

Interaction Regularity of Biomolecules on Mg and Mg-Based Alloy Surfaces: A First-Principles Study


Abstract
:1. Introduction
2. Computational Methods
3. Results and Discussion
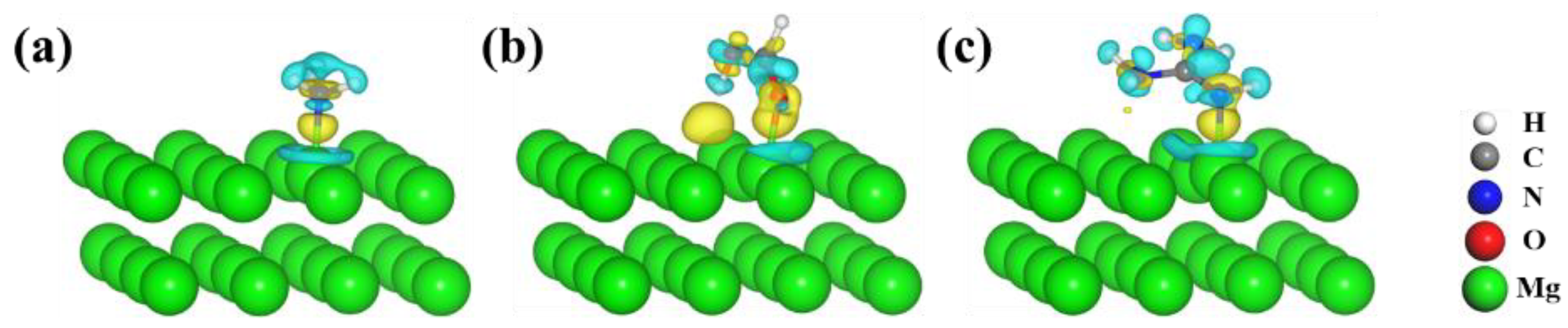
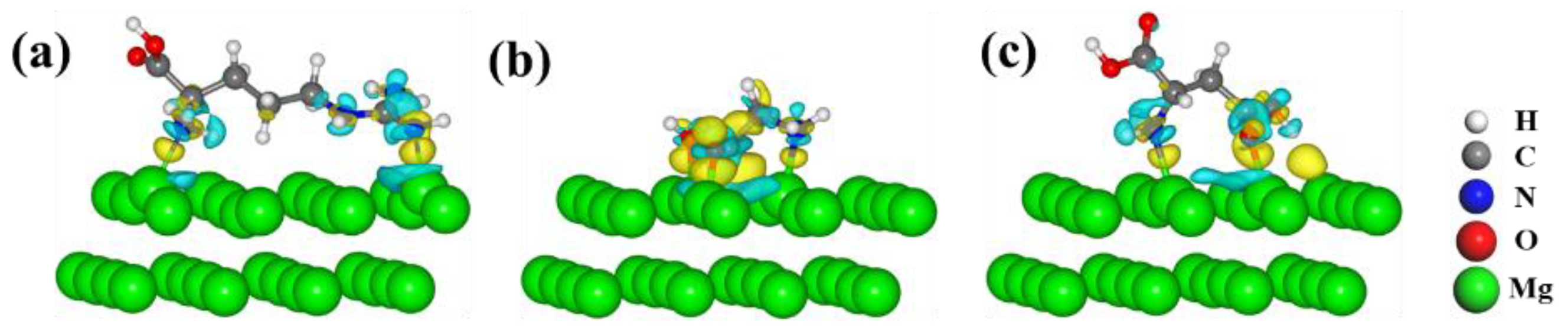
3.1. Adsorption Properties of Molecules on Mg(0001) Surfaces
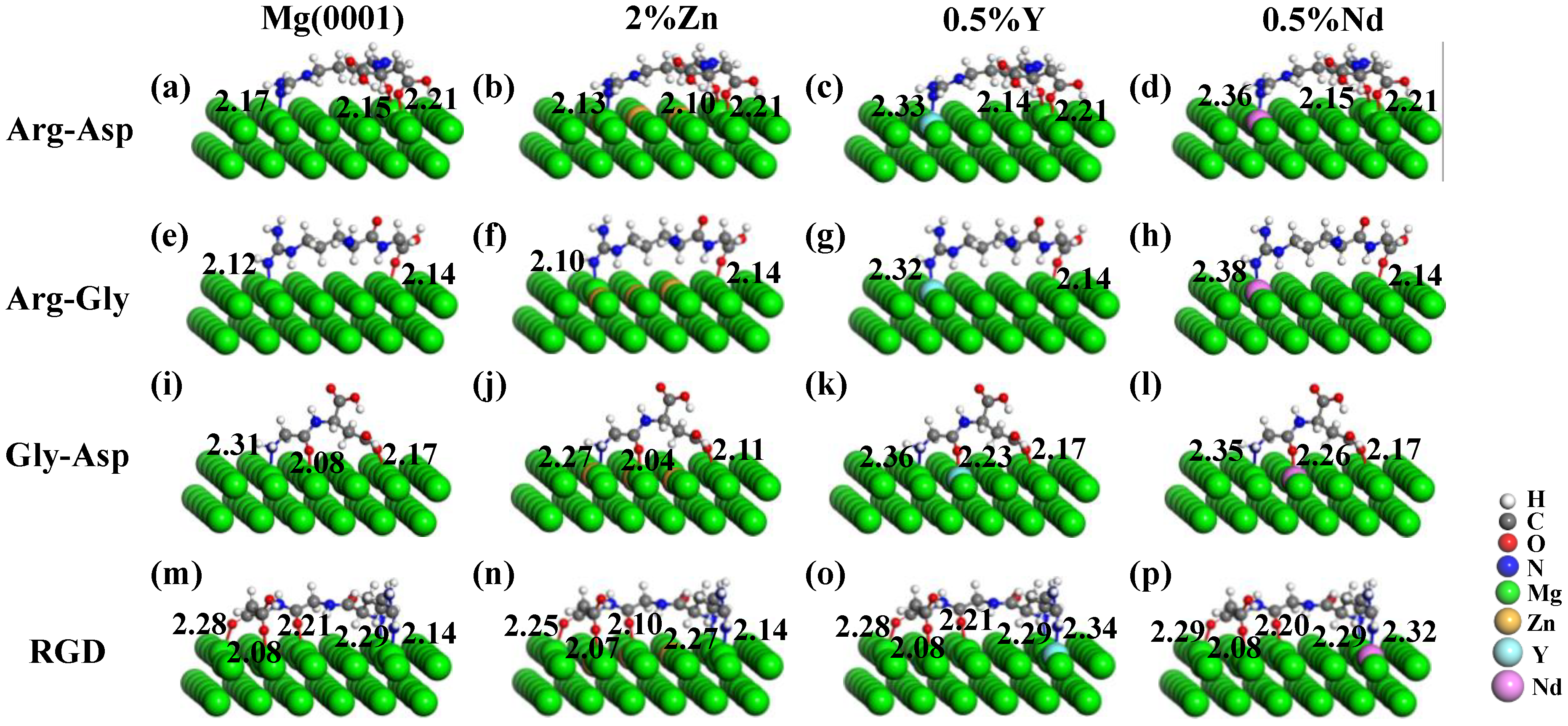
3.2. Effect of Alloying Elements on the Adsorption of Molecules on Mg(0001) Surfaces
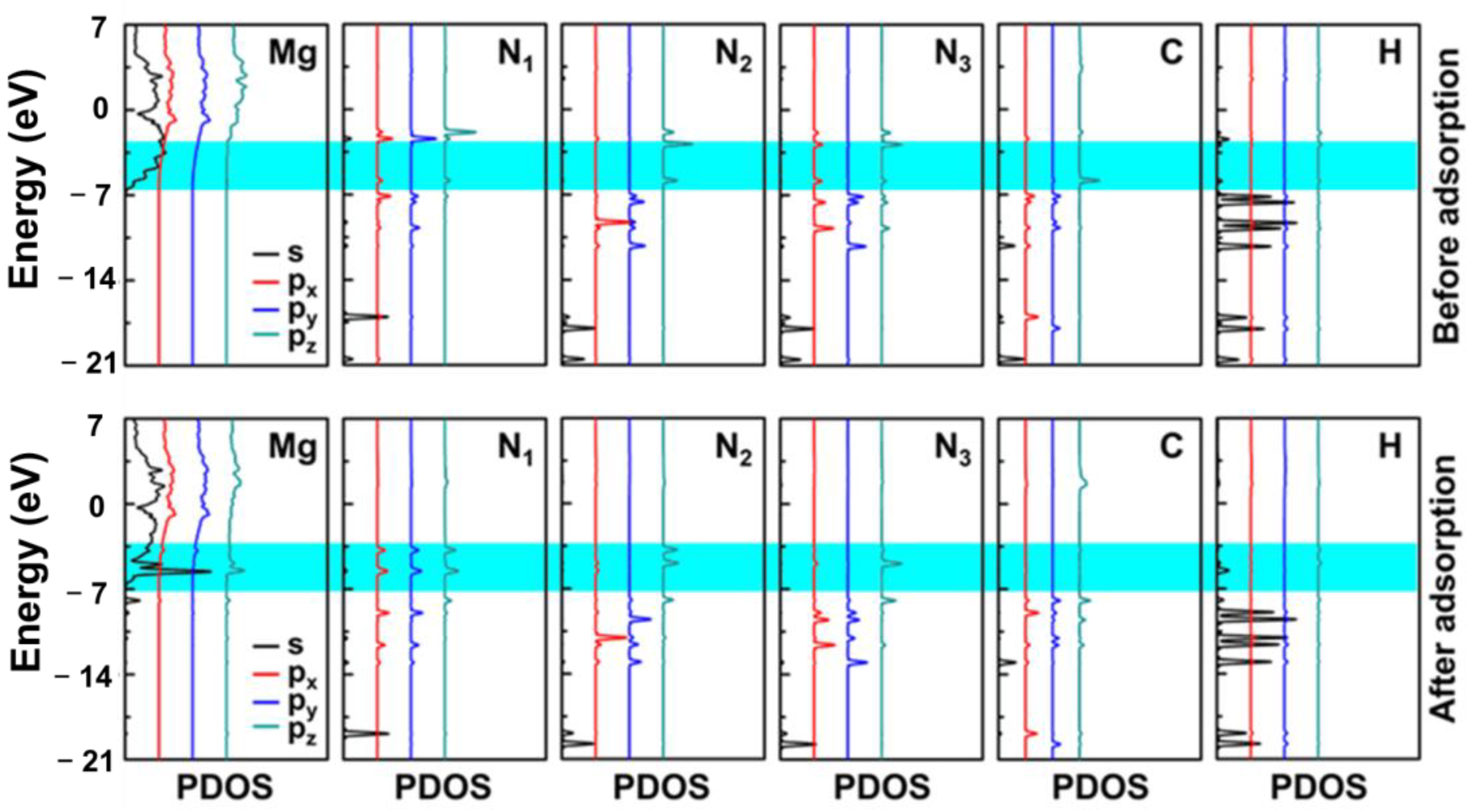
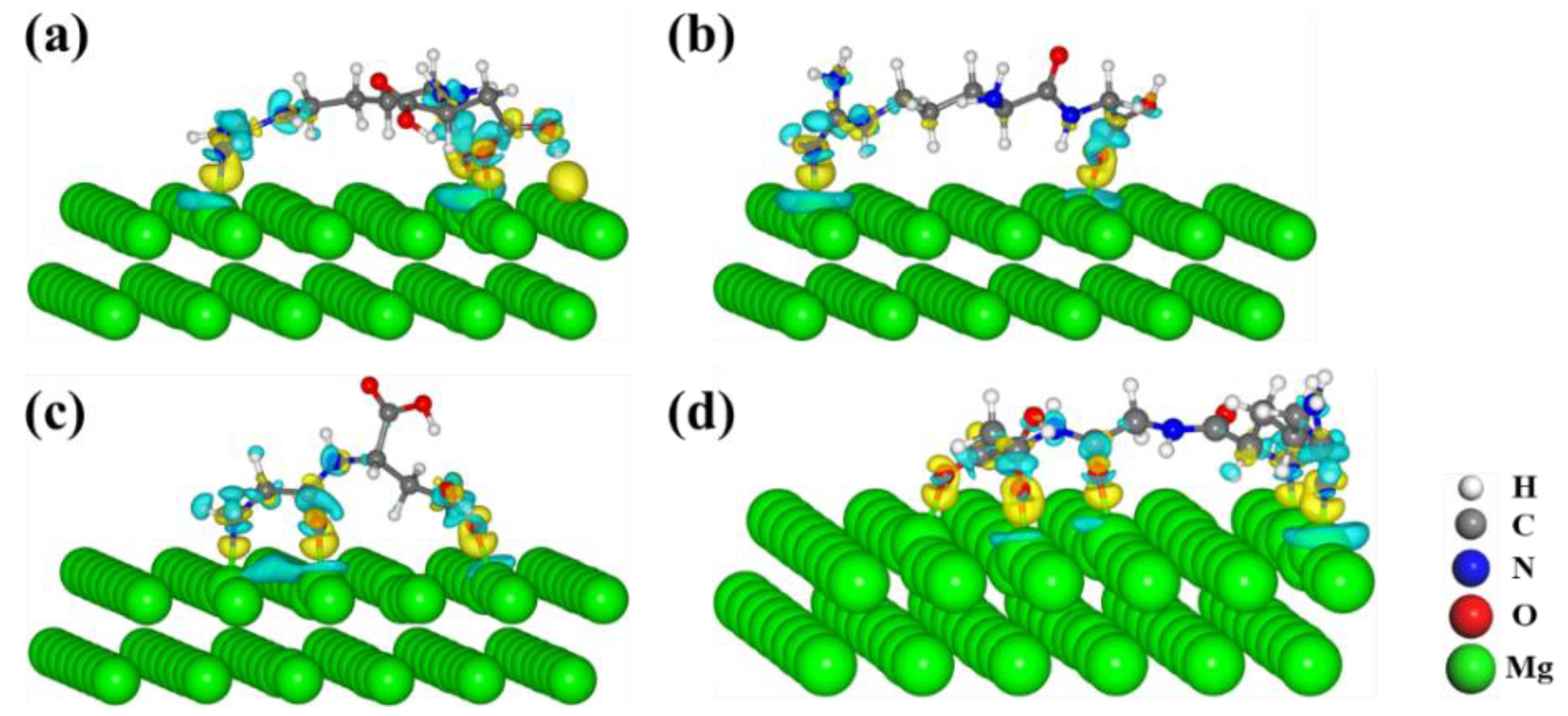
3.3. Electronic Properties of Molecules on the Mg(0001) Surface
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hao, D.K.; Lin, J.; Liu, R.W.; Pivetti, C.; Yamashiro, K.; Schutzman, L.M.; Sageshima, J.; Kwong, M.; Bahatyrevich, N.; Farmer, D.L.; et al. A bio-instructive parylene-based conformal coating suppresses thrombosis and intimal hyperplasia of implantable vascular devices. Bioact. Mater. 2023, 28, 467–479. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.Y.; Thangam, R.; Kang, N.Y.; Hong, H.; Kim, C.; Lee, S.K.Y.; Son, S.; Lee, H.J.; Tag, K.R.; Min, S.H.; et al. Modulation of macrophages by in situ ligand bridging. Adv. Funct. Mater. 2023, 33, 2215166. [Google Scholar] [CrossRef]
- Sung, T.C.; Wang, T.; Liu, Q.; Ling, Q.D.; Subbiah, S.K.; Renuka, R.R.; Hsu, S.T.; Umezawa, A.; Higuchi, A. Cell-binding peptides on the material surface guide stem cell fate of adhesion, proliferation and differentiation. J. Mater. Chem. B 2023, 11, 1389–1415. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.Q.; Wang, H.Y.; Wang, L.; Chen, X.B.; Guan, S.K.; Lin, C.G.; Zeng, R.C. Protein conformation and electric attraction adsorption mechanisms on anodized magnesium alloy by molecular dynamics simulations. J. Magnes. Alloys 2022, 10, 3143–3155. [Google Scholar] [CrossRef]
