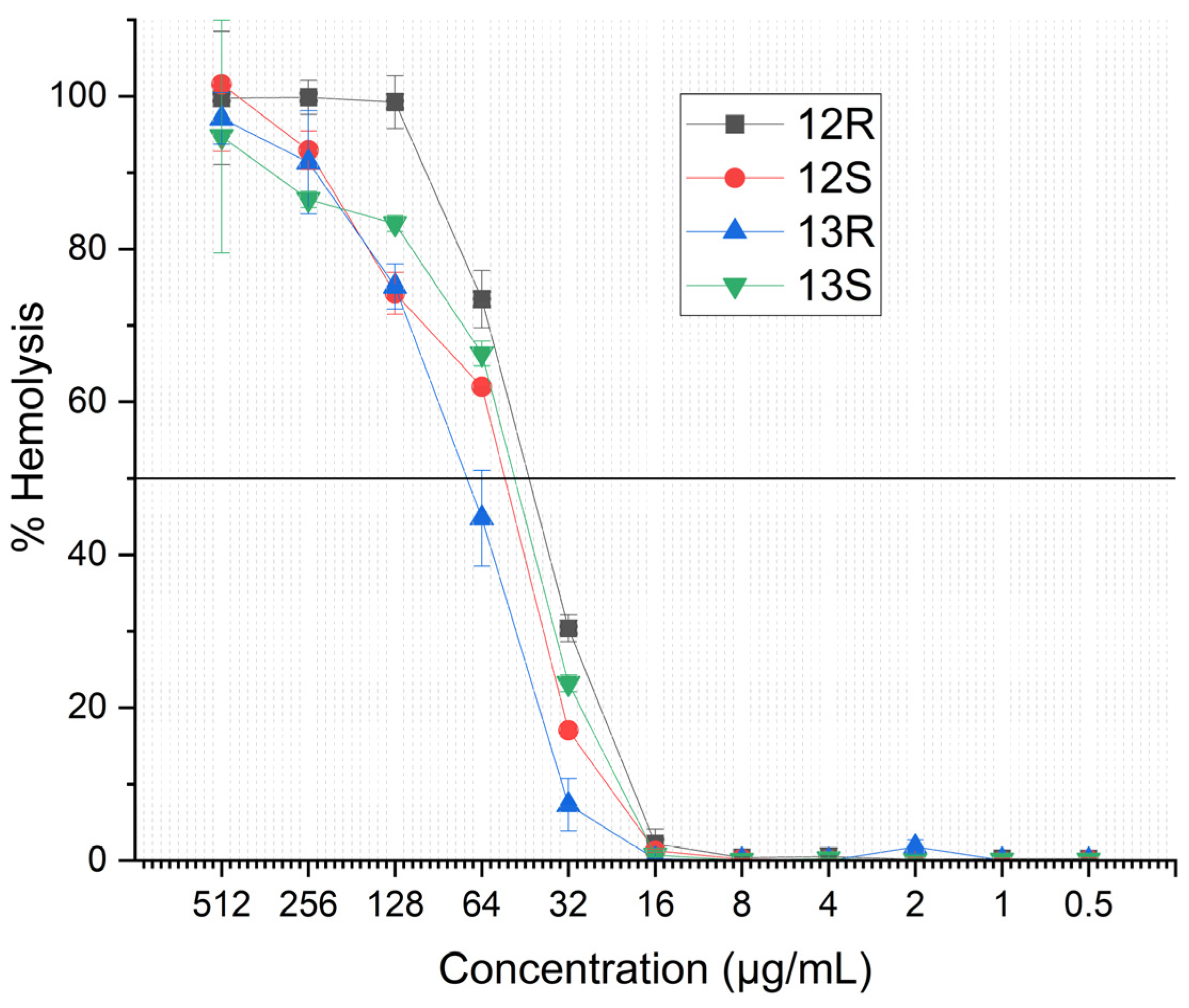
2.5. Hemolytic Activity
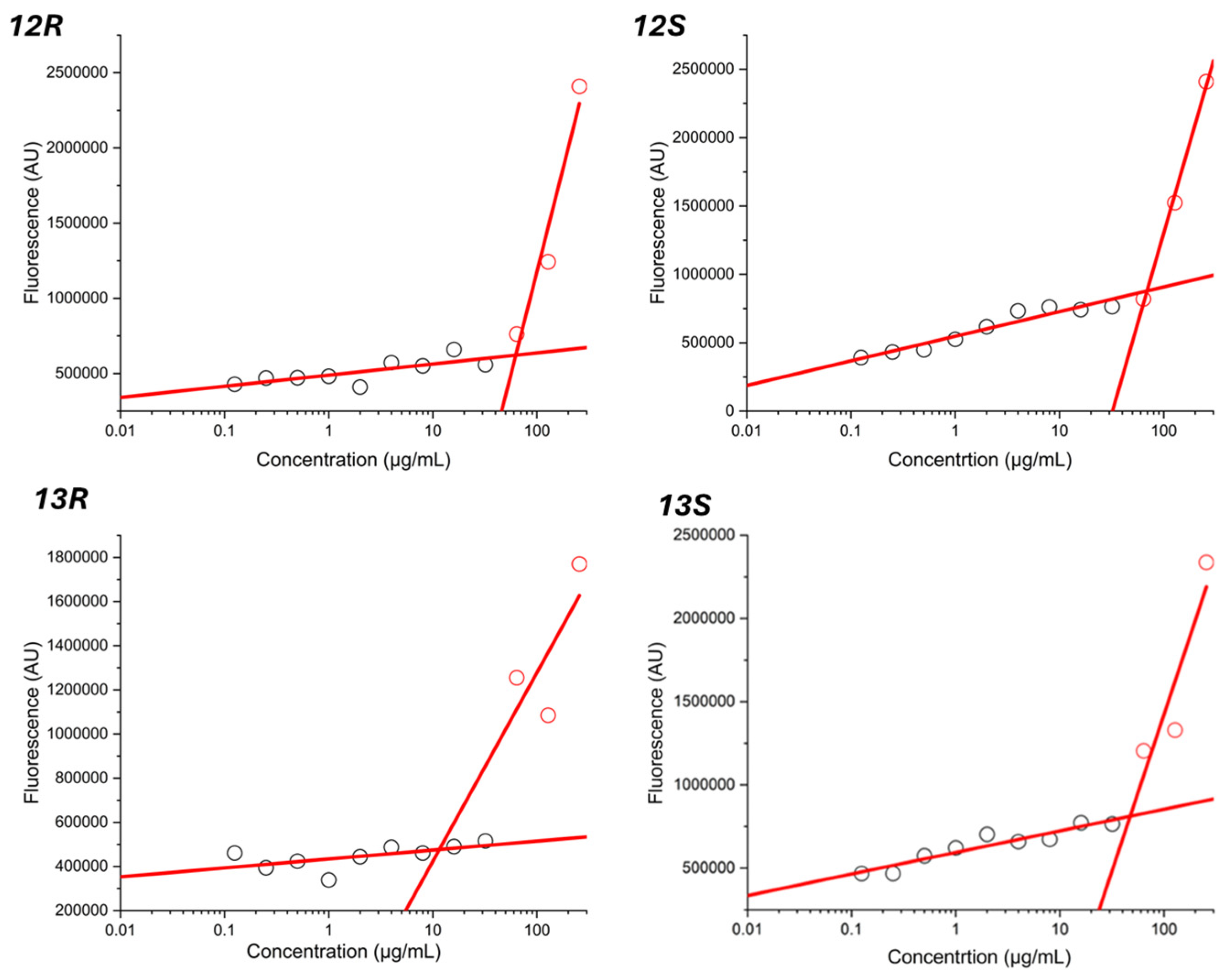
2.6. Determination of the Critical Micelle Concentration
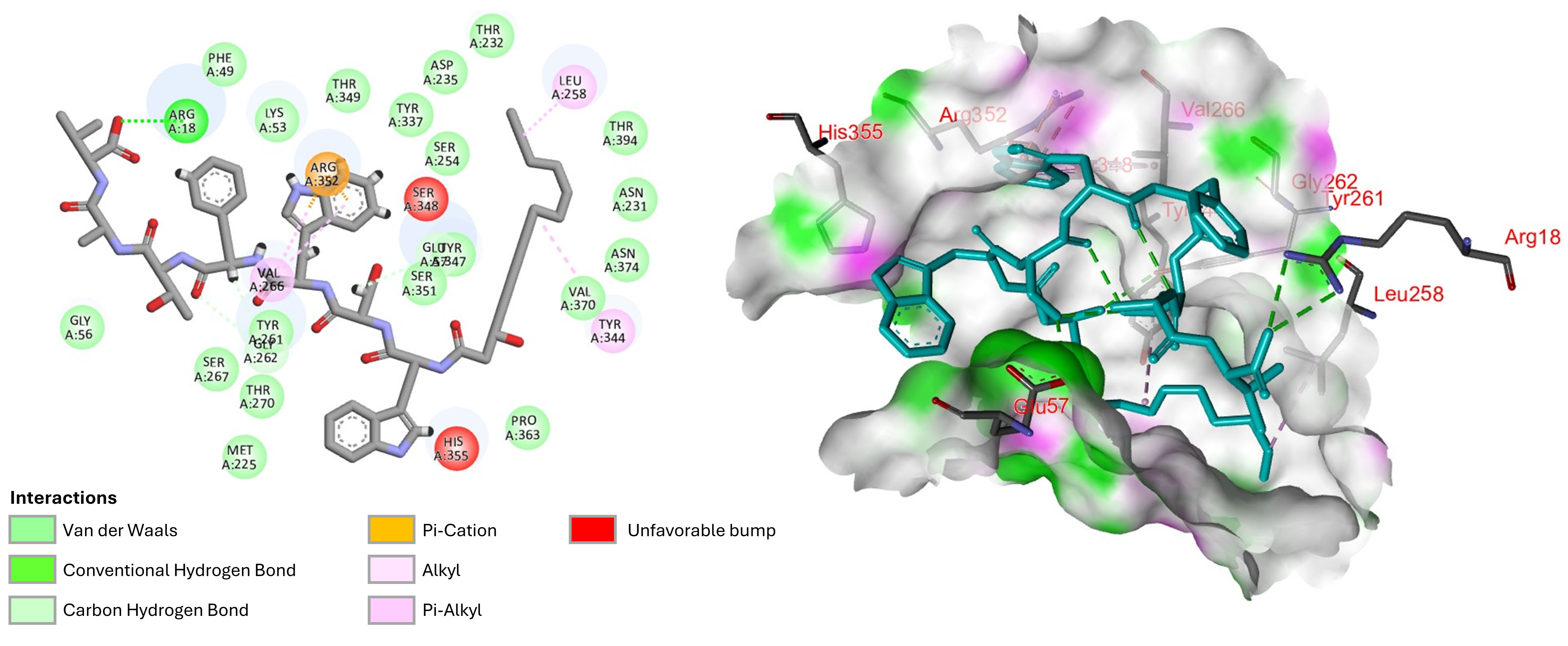
2.7. Molecular Docking and Molecular Dynamics Simulation
3. Discussion
4. Materials and Methods
4.1. Solid-Phase Peptide Synthesis, Purification, and Analysis
4.2. Antimicrobial Activity Assays
4.2.1. Bacterial Strains and Culture Medias
4.2.2. Minimal Inhibitory Concentration and Agar Diffusion Assays
4.3. Bacterial Growth Experiments
4.4. Hemolytic Activity Assay
4.5. Critical Micelle Concentration Determination
4.6. Molecular Docking Study
4.7. Molecular Dynamics Simulation
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dayan, G.H.; Mohamed, N.; Scully, I.L.; Cooper, D.; Begier, E.; Eiden, J.; Jansen, K.U.; Gurtman, A.; Anderson, A.S. Staphylococcus aureus: The current state of disease, pathophysiology and strategies for prevention. Expert Rev. Vaccines 2016, 15, 1373–1392. [Google Scholar] [CrossRef] [PubMed]
- Gonçalves, J.L.; Lee, S.H.I.; Camargo, C.H.; Zanella, R.C.; Silva, N.C.C.; Rall, V.L.M.; Cue, R.I.; dos Santos, M.V. Molecular characterization of persistent subclinical mastitis-causing Staphylococcus aureus from dairy farms. Braz. J. Microbiol. 2023, 54, 1181–1189. [Google Scholar] [CrossRef] [PubMed]
- Zhen, X.; Lundborg, C.S.; Sun, X.; Hu, X.; Dong, H. Economic burden of antibiotic resistance in ESKAPE organisms: A systematic review. Antimicrob. Resist. Infect. Control 2019, 8, 137. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Zhang, J.; He, Y.; Lv, Z.; Liang, Z.; Chen, J.; Li, P.; Liu, J.; Yang, H.; Tao, A.; et al. Exploring the Role of Staphylococcus aureus in Inflammatory Diseases. Toxins 2022, 14, 464. [Google Scholar] [CrossRef]
- Beavin, L.; Salunkhe, V.; Singh, S.; Gana, A.; Sekaran, B.; Tella, M.A.; Furmanek, S.P.; Arnold, F.W. Compliance with Guidelines for Treatment of Staphylococcus aureus Bacteremia is Associated with Decreased Mortality in Patients Hospitalized for Community-Acquired Pneumonia with Staphylococcus aureus Bacteremia. Univ. Louisville J. Respir. Infect. 2022, 6, 5. [Google Scholar]
