Abstract
Developing an efficient approach to improve the luminescence of the phosphors without heating processing is a challenge, but attracts much attention. In the present paper, prismatic microcrystals of RE4O(OH)9NO3 (RE = Y, Eu) were synthesized by a hydrothermal reaction at 180 °C for 24 h. The reaction with VO3− did not change the crystal structure of the microcrystals and VO3− substituting for NO3− anions did not take place. However, it contributed to the formation of amorphous particles containing VO3− on the surface of a prism, which is similar to the surface corrosion of a metal, called “surface eroding”. Therefore, surface modification was successfully achieved by eroding the surface of the microcrystals through the reaction with vanadate ions. As a result of VO3−→Eu3+ energy transfer and the light-harvesting ability of VO3−, the red emission intensity at 617 nm of the modified microcrystals greatly increased. Eroding the surface of rare earth microcrystals recommends a new paradigm for luminescence improvement of rare earth compounds.
1. Introduction
Directly exciting RE3+ ions upon irradiation for luminescence is not an efficient method, because the f–f electronic transitions of the rare earth ions (RE3+) are usually forbidden by the spin selection rules []. Therefore, doping RE3+ ions with a low concentration into an appropriate host lattice is widely used to produce solid-solution phosphor and obtain desirable luminescence []. In recent years, much attention has been paid to RE3+-doped inorganic materials with a uniform size and specific morphology, because they allow for the attainment of functionalities, not only from the constituent substance but also from the special structure [,,,]. For rare earth oxide phosphors, calcining the precursors is widely used to obtain the phosphors with a variety of novel structures, since the precursors and their calcined products tend to exhibit generic relationships [,,,,,]. RE2(OH)5NO3·nH2O layered rare earth hydroxide (LRH), RE4O(OH)9NO3 oxy-hydroxyl nitrate, RE(OH)2.94(NO3)0.06·nH2O hydroxyl nitrate, and RE(OH)3 hydroxide are the reported products synthesized from the hydrothermal reaction system [,,,], and their morphology and size could be easily regulated by varying the synthesis conditions, including the pH value, reaction temperature, and reaction time. Therefore, they are the desirable precursors for rare earth oxide phosphors. Therefore, there are a lot of investigations on these precursors. However, abundant water molecules or hydroxyls directly coordinate to the rare earth ions, which result in a serious luminescence quenching [,]. Thus, there is the question of how to enhance the emission intensity of the rare earth phosphors. Of course, the thermal decomposition of the precursors is an efficient method to synthesize rare earth oxides, and it could remove the water molecules/dehydroxyl and thus enhance the luminescence intensity []. However, morphological damage and crystal-structure collapse would take place during the annealing process, which may significantly affect the final performance []. Therefore, developing an efficient approach to improve the luminescence of the phosphors without heating processing is a challenge, but attracts much attention.
Recently, it has been reported that an antenna effect could give rise to an enhanced photoluminescence upon light irradiation, because an effective intramolecular energy transfer from coordinated ligands to the activated RE3+ ions can enhance the absorption of the optical excitation. This antenna effect effectively promotes the energy transfer from coordinated ligands to the activated RE3+ ions, and thus enhances the emission intensity []. This light-harvesting ability was successfully applied onto the layered rare earth hydroxide nanosheets [,], hydroxyl nitrate square nanoplates [], and rare earth nanoparticles [,], through the grafting of organic ligands, including picolinic acid and a rare earth complex, on the surface of the nanocrystals. Indeed, a considerable improvement of luminescence was observed for these rare earth crystals. However, the organic ligands were indirectly linked to the surface of the nanocrystals by hydrogen bonding, indicating that the hybrid phosphors grafted organic ligands were unstable. In addition, the hydrogen bonding connection made the energy transfer from the ligands to the activated RE3+ ions difficult [,], so it is not the most effective way for energy transfer, compared to the directly connecting coordination.
In the present work, prismatic microcrystals of RE4O(OH)9NO3 (RE = Y, Eu) were synthesized by the hydrothermal reaction. The surface modification of the microcrystals was successfully achieved by eroding the surface of the microcrystals through vanadate ions. The main means of sample characterization are X-ray diffraction (XRD), transmission electron microscopy (TEM), selected area electron diffraction (SAED), scanning transmission electron microscopy (STEM), photoluminescence (PL)/photoluminescence excitation (PLE) spectroscopy, and fluorescence decay curve analysis. The reaction with VO3− contributed to the formation of amorphous particles containing VO3− on the surface of the prism, which lead to a great enhancement of luminescence. The usage of the energy transfer to the activated RE3+ ions through surface eroding processing, paves a new paradigm for luminescence improvement of the rare earth compounds.