- Raikar, A.S.; Priya, S.; Bhilegaonkar, S.P.; Somnache, S.N.; Kalaskar, D.M. Surface Engineering of Bioactive Coatings for Improved Stent Hemocompatibility: A Comprehensive Review. Materials 2023, 16, 6940. [Google Scholar] [CrossRef]
- Chen, J.; Liu, X.H.; Hong, Q.X.; Meng, L.J.; Ji, Y.; Wang, L.T.; Zhang, Q.Y.; Lin, J.F.; Pan, C.J. Synthesizing a multifunctional polymer to construct the catalytically NO-generating coating for improving corrosion resistance and biocompatibility of the magnesium alloy stent materials. Prog. Org. Coat. 2023, 186, 108058. [Google Scholar] [CrossRef]
- Fang, H.; Wang, C.X.; Zhou, S.C.; Zheng, Z.; Lu, T.; Li, G.; Tian, Y.H.; Suga, T. Enhanced adhesion and anticorrosion of silk fibroin coated biodegradable Mg-Zn-Ca alloy via a two-step plasma activation. Corros. Sci. 2020, 168, 108466. [Google Scholar] [CrossRef]
- Li, H.T.; Si, S.H.; Yang, K.; Mao, Z.A.; Sun, Y.H.; Cao, X.R.; Yu, H.T.; Zhang, J.W.; Ding, C.; Liang, H.X.; et al. Hexafluoroisopropanol based silk fibroin coatings on AZ31 biometals with enhanced adhesion, corrosion resistance and biocompatibility. Prog. Org. Coat. 2023, 184, 107881. [Google Scholar] [CrossRef]
- Li, D.; Dai, D.N.; Xiong, G.G.; Lan, S.Q.; Zhang, C. Composite nanocoatings of biomedical magnesium alloy implants: Advantages, mechanisms, and design strategies. Adv. Sci. 2023, 10, 2300658. [Google Scholar] [CrossRef]
- Shahed, C.A.; Ahmad, F.; Günister, E.; Foudzi, F.M.; Ali, S.; Malik, K.; Harun, W.S.W. Antibacterial mechanism with consequent cytotoxicity of different reinforcements in biodegradable magnesium and zinc alloys: A review. J. Magnes. Alloys 2023, 11, 3038–3058. [Google Scholar] [CrossRef]