- Jannat, H.; Ahmad, H. Prosthetic Joint Infection Complicated by Staphylococcus aureus Bacteremia and Tricuspid Valve Infective Endocarditis: A Novel Case Report. Cureus 2024, 16, e64821. [Google Scholar] [CrossRef]
- Bai, A.D.; Lo, C.K.L.; Komorowski, A.S.; Suresh, M.; Guo, K.; Garg, A.; Tandon, P.; Senecal, J.; Del Corpo, O.; Stefanova, I.; et al. Staphylococcus aureus bacteraemia mortality: A systematic review and meta-analysis. Clin. Microbiol. Infect. 2022, 28, 1076–1084. [Google Scholar] [CrossRef]
- Stanek, P.; Żółkiewski, P.; Januś, E. A Review on Mastitis in Dairy Cows Research: Current Status and Future Perspectives. Agriculture 2024, 14, 1292. [Google Scholar] [CrossRef]
- Hogeveen, H.; Steeneveld, W.; Wolf, C.A. Production Diseases Reduce the Efficiency of Dairy Production: A Review of the Results, Methods, and Approaches Regarding the Economics of Mastitis. Annu. Rev. Resour. Econ. 2019, 11, 289–312. [Google Scholar] [CrossRef]
- Jones, T.; Yeaman, M.R.; Sakoulas, G.; Yang, S.-J.; Proctor, R.A.; Sahl, H.-G.; Schrenzel, J.; Xiong, Y.Q.; Bayer, A.S. Failures in clinical treatment of Staphylococcus aureus infection with daptomycin are associated with alterations in surface charge, membrane phospholipid asymmetry, and drug binding. Antimicrob. Agents Chemother. 2008, 52, 269–278. [Google Scholar]
- Al-Mebairik, N.F.; El-Kersh, T.A.; Al-Sheikh, Y.A.; Marie, M.A.M. A review of virulence factors, pathogenesis, and antibiotic resistance in Staphylococcus aureus. Rev. Res. Med. Microbiol. 2016, 27, 50–56. [Google Scholar] [CrossRef]
- Chen, E.H.L.; Wang, C.-H.; Liao, Y.-T.; Chan, F.-Y.; Kanaoka, Y.; Uchihashi, T.; Kato, K.; Lai, L.; Chang, Y.-W.; Ho, M.-C.; et al. Visualizing the membrane disruption action of antimicrobial peptides by cryo-electron tomography. Nat. Commun. 2023, 14, 5464. [Google Scholar] [CrossRef]
- Miller, W.R.; Bayer, A.S.; Arias, C.A. Mechanism of Action and Resistance to Daptomycin in Staphylococcus aureus and Enterococci. Cold Spring Harb. Perspect. Med. 2016, 6, a026997. [Google Scholar] [CrossRef] [PubMed]