2. Experimental Section
2.1. Synthesis
Preparation of oxy-hydroxyl nitrate microcrystals. The starting rare earth sources are rare earth oxides, which are Y2O3 and Eu2O3, all 99.99% pure products from Huizhou Ruier Rare-Chem, Hi-Tech, Co., Ltd. (Huizhou, China). Dissolving the rare earth oxide in hot nitric acid yielded a rare earth nitrate solution, which was then treated by evaporation at 90 °C to remove the excess acid. The synthesis of Y/Eu binary microcrystals (Y:Eu molar ratio of 0.95:0.05) was conducted via a hydrothermal reaction at 180 °C for 24 h with a pH value of 10–11, which was described in our previous work [].
Erosion of the microcrystal surface. In typical processing, 2 mmol of RE4O(OH)9NO3 microcrystals was dispersed in 60 mL of water containing a proper amount of NH4VO3 and NH4OH with the pH value of 10–11. After being stirred for 30 min, the resultant suspension was transferred into a Teflon-lined stainless-steel autoclave of 100 mL capacity. The autoclave was tightly sealed and was put in an electric oven preheated to 200 °C. After a 12 h reaction, the autoclave was left to cool naturally to room temperature, and then the product was collected via centrifugation. The wet precipitate was washed with distilled water 3 times, rinsed with absolute ethanol, and was finally dried in air at 60 °C for a day. The oxy-hydroxyl nitrate microcrystals and the microcrystals eroded by VO3− are denoted as MC and MC-RV hereafter, with the R-fold VO3− in the reaction system.
2.2. Characterization Techniques
Phase identification was performed by X-ray diffraction (XRD, Model SmartLab, Rigaku, Tokyo, Japan), operating at 40 kV/40 mA using nickel-filtered Cu Kα radiation and a scanning speed of 6.0° 2θ/min. The product morphology was analyzed by field emission scanning electron microscopy (FE-SEM, Model JSM-7001F, JEOL, Tokyo, Japan) and transmission electron microscopy (TEM, Model JEM-2000FX, JEOL, Tokyo, Japan). Elemental mapping was performed using scanning transmission electron microscopy (STEM, Model JEM-2000FX, JEOL, Tokyo, Japan). Fourier transform infrared spectroscopy experiments (FT-IR, Nicolet iS5, Thermal Fisher Scientific, NY, USA) were under taken using the standard KBr method. Photoluminescence (PL), photoluminescence excitation (PLE), and fluorescence decay were analyzed at room temperature using an FP-8600 fluorospectrophotometer (JASCO, Tokyo, Japan) equipped with a Φ60 mm integrating sphere and a 150-W Xe-lamp. The slit widths were 5 nm for both the excitation and emission sides. The spectral responses were corrected in the range 220–850 nm with a Rhodamine-B solution (5.5 g/L in ethylene glycol, for 220–600 nm range) and with a standard light source unit (ECS-333, JASCO, for 350–850 nm) as references. The external quantum efficiency (εex, the total number of emitted photons divided by the total number of excitation photons) and the internal quantum efficiency (εin, the total number of emitted photons divided by the number of photons absorbed by the sample) of the phosphors are derived from the following equations [], using the built-in analysis software:
where E(λ)/hv, R(λ)/hv, and P(λ)/hv are the number of photons in the excitation, reflectance, and emission spectra of the samples, respectively. The reflection spectrum of the spectral on diffusive white standards was used for calibration.