- Fan, Z.W.; Zou, Y.B.; Liu, C.L.; Xiang, S.C.; Zhang, Z.J. Hydrogen-Bonded organic frameworks: Functionalized construction strategy by nitrogen-containing functional group. Chem.—A Eur. J. 2022, 28, e202200422. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.K.; Dong, Y.; Wang, Y.Y. Collagen-based biomaterials for tissue engineering. ACS Biomater. Sci. Eng. 2023, 9, 1132–1150. [Google Scholar] [CrossRef] [PubMed]
- Acet, Ö.; Shcharbin, D.; Zhogla, V.; Kirsanov, P.; Halets-Bui, I.; Acet, B.; Gök, T.; Bryszewska, M.; Odabasi, M. Dipeptide nanostructures: Synthesis, interactions, advantages and biomedical applications. Colloids Surf. B-Biointerfaces 2023, 222, 113031. [Google Scholar] [CrossRef] [PubMed]
- Li, L.C.; Xie, L.; Zheng, R.L.; Sun, R.Q. Self-assembly dipeptide hydrogel: The structures and properties. Front. Chem. 2021, 9, 739791. [Google Scholar] [CrossRef]
- Shams, E.; Barzad, M.S.; Mohamadnia, S.; Tavakoli, O.; Mehrdadfar, A. A review on alginate-based bioinks, combination with other natural biomaterials and characteristics. J. Biomater. Appl. 2022, 37, 355–372. [Google Scholar] [CrossRef]
- Yang, F.; Guo, G.Y.; Wang, Y.B. Inflammation-directed nanozyme-eluting hydrogel coating promotes vascular tissue repair by restoring reactive oxygen species homeostasis. Chem. Eng. J. 2023, 454, 140556. [Google Scholar] [CrossRef]
- Chen, J.J.; Hu, G.S.; Li, T.J.; Chen, Y.H.; Gao, M.; Li, Q.T.; Hao, L.J.; Jia, Y.G.; Wang, L.; Wang, Y.J. Fusion peptide engineered “statically-versatile” titanium implant simultaneously enhancing anti-infection, vascularization and osseointegration. Biomaterials 2021, 264, 120446. [Google Scholar] [CrossRef]
- Liang, X.Y.; Yang, L.; Lei, Y.; Zhang, S.M.; Chen, L.; Hu, C.; Wang, Y.B. Biomimetic-modified bioprosthetic heart valves with Cysteine-Alanine-Glycine peptide for anti-thrombotic, endothelialization and anti-calcification. Int. J. Biol. Macromol. 2023, 250, 126244. [Google Scholar] [CrossRef]