- Ji, S.; An, F.; Zhang, T.; Lou, M.; Guo, J.; Liu, K.; Zhu, Y.; Wu, J.; Wu, R. Antimicrobial peptides: An alternative to traditional antibiotics. Eur. J. Med. Chem. 2024, 265, 116072. [Google Scholar] [CrossRef] [PubMed]
- Yu, G.; Baeder, D.Y.; Regoes, R.R.; Rolff, J. Predicting drug resistance evolution: Insights from antimicrobial peptides and antibiotics. Proc. R. Soc. B Biol. Sci. 2018, 285, 20172687. [Google Scholar] [CrossRef]
- Decker, T.; Rautenbach, M.; Khan, S.; Khan, W. Antibacterial efficacy and membrane mechanism of action of the Serratia-derived non-ionic lipopeptide, serrawettin W2-FL10. Microbiol. Spectr. 2024, 12, e0295223. [Google Scholar]
- Desmyttere, H.; Deweer, C.; Muchembled, J.; Sahmer, K.; Jacquin, J.; Coutte, F.; Jacques, P. Antifungal Activities of Bacillus subtilis Lipopeptides to Two Venturia inaequalis Strains Possessing Different Tebuconazole Sensitivity. Front. Microbiol. 2019, 10, 2327. [Google Scholar] [CrossRef]
- Bird, G.H.; Patten, J.J.; Zavadoski, W.; Barucci, N.; Godes, M.; Moyer, B.M.; Owen, C.D.; DaSilva-Jardine, P.; Neuberg, D.S.; Bowen, R.A.; et al. A stapled lipopeptide platform for preventing and treating highly pathogenic viruses of pandemic potential. Nat. Commun. 2024, 15, 274. [Google Scholar] [CrossRef]
- Chu, J.; Vila-Farres, X.; Inoyama, D.; Ternei, M.; Cohen, L.J.; Gordon, E.A.; Reddy, B.V.; Charlop-Powers, Z.; Zebroski, H.A.; Gallardo-Macias, R.; et al. Discovery of MRSA active antibiotics using primary sequence from the human microbiome. Nat. Chem. Biol. 2016, 12, 1004–1006. [Google Scholar] [CrossRef]
- Yu, Z.; Sun, Z.; Yin, J.; Qiu, J. Enhanced Production of Polymyxin E in Paenibacillus polymyxa by Replacement of Glucose by Starch. BioMed Res. Int. 2018, 2018, 1934309. [Google Scholar] [CrossRef]
- McIntyre, J.J.; Bull, A.T.; Bunch, A.W. Vancomycin production in batch and continuous culture. Biotechnol. Bioeng. 1996, 49, 412–420. [Google Scholar] [CrossRef] [PubMed]