3. Results and Discussion
In the present study, the RE4O(OH)9NO3 (RE = Y, Eu) microcrystals (termed as MC) was chosen as an example for surface modification. As reported in the literature [], Y4O(OH)9NO3 is a monoclinic crystal structure, which is a three-dimensional framework with one-dimensional channels containing NO3−. The NO3− is indirectly linked to Y3+ rather than forming a direct connection. There are 4 trivalent yttrium ions in the asymmetric unit, with 3 in a 7-coordinated environment with a capped trigonal prismatic geometry and 1 in a 9-coordinated environment with a tricapped trigonal prismatic geometry. The trivalent yttrium ions are linked through hydroxide anions forming the framework around the channels. In the channel, the nitrate ion is indirectly linked to Y3+ through the hydrogen bonding. As a result of the one-dimensional channels, Y4O(OH)9NO3 always crystallizes into prismatic and wire-like crystallites []. Here, the incorporation of Eu3+ in Y4O(OH)9NO3 does not significantly affect its crystal structure, because the diffractions of MC are indexed to the monoclinic Y4O(OH)9NO3 (JCPDS File no. 79-1352), except for slight spectral shifts to the lower angle side (Figure 1c). The replacement of Y3+ with larger Eu3+ ions (for 8-fold coordination, = 0.1019 nm, = 0.1066 nm) contributed to the lattice expansion, thus resulting in the diffraction shifts []. Figure 1d shows the FE-SEM image of MC, and pure hexagonal prisms with a diameter of ~0.3–0.5 μm and a length of ~1.5–2.5 μm are found in the observation.
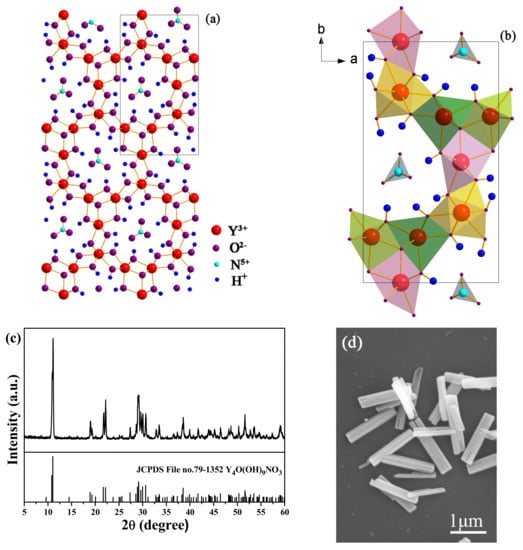
Figure 1.
(a,b) Crystal structure of Y4O(OH)9NO3, (c) XRD patterns, and (d) FE-SEM image of MC.
Figure 2 shows the XRD patterns of MC after the reaction with VO3−. Evidently, the reaction products are the same as MC, because all the diffractions are indexed to the monoclinic Y4O(OH)9NO3 (JCPDS File no. 79-1352), indicating that the reaction with VO3− did not result in a phase transformation. Increasing the R value from 0 to 1.5 (R, the molar ratio of VO3− to MC) induced a small shift of the diffraction positions. Since VO3− is smaller than NO3− [], VO3− substituting for NO3− anions may induce the diffraction shift to the higher angle side arising from the lattice contraction. Indeed, NO3− is indirectly coordinated in Y4O(OH)9NO3 rather than the free anion, so it cannot be easily replaced by other anions through ion exchange. After the reaction with VO3−, the products mainly remain in the original morphology of MC (Figure 3). However, the surface of MC-RV (microcrystals reacted with VO3−, with R-fold VO3− in the reaction system) is not smooth, with nano-sized crystals on the prism surface. Increasing the R value from 0.25 to 1.5 contributed to a rougher particle surface for MC-RV. Since the above phenomenon is similar to that for the surface corrosion of metal, in the present paper, it is called “surface eroding” for the reaction with VO3−. However, there are not any other impurity phases in the XRD patterns, indicating that the nano-sized crystals are amorphous.
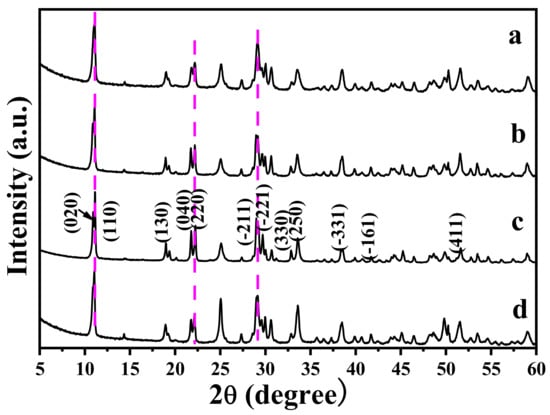
Figure 2.
XRD patterns of MC-RM, with (a) R = 0.25, (b) R = 0.5, (c) R = 1.0, and (d) R =1.5.