- Yan, L.W.; Zhou, T.; Ni, R.C.; Jia, Z.R.; Jiang, Y.A.; Guo, T.L.; Wang, K.F.; Chen, X.; Han, L.; Lu, X. Adhesive Gelatin-Catechol complex reinforced poly(Acrylic Acid) hydrogel with enhanced toughness and cell affinity for cartilage regeneration. ACS Appl. Bio Mater. 2022, 5, 4366–4377. [Google Scholar] [CrossRef]
- Aghaei, M.; Ramezanitaghartapeh, M.; Javan, M.; Hoseininezhad-Namin, M.S.; Mirzaei, H.; Rad, A.S.; Soltani, A.; Sedighi, S.; Lup, A.N.K.; Khori, V.; et al. Investigations of adsorption behavior and anti-inflammatory activity of glycine functionalized Al12N12 and Al12ON11 fullerene-like cages. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2021, 246, 119023. [Google Scholar] [CrossRef]
- Wang, Z.Q.; Song, T.; Guo, Z.W.; Cao, K.K.; Chen, C.; Feng, Y.G.; Wang, H.; Yin, F.K.; Zhou, S.; Dai, J.; et al. Targeting the allosteric pathway that interconnects the core- functional scaffold and the distal phosphorylation sites for specific dephosphorylation of Bcl-2. J. Med. Chem. 2020, 63, 13733–13744. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Jiang, M.; Zhu, D.Z.; Zhang, J.M.; Wei, G. Metal-organic frameworks functionalized with nucleic acids and amino acids for structure- and function-specific applications: A tutorial review. Chem. Eng. J. 2022, 428, 131118. [Google Scholar] [CrossRef]
- Schweitzer-Stenner, R. The relevance of short peptides for an understanding of unfolded and intrinsically disordered proteins. Phys. Chem. Chem. Phys. 2023, 25, 11908–11933. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.L.; Yi, J.H.; Gao, Q.; Wang, X.D.; Chen, Y.S.; Liu, P. Carboxymethyl chitosan functionalization of CPED-treated magnesium alloy via polydopamine as intermediate layer. Surf. Coat. Technol. 2014, 258, 664–671. [Google Scholar] [CrossRef]