- Chu, J.; Vila-Farres, X.; Inoyama, D.; Gallardo-Macias, R.; Jaskowski, M.; Satish, S.; Freundlich, J.S.; Brady, S.F. Human Microbiome Inspired Antibiotics with Improved β-Lactam Synergy against MDR Staphylococcus aureus. ACS Infect. Dis. 2018, 4, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Rubino, F.A.; Mendoza, A.G.; Ruiz, N. The bacterial lipid II flippase MurJ functions by an alternating-access mechanism. J. Biol. Chem. 2019, 294, 981–990. [Google Scholar] [CrossRef] [PubMed]
- Tancer, R.J.; Baynes, K.; Wiedman, G.R. Synergy among humimycins against methicillin-resistant Staphylococcus aureus. Pept. Sci. 2020, 113, e24197. [Google Scholar] [CrossRef]
- Casillas-Vargas, G.; Ocasio-Malave, C.; Medina, S.; Morales-Guzman, C.; Del Valle, R.G.; Carballeira, N.M.; Sanabria-Rios, D.J. Antibacterial fatty acids: An update of possible mechanisms of action and implications in the development of the next-generation of antibacterial agents. Prog. Lipid Res. 2021, 82, 101093. [Google Scholar] [CrossRef]
- Yuyama, K.T.; Rohde, M.; Molinari, G.; Stadler, M.; Abraham, W.R. Unsaturated Fatty Acids Control Biofilm Formation of Staphylococcus aureus and Other Gram-Positive Bacteria. Antibiotics 2020, 9, 788. [Google Scholar] [CrossRef]
- Pushparaj Selvadoss, P.; Nellore, J.; Balaraman Ravindrran, M.; Sekar, U.; Tippabathani, J. Enhancement of antimicrobial activity by liposomal oleic acid-loaded antibiotics for the treatment of multidrug-resistant Pseudomonas aeruginosa. Artif. Cells Nanomed. Biotechnol. 2018, 46, 268–273. [Google Scholar] [CrossRef]
- Kim, Y.G.; Lee, J.H.; Lee, J. Antibiofilm activities of fatty acids including myristoleic acid against Cutibacterium acnes via reduced cell hydrophobicity. Phytomedicine 2021, 91, 153710. [Google Scholar] [CrossRef]
- Ballantine, R.D.; Al Ayed, K.; Bann, S.J.; Hoekstra, M.; Martin, N.I.; Cochrane, S.A. Linearization of the Brevicidine and Laterocidine Lipopeptides Yields Analogues That Retain Full Antibacterial Activity. J. Med. Chem. 2023, 66, 6002–6009. [Google Scholar] [CrossRef]
- Bennett, S.; Ben Said, L.; Lacasse, P.; Malouin, F.; Fliss, I. Susceptibility to Nisin, Bactofencin, Pediocin and Reuterin of Multidrug Resistant Staphylococcus aureus, Streptococcus dysgalactiae and Streptococcus uberis Causing Bovine Mastitis. Antibiotics 2021, 10, 1418. [Google Scholar] [CrossRef]