Figure 3.
FE-SEM micrographs of MC (a) and MC-RM (b–d), with (b) R = 0.25, (c) R = 1.0, and (d) R =1.5.
Figure 4 shows the FT-IR spectra for the samples after the reaction with different VO3− contents. The MC exhibits splitting absorption peaks in the range of 3350–3750 cm−1, with intense absorptions at 3401, 3487, and 3615 cm−1, which arise from hydroxyl (OH−) groups [,]. This is in compliance with the derived chemical formula of RE4O(OH)9NO3. The absorption peaks around 1364 and 1407 cm−1 are assignable to the v3 vibration mode of NO3− and the v4 asymmetric stretch of O-NO2, respectively [,]. It is clearly seen that the absorptions of NO3− in MC are different from those of the interlaye- free NO3− in layered rare earth hydroxide. After the reaction with VO3−, NO3− still exists in the FT-IR spectra and is not significantly affected by the reaction. However, the absorption for VO3− at 792 cm−1 appeared after the reaction with VO3− [,,], and more intense absorption is found at a higher R value.
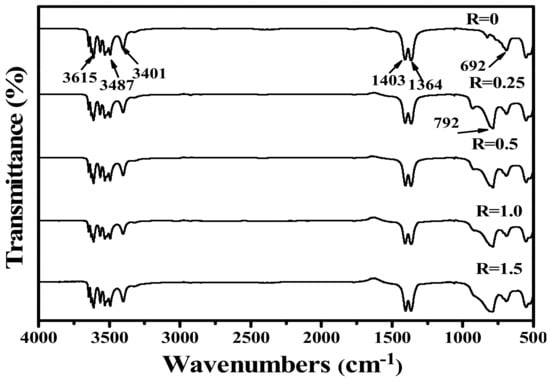
Figure 4.
FT-IR spectra for MC (R = 0) and MC-RM after the reaction with VO3−.
Figure 5 shows the elemental mapping distribution of MC, and the results indicate that MC is a homogeneous solid solution, because all the elements of Y, Eu, and N are distributed among the particles. In addition, the close observation of MC through the TEM image found that MC is well crystallized, with sharp edges and corners, and the surface is smooth. Selected area electron diffraction (SAED) found that the MC is a single crystalline. The calculated planar spacings of ~0.352 nm and ~0.268 nm correspond well with the (230) and (-141) planes of Y4O(OH)9NO3. After the reaction with VO3−, V is distributed among the particles, except for the original component elements of Y, Eu, and N (Figure 6 and Figure 7). The TEM images of MC-RV confirmed that there are nano-sized crystals on the surface of the prisms, and they grow up and tend to dendritic growth. Evidently, the materials needed for growth are obtained from the dissolution of the MC surface, similar to the surface corrosion of metal. The diameter of the prisms became slimmer at a higher R value, further presenting direct evidence. Selected area electron diffraction (SAED) yields circular patterns, suggesting the MC-RV consists of polycrystalline (Figure 6). The calculated planar spacings of ~0.374 nm, ~0.301 nm, ~0.287 nm, and ~0.184 nm correspond well with the (140), (310), (320), and (500) planes of Y4O(OH)9NO3, i.e., d(140) = 0.374026 nm, d(310) = 0.301376 nm, d(320) = 0.287141 nm, and d(500) = 0.183968 nm (JCPDS File no. 79-1352). Evidently, amorphous diffraction circular patterns were found in the SAED patterns, confirming the existence of amorphous particles on the surface of the prism. Collectively, the results from these analyses confirmed that the reaction with VO3− contributed to the formation of amorphous particles containing VO3− on the surface of the prism, which is similar to the surface corrosion of metal, called “surface eroding”. Therefore, it can be said that the surface modification of MC was successfully achieved by eroding the surface of MC through vanadate ions.

Figure 5.
(a–c) Elemental mapping, (d) TEM image, and (e) SAED for MC.

Figure 6.
(a–d) Elemental mapping, (e) TEM image, and (f) SAED for MC-0.25V.

Figure 7.
(a–d) Elemental mapping and (e) TEM image for MC-1.5V.