- Guo, X.; Hu, Y.P.; Yuan, K.Z.; Qiao, Y. Review of the Effect of Surface Coating Modification on Magnesium Alloy Biocompatibility. Materials 2022, 15, 3291. [Google Scholar] [CrossRef] [PubMed]
- Lin, Z.S.; Wang, T.L.; Yu, X.M.; Sun, X.T.; Yang, H.Z. Functionalization treatment of micro-arc oxidation coatings on magnesium alloys: A review. J. Alloys Compd. 2021, 879, 160453. [Google Scholar] [CrossRef]
- Farwa, U.; Lee, H.Y.; Lim, H.; Park, I.; Park, S.; Moon, B.G.; Lee, B.T. Poly(L-lactide)/polycaprolactone based multifunctional coating to deliver paclitaxel/VEGF and control the degradation rate of magnesium alloy stent. Int. J. Biol. Macromol. 2023, 250, 126218. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.; Dai, B.; Cui, X.F.; Li, P. The effects of electrodeposition temperature on morphology and corrosion resistance of calcium phosphorus coatings on magnesium alloy: Comparative experimental and molecular dynamics simulation studies. RSC Adv. 2021, 13, 34145–34156. [Google Scholar] [CrossRef]
- Wang, Y.W.; Tang, Q.L.; Xu, X.C.; Weng, P.L.; Ying, T.; Yang, Y.; Zeng, X.Q.; Zhu, H. Accelerated discovery of magnesium intermetallic compounds with sluggish corrosion cathodic reactions through active learning and DFT calculations. Acta Mater. 2023, 255, 119063. [Google Scholar] [CrossRef]
- Mamad, D.M.; Omer, R.A.; Othman, K.A. Quantum chemical analysis of amino acids as anti-corrosion agents. Corros. Rev. 2023, 41, 703–717. [Google Scholar] [CrossRef]
- Méthivier, C.; Cornette, P.; Costa, D.; Landoulsi, J. Electrospray ion beam deposition of small peptides on solid surfaces: A molecular level description of the glutathione/copper interface. Appl. Surf. Sci. 2023, 612, 155895. [Google Scholar] [CrossRef]