- Ye, X.; Zhang, H.; Luo, X.; Huang, F.; Sun, F.; Zhou, L.; Qin, C.; Ding, L.; Zhou, H.; Liu, X.; et al. Characterization of the Hemolytic Activity of Mastoparan Family Peptides from Wasp Venoms. Toxins 2023, 15, 591. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Mollo, A.; Kahne, D.; Ruiz, N. The Bacterial Cell Wall: From Lipid II Flipping to Polymerization. Chem. Rev. 2022, 122, 8884–8910. [Google Scholar] [CrossRef] [PubMed]
- Dominguez, C.; Boelens, R.; Bonvin, A.M.J.J. HADDOCK: A Protein−Protein Docking Approach Based on Biochemical or Biophysical Information. J. Am. Chem. Soc. 2003, 125, 1731–1737. [Google Scholar] [CrossRef] [PubMed]
- Santos, K.B.; Guedes, I.A.; Karl, A.L.M.; Dardenne, L.E. Highly Flexible Ligand Docking: Benchmarking of the DockThor Program on the LEADS-PEP Protein–Peptide Data Set. J. Chem. Inf. Model. 2020, 60, 667–683. [Google Scholar] [CrossRef]
- Vanier, G.S. Microwave-assisted solid-phase peptide synthesis based on the Fmoc protecting group strategy (CEM). Methods Mol. Biol. 2013, 1047, 235–249. [Google Scholar] [CrossRef]
- Khemaissa, S.; Walrant, A.; Sagan, S. Tryptophan, more than just an interfacial amino acid in the membrane activity of cationic cell-penetrating and antimicrobial peptides. Q. Rev. Biophys. 2022, 55, e10. [Google Scholar] [CrossRef]
- Mishra, A.K.; Choi, J.; Moon, E.; Baek, K.H. Tryptophan-Rich and Proline-Rich Antimicrobial Peptides. Molecules 2018, 23, 815. [Google Scholar] [CrossRef]
- Shahmiri, M.; Mechler, A. The role of C-terminal amidation in the mechanism of action of the antimicrobial peptide aurein 1.2. EuroBiotech J. 2020, 4, 25–31. [Google Scholar] [CrossRef]
- Strandberg, E.; Tiltak, D.; Ieronimo, M.; Kanithasen, N.; Wadhwani, P.; Ulrich, A.S. Influence of C-terminal amidation on the antimicrobial and hemolytic activities of cationic α-helical peptides. Pure Appl. Chem. 2007, 79, 717–728. [Google Scholar] [CrossRef]