Figure 8 shows the PLE and PL spectra for MC. By monitoring the 5D0→7F2 emission at 617 nm, a series of sharp lines in the PLE spectrum ranging from 300 nm to 500 nm can be ascribed to the transitions within the Eu3+ 4f6 electronic configuration. Different from other rare earth hydroxide precursors, MC exhibited O2−-Eu3+ charge transfer (CT) transitions at ~255 nm, as were commonly found for Eu3+-activated Y2O3 [,,,]. Upon excitation at 395 nm (intra-4f6 transition of Eu3+), the PL spectrum displayed the typical 5D0→7FJ(J = 0–4) transitions of Eu3+. The relative intensities of the transitions to different J levels depended on the symmetry of the Eu3+ environment and can be described in terms of the Judd–Ofelt theory []. The Judd–Ofelt parity law predicts that the magnetic dipole 5D0→7F1 transition is permitted while the electric dipole 5D0→7F2 transition is forbidden, and the latter is allowed only on the condition that the Eu3+ ions occupy the asymmetric site [,,]. The MC, having the composition of (Y0.95Eu0.05)4O(OH)9NO3, has a monoclinic structure and 2 kinds of Eu3+ ions, which are 7-coordinated Eu3+ ions in C2v non-centrosymmetric sites and 9-coordinated Eu3+ ions in D3h centrosymmetric sites []. Since the molar ratio of C2v occupancy to that of D3h is 3, most Eu3+ ions occupy the asymmetric site, and thus the 5D0→7F2 transition at 617 nm is stronger than the 5D0→7F1 transition at 595 nm []. However, the MC did not output a strong red light, mainly due to the fact that the hydroxyls directly coordinated to the rare earth ions Eu3+, which resulted in serious luminescence quenching [].
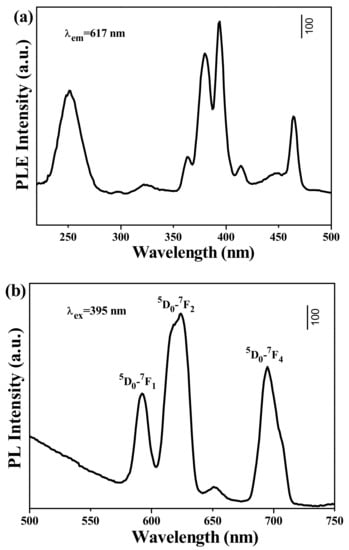
Figure 8.
(a) PLE and (b) PL spectra for MC at room temperature.
Figure 9 shows the PLE and PL spectra for MC-RV, which was eroded by vanadate ions. The excitation spectrum consisted of a strong and broad absorption band ranging from 200 to 350 nm, which was assigned to the energy transfer from VO3− to Eu3+. The overlapped excitation was from 2 individual bands located at ~275 nm and ~323 nm, which corresponded to the transitions of 1A2(1T1)→1E (1T2) and 1A2(1T2)→1A1 (1E) of V5+, respectively [,]. The band at ~255 nm was the contribution of the O2−-Eu3+ charge transfer [,,], while the very weak transitions of 7F0,1→5L6 and 7F0,1→5D2 at 395 nm and 463 nm for Eu3+ were observed in the excitation spectra [,,]. Since the strongest excitation was located at ~323 nm, the excitation wavelength was chosen as 323 nm (1A2(1T2)→1A1 (1E) transition of V5+). Upon UV excitation at 323 nm, the PL spectra displayed strong emissions at 540 nm, 590 nm, 617 nm, 650 nm, and 702 nm, which were assigned to 5D1→7F0,1, 5D0→7F1, 5D0→7F2, 5D0→7F3, and 5D0→7F4 transitions of Eu3+, respectively. The emission at 617 nm attained the dominate role. This further confirmed the existence of VO3−→Eu3+ energy transfer. Interestingly, the emission intensity at 617 nm was greatly enhanced by increasing the R value from 0 to 0.25, indicating that the VO3−→Eu3+ energy transfer contributed to the improved luminescence. The external/internal quantum efficiencies for R = 0 were 6 ± 1%/11 ± 1%, and the external/internal quantum efficiencies for R = 0.25 were 36 ± 1%/65 ± 1%, directly confirm the great enhancement of luminescence. However, a higher R value induced a rougher particle surface, which contributed to the light scattering and quenching of the luminescence. Therefore, increasing the R value further resulted in the luminescent decay. The external/internal quantum efficiencies for R = 0.5, R = 1.0, and R = 1.5 were 25 ± 1%/49 ± 2%, 18 ± 2%/43 ± 1%, and 10 ± 1%/37 ± 2%, respectively. However, the emission intensity for MC-RV is evidently stronger than that for MC on the whole, indicating that the erosion of the surface of rare earth microcrystals through vanadate ions can contribute to the considerable improvement to luminescence (Figure 10).