- Fang, Z.; Wang, J.F.; Yang, X.F.; Sun, Q.; Jia, Y.; Liu, H.R.; Xi, T.F.; Guan, S.K. Adsorption of arginine, glycine and aspartic acid on Mg and Mg-based alloy surfaces: A first-principles study. Appl. Surf. Sci. 2017, 409, 149–155. [Google Scholar] [CrossRef]
- Fang, Z.; Wang, J.F.; Zhu, S.J.; Yang, X.F.; Jia, Y.; Sun, Q.; Guan, S.K. A DFT study of the adsorption of short peptides on Mg and Mg-based alloy surfaces. Phys. Chem. Chem. Phys. 2018, 20, 3602–3607. [Google Scholar] [CrossRef] [PubMed]
- Fang, Z.; Zhao, Y.; Wang, H.Y.; Wang, J.F.; Zhu, S.J.; Jia, Y.; Cho, J.Y.; Guan, S.K. Influence of surface charge density on ligand-metal bonding: A DFT study of NH3 and HCOOH on Mg (0001) surface. Appl. Surf. Sci. 2019, 470, 893–898. [Google Scholar] [CrossRef]
- Fang, Z.; Wei, W.T.; Qiao, H.J.; Liang, E.R.; Jia, Y.; Guan, S.K. Effect of vacancy defects and hydroxyl on the adsorption of Glycine on Mg(0001): A first-principles study. Coatings 2023, 13, 1684. [Google Scholar] [CrossRef]
- Kresse, G.; Hafner, J. Ab-initio molecular-dynamics simulation of the liquid-metal amorphous-semicinductor transition in germanium. Phys. Rev. B 1994, 49, 14251–14269. [Google Scholar] [CrossRef] [PubMed]
- Kresse, G.; Furthmuller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef] [PubMed]
- Kresse, G.; Furthmuller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Blochl, P.E. Projector augmented-wave method. Phys. Rev. B 1994, 50, 17953–17979. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1997, 78, 1396. [Google Scholar] [CrossRef]
- Lüder, J.; Sanyal, B.; Eriksson, O.; Puglia, C.; Brena, B. Comparison of van der Waals corrected and sparse-matter density functionals for the metal-free phthalocyanine/gold interface. Phys. Rev. B 2014, 89, 045416. [Google Scholar] [CrossRef]
- Sowmiya, M.; Senthilkumar, K. Adsorption of RGD tripeptide on anatase (001) surface—A first principle study. Comput. Mater. Sci. 2015, 104, 124–129. [Google Scholar] [CrossRef]