- Youssef, N.H.; Wofford, N.; McInerney, M.J. Importance of the long-chain fatty acid beta-hydroxylating cytochrome P450 enzyme YbdT for lipopeptide biosynthesis in Bacillus subtilis strain OKB105. Int. J. Mol. Sci. 2011, 12, 1767–1786. [Google Scholar] [CrossRef]
- Vijayarohini, P.; Kavitha, G.; Bangaru Sudarsan Alwar, S.; Andrew Swamidoss, C.M. Antimicrobial activity of selective transition metal co-ordination complexes of myristic acid. Mater. Today Proc. 2020, 33, 4198–4205. [Google Scholar] [CrossRef]
- Okukawa, M.; Yoshizaki, Y.; Yano, S.; Nonomura, Y. The Selective Antibacterial Activity of the Mixed Systems Containing Myristic Acid against Staphylococci. J. Oleo Sci. 2021, 70, 1239–1246. [Google Scholar] [CrossRef] [PubMed]
- Saint Jean, K.D.; Henderson, K.D.; Chrom, C.L.; Abiuso, L.E.; Renn, L.M.; Caputo, G.A. Effects of Hydrophobic Amino Acid Substitutions on Antimicrobial Peptide Behavior. Probiotics Antimicrob. Proteins 2018, 10, 408–419. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Wang, X.; Shukla, R.; Kumar, R.; Weingarth, M.; Breukink, E.; Kuipers, O.P. Brevibacillin 2V, a Novel Antimicrobial Lipopeptide With an Exceptionally Low Hemolytic Activity. Front. Microbiol. 2021, 12, 693725. [Google Scholar] [CrossRef]
- Fliss, O.; Guay, L.D.; Fliss, I.; Biron, É. Synthesis and structure-activity study of the antimicrobial lipopeptide brevibacillin. RSC Med. Chem. 2024, 15, 4168–4179. [Google Scholar] [CrossRef]
- Oliveras, À.; Baró, A.; Montesinos, L.; Badosa, E.; Montesinos, E.; Feliu, L.; Planas, M. Antimicrobial activity of linear lipopeptides derived from BP100 towards plant pathogens. PLoS ONE 2018, 13, e0201571. [Google Scholar] [CrossRef]
- Sikorska, E.; Stachurski, O.; Neubauer, D.; Małuch, I.; Wyrzykowski, D.; Bauer, M.; Brzozowski, K.; Kamysz, W. Short arginine-rich lipopeptides: From self-assembly to antimicrobial activity. Biochim. Biophys. Acta (BBA)-Biomembr. 2018, 1860, 2242–2251. [Google Scholar] [CrossRef]
- Patil, R.; Das, S.; Stanley, A.; Yadav, L.; Sudhakar, A.; Varma, A.K. Optimized Hydrophobic Interactions and Hydrogen Bonding at the Target-Ligand Interface Leads the Pathways of Drug-Designing. PLoS ONE 2010, 5, e12029. [Google Scholar] [CrossRef]