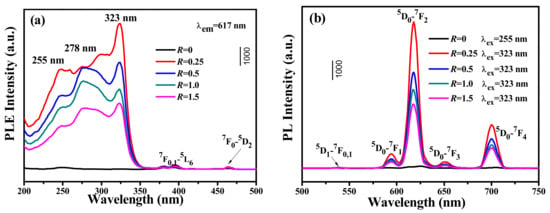
Figure 9.
(a) PLE and (b) PL spectra for MC and MC-RV at room temperature.
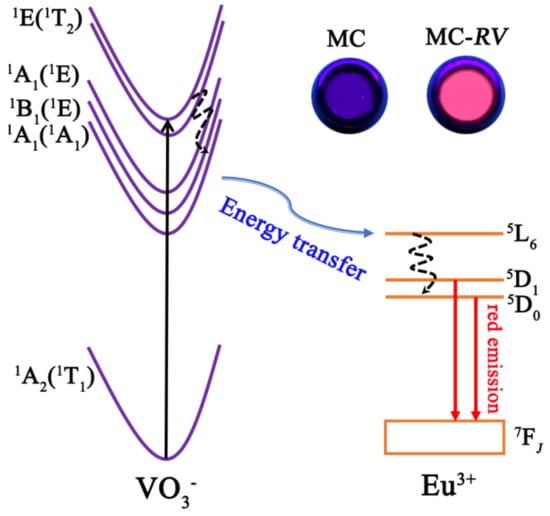
Figure 10.
Schematic illustration of VO3−→Eu3+ energy transfer.
The decay kinetics of the 5D0→7F2 transition at 617 nm for MC and MC-RVre a investigated in Figure 11. All the fluorescence decay curves can be fitted to single exponentials. The average lifetimes of the MC, MC-0.25V, MC-0.50V, MC-1.0V, and MC-1.5V samples determined in this work are ~0.67 ms, ~1.38 ms, ~1.21 ms, ~1.14 ms, and ~1.12 ms, respectively. Evidently, the lifetime for MC-RV is longer than that for MC, since the energy transfer of VO3− to Eu3+ is more time consuming.
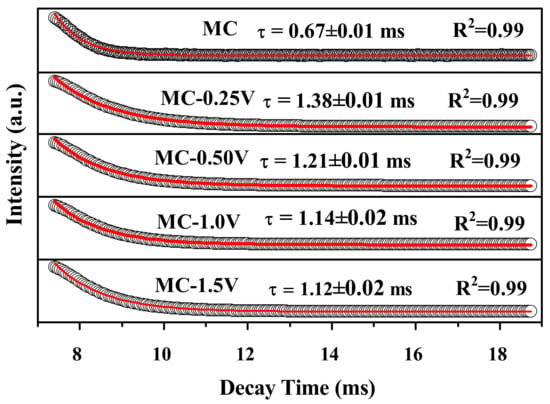
Figure 11.
Fluorescence decay curves for the 617 nm emission of MC and MC-RV.
4. Conclusions
In the present study, prismatic microcrystals of RE4O(OH)9NO3 (RE = Y, Eu) were synthesized by a hydrothermal reaction (180 °C for 24 h, pH = 10–11). The surface modification of the microcrystals was successfully achieved by eroding the surface of the microcrystals through vanadate ions. The main means of sample characterization were XRD, TEM, SAED, STEM, PLE/PL spectroscopy, and fluorescence decay curve analysis. The reaction with VO3− contributed to the formation of amorphous particles containing VO3− on the surface of the prism, which was similar to the surface corrosion of metal, called “surface eroding”. Upon UV excitation at 323 nm (1A2(1T2)→1A1 (1E) transition of V5+), the modified microcrystals displayed a strong red emission at 617 nm (5D0→7F2 transition of Eu3+). As a result of the VO3−→Eu3+ energy transfer, the emission intensity at 617 nm for the modified microcrystals increased greatly after modification by vanadate ions. Additionally, due to the VO3−→Eu3+ energy transfer, the modified microcrystals exhibited longer lifetimes. The usage of the energy transfer to the activated RE3+ ions through surface eroding processing for rare earth compounds recommends a new paradigm for luminescence improvement.
Author Contributions
Writing—original draft preparation, data curation, J.Q.; Formal analysis, investigation, J.L.; Conceptualization, writing—review and editing, visualization, Q.Z. All authors have read and agreed to the published version of the manuscript.
Funding
This research was founded by the Natural Science Foundation of Liaoning Province (Grant 2020-MS-081) and the National Natural Science Foundation of China (Grant 51302032).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data sharing is not applicable to this article.
Conflicts of Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- Blasse, G.; Grabmaier, B.C. Luminescent Materials; Springer: Berlin/Heidelberg, Germany, 1994. [Google Scholar]
- Gai, S.L.; Li, C.X.; Yang, P.P.; Lin, J. Recent progress in rare earth micro/nanocrystals: Soft chemical synthesis, luminescent properties, and biomedical applications. Chem. Rev. 2014, 114, 2343–2389. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Q.; Li, J.-G.; Ma, R.; Sasaki, T.; Yang, X.; Li, X.D.; Sun, X.D.; Sakka, Y. Well-defined crystallites autoclaved from the nitrate/NH4OH reaction system as the precursor for (Y,Eu)2O3 red phosphor: Crystallization mechanism, phase and morphology control, and luminescent property. J. Solid State Chem. 2012, 192, 229–237. [Google Scholar] [CrossRef]
- Zhang, F.; Zhao, D.Y. Synthesis of uniform rare earth fluoride (NaMF4) nanotubes by in situ ion-exchange from their hydroxide [M(OH)3] parents. ACS Nano 2009, 3, 159–164. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.-H.; Wang, J.; Qiu, G.-H.; Li, X.-B.; Huang, W.-T.; Zhang, R.-R.; Lin, T.; Wang, L.-X.; Zhang, Q.-T. A strategy to achieve efficient green-emission dual-mode luminescence of Yb3+, Er3+ doped NaBiF4. Rare Met. 2021, 40, 2040–2048. [Google Scholar] [CrossRef]
- Fu, R.; Hu, Y.-Y.; Qiao, H.-N.; Yang, C.-L.; Yin, H.; Qu, M.-G. Luminescence property and magnetic resonance imaging of Gd2O3:Tb3+ nanocrystals doped with Zn2+, Li+. Rare Met. 2021, 40, 2049–2058. [Google Scholar] [CrossRef]
- Geng, F.X.; Ma, R.Z.; Sasaki, T. Anion-exchangeable layered materials based on rare-earth phosphors: Unique combination of rare-earth host and exchangeable anions. Acc. Chem. Res. 2010, 43, 1177–1185. [Google Scholar] [CrossRef]
- Xu, Z.H.; Kang, X.J.; Li, C.X.; Hou, Z.Y.; Zhang, C.M.; Yang, D.M.; Li, G.G.; Lin, J. Ln3+ (Ln=Eu, Dy, Sm, and Er) ion-doped YVO4 nano/microcrystals with multiform morphologies: Hydrothermal synthesis, growing mechanism, and luminescent properties. Inorg. Chem. 2010, 49, 6706–6715. [Google Scholar] [CrossRef]
- Li, G.C.; Chao, K.; Peng, H.R.; Chen, K.Z. Hydrothermal synthesis and characterization of YVO4 and YVO4:Eu3+ nanobelts and polyhedral micron crystals. J. Phys. Chem. C 2008, 112, 6228–6231. [Google Scholar] [CrossRef]
- Li, C.X.; Quan, Z.W.; Yang, J.; Yang, P.P.; Lin, J. Highly uniform and monodisperse β-NaYF4:Ln3+ (Ln = Eu, Tb, Yb/Er, and Yb/Tm) hexagonal microprism crystals: Hydrothermal synthesisand luminescent properties. Inorg. Chem. 2007, 46, 6329–6337. [Google Scholar] [CrossRef]
- Selvi, G.T.; Grace, A.N.; Jeong, S.K. Synthesis of rare earth hydroxycarbonate (LaOHCO3) nanocrystals with tuneable morphology and luminescence properties. Adv. Powder Technol. 2020, 31, 2366–2378. [Google Scholar] [CrossRef]