- García-Vela, S.; Guay, L.-D.; Rahman, M.R.T.; Biron, E.; Torres, C.; Fliss, I. Antimicrobial Activity of Synthetic Enterocins A, B, P, SEK4, and L50, Alone and in Combinations, against Clostridium perfringens. Int. J. Mol. Sci. 2024, 25, 1597. [Google Scholar] [CrossRef]
- Reyher, K.K.; Dufour, S.; Barkema, H.W.; Des Côteaux, L.; Devries, T.J.; Dohoo, I.R.; Keefe, G.P.; Roy, J.P.; Scholl, D.T. The National Cohort of Dairy Farms-A data collection platform for mastitis research in Canada. J. Dairy Sci. 2011, 94, 1616–1626. [Google Scholar] [CrossRef]
- Zhang, L.; Ben Said, L.; Diarra, M.S.; Fliss, I. Inhibitory Activity of Natural Synergetic Antimicrobial Consortia Against Salmonella enterica on Broiler Chicken Carcasses. Front. Microbiol. 2021, 12, 656956. [Google Scholar] [CrossRef] [PubMed]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Dundas, J.; Ouyang, Z.; Tseng, J.; Binkowski, A.; Turpaz, Y.; Liang, J. CASTp: Computed atlas of surface topography of proteins with structural and topographical mapping of functionally annotated residues. Nucleic Acids Res. 2006, 34, W116–W118. [Google Scholar] [CrossRef] [PubMed]
- Schrodinger, L. The PyMOL molecular graphics system. Version 2015, 1, 8. [Google Scholar]
- BIOVIA. Dassault Systèmes, Discovery Studio Visualizer; BIOVIA: San Diego, CA, USA, 2021. [Google Scholar]


| Pathogens | Peptide | ||||
|---|---|---|---|---|---|
| 1 | 12R | 12S | 13R | 13S | |
| Staphylococcus aureus ATCC 29213 | 8 | 16 | 8 | 4 | 2 |
| Staphylococcus aureus ATCC 43300 a | 16 | 16 | 16 | 128 | 64 |
| Bacillus subtilis ATCC 23857 | - b | - | - | - | - |
| Escherichia coli ATCC 25922 | - | - | - | - | - |
| Pseudomonas aeruginosa ATCC 27853 | - | - | - | - | - |
| Salmonella enterica ATCC 14028 | - | - | - | - | - |
| S. aureus Strain | Antibiotic Resistance Profile a | 12R | 12S | 13R | 13S |
|---|---|---|---|---|---|
| 4-121 | 32 | 32 | 256 | 256 | |
| 4-131 | >256 | >256 | >256 | >256 | |
| 4-133 | PEN-CEF-CTX-FOX-CIP-CHL-KAN-GEN | 256 | 256 | >256 | >256 |
| 4-134 | PEN-VAN-CEF-CTX-FOX-ERY-CC-TET-KAN-GEN-STR | >256 | >256 | >256 | >256 |
| 4-138 | 4 | 4 | 16 | 16 | |