- Zhang, Y.; Wu, Z.; Geng, D.; Kang, X.; Shang, M.; Li, X.; Lian, H.; Cheng, Z.; Lin, J. Full color emission in ZnGa2O4: Simultaneous control of the spherical morphology, luminescent, and electric properties via hydrothermal approach. Adv. Funct. Mater. 2014, 24, 6581–6593. [Google Scholar] [CrossRef]
- Allendorf, M.D.; Bauer, C.A.; Bhakta, R.K.; Houk, R.J.T. Luminescent metal-organic frameworks. Chem. Soc. Rev. 2009, 38, 1330–1352. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Q.; Li, S.Y.; Wang, Q.; Qi, Y.; Li, X.D.; Sun, X.D.; Li, J.-G. Grafting of terbium (III) complexes onto layered rare-earth hydroxide nanosheets to fabricate novel optical fiber temperature sensors. Nanoscale 2019, 11, 2795–2804. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Q.; Ding, S.N.; Xiahou, J.Q.; Li, S.Y.; Xu, X.D.; Li, J.-G. A groundbreaking strategy for fabricating YAG:Ce3+ transparent ceramic film via sintering of LRH nanosheets on sapphire. Chem. Commun. 2020, 56, 12761–12764. [Google Scholar] [CrossRef]
- Zhu, Q.; Liu, J.J.; Li, X.D.; Li, J.-G. Grafting organic antenna onto rare earth hydroxynitrate nanosheets for excitation-dependent and greatly enhanced photoluminescence by multi-modal energy transfer. Appl. Surf. Sci. 2019, 489, 142–148. [Google Scholar] [CrossRef]
- Zhu, Q.; Song, C.Y.; Tong, W.P.; Li, J.-G. Controllable phase/morphology tailoring of REF3 and NaREF4 (RE=La-Lu, Y), and insights into the up-conversion luminescence of GdF3:Yb3+/Tm3+ spheres. Adv. Powder Technol. 2020, 31, 2235–2243. [Google Scholar] [CrossRef]
- Fan, X.F.; Gu, L.Q.; Hu, Y.L.; Zhu, Q. Wearing an organic “coat” on nanocrystals of LaF3:Eu3+ to generate dynamic luminescence for optical anti-counterfeit. Adv. Powder Technol. 2021, 32, 2645–2653. [Google Scholar] [CrossRef]
- Okubo, K.; Shigeta, T. Absolute fluorescent quantum efficiency of NBS phosphor standard sample. J. Illum. Eng. Inst. Jpn. 1999, 83, 87–93. [Google Scholar] [CrossRef] [Green Version]
- Shannon, R.D. Revised effective ionic radii and systematic studies of interatomic distances in halides and chalcogenides. Acta Cryst. 1976, A32, 751–767. [Google Scholar] [CrossRef]
- Jeong, H.; Lee, B.-I.; Byeon, S.-H. Antenna effect on the organic spacer-modified Eu-doped layered gadolinium hydroxide for the detection of vanadate ions over a wide pH range. ACS Appl. Mater. Interfaces 2016, 8, 10946–10953. [Google Scholar] [CrossRef]
- Nakamoto, K. Infrared Spectra of Inorganic and Coordination Compounds; John Wiley & Sons: New York, NY, USA, 1963. [Google Scholar]
- Gadsden, J.A. Infrared Spectra of Minerals and Related Inorganic Compounds; Butterworths: London, UK, 1975. [Google Scholar]
- Zhang, L.Z.; Wang, Z.H.; Huang, J.; Tang, F. Fabrication and photoluminescence properties of Eu3+ doped Y2O3 ceramic fiber with high aspect ratio. Chin. J. Lumin. 2021, 4, 1891–1899. [Google Scholar] [CrossRef]
- Huang, S.; Wang, Z.H.; Zhu, Q.; Shi, X.F.; Wang, X.J.; Li, X.D.; Sun, X.D.; Li, J.-G. A new protocol for templated synthesis of YVO4: Ln luminescent crystallites (Ln=Eu, Dy, Sm). J. Alloys Compd. 2019, 776, 773–781. [Google Scholar] [CrossRef]
- Hsu, C.; Powell, R.C. Energy transfer in europium doped yttrium vanadate crystals. J. Lumin. 1975, 10, 273–293. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).