| 4-139 | PEN-ERY-CC-TET-KAN | 4 | 8 | 8 | 8 |
| 4-140 | TET | 8 | 16 | 16 | 16 |
| 4-144 | PEN-AMC-CEF-CTX-FOX-CIP-CHL-ERY-TET | 8 | 16 | 8 | 16 |
| 4-145 | 16 | 32 | 16 | 32 | |
| 4-153 | 16 | 16 | 32 | 32 | |
| 4-156 | 4 | 8 | 16 | 8 | |
| 4-157 | 16 | 16 | 32 | 32 | |
| 4-159 | 8 | 16 | 16 | 8 | |
| 4-162 | >256 | >256 | >256 | >256 | |
| A-165 | PEN-CEF-CTX-FOX-CIP-CC-KAN-GEN-STR-P/N | 256 | 256 | >256 | >256 |
| 4-170 | 4 | 8 | 8 | 8 | |
| 4-171 | PEN-AMC-CEF-CTX-FOX | 2 | 1 | 0.5 | 8 |
| 4-172 | PEN-AMC-CEF-CTX-FOX | 8 | 8 | 8 | 8 |
| 4-173 | 8 | 16 | 8 | 8 |
| Analog | Docking Score by Hepdoc | DockThor | |||
|---|---|---|---|---|---|
| Affinity | Total Energy (kcal/mol) | van der Waals Energy (kcal/mol) | Electrostatic Energy (kcal/mol) | ||
| 12R | −193.350 | −11.035 | 139.473 | −38.426 | −34.328 |
| 12S | −174.120 | −10.287 | 144.212 | −33.921 | −35.708 |
| 13R | −187.006 | −11.170 | 150.169 | −37.841 | −18.467 |
| 13S | −191.760 | −10.618 | 150.818 | −52.443 | −12.309 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rahman, M.R.T.; Guay, L.-D.; Fliss, I.; Biron, E. Structure–Activity Study of the Antimicrobial Lipopeptide Humimycin A and Screening Against Multidrug-Resistant Staphylococcus aureus. Antibiotics 2025, 14, 385. https://doi.org/10.3390/antibiotics14040385
Rahman MRT, Guay L-D, Fliss I, Biron E. Structure–Activity Study of the Antimicrobial Lipopeptide Humimycin A and Screening Against Multidrug-Resistant Staphylococcus aureus. Antibiotics. 2025; 14(4):385. https://doi.org/10.3390/antibiotics14040385
Chicago/Turabian StyleRahman, Md Ramim Tanver, Louis-David Guay, Ismail Fliss, and Eric Biron. 2025. "Structure–Activity Study of the Antimicrobial Lipopeptide Humimycin A and Screening Against Multidrug-Resistant Staphylococcus aureus" Antibiotics 14, no. 4: 385. https://doi.org/10.3390/antibiotics14040385
APA StyleRahman, M. R. T., Guay, L.-D., Fliss, I., & Biron, E. (2025). Structure–Activity Study of the Antimicrobial Lipopeptide Humimycin A and Screening Against Multidrug-Resistant Staphylococcus aureus. Antibiotics, 14(4), 385. https://doi.org/10.3390/antibiotics14040385