- Swanson, H.E.; McMurdie, H.F.; Morris, M.C.; Evans, E.H. Standard X-ray Diffraction Powder Patterns. In National Bureau of Standards Monograph 25—Section 6; U.S. Department of Commerce, National Bureau of Standards: Gaithersburg, MD, USA, 1968. [Google Scholar]
- Madden, D.C.; Temprano, I.; Sacchi, M.; Jenkins, S.J. Spontaneous local symmetry breaking: A conformational study of Glycine on Cu{311}. J. Phys. Chem. C 2015, 119, 13041–13049. [Google Scholar] [CrossRef]
- Höffling, B.; Ortmann, F.; Hannewald, K.; Bechstedt, F. Single cysteine adsorption on Au(110): A first-principles study. Phys. Rev. B 2010, 81, 045407. [Google Scholar] [CrossRef]
- Sargent, W. Table of Periodic Properties of the Elements; Sargent-Welch Scientific: Skokie, IL, USA, 1980. [Google Scholar]
- Nilsson, A.; Pettersson, L.G.M. Chemical bonding on surfaces probed by X-ray emission spectroscopy and density functional theory. Surf. Sci. Rep. 2004, 55, 49–167. [Google Scholar] [CrossRef]
- Jiang, Z.; Qin, P.; Fang, T. Decomposition mechanism of formic acid on Cu (111) surface: A theoretical study. Appl. Surf. Sci. 2017, 396, 857–864. [Google Scholar] [CrossRef]
- Zhao, N.; Zhu, D.H. Endothelial responses of magnesium and other alloying elements in magnesium-based stent materials. Metallomics 2015, 7, 113–123. [Google Scholar] [CrossRef]
- Wang, B.; Guan, S.K.; Wang, J.; Wang, L.G.; Zhu, S.J. Effects of Nd on microstructures and properties of extruded Mg-2Zn-0.46Y-xNd alloys for stent application. Mater. Sci. Eng. B-Adv. Funct. Solid-State Mater. 2011, 176, 1673–1678. [Google Scholar] [CrossRef]
- Allred, A.L. Electronegativity values from thermochemical data. J. Inorg. Nucl. Chem. 1961, 17, 215–221. [Google Scholar] [CrossRef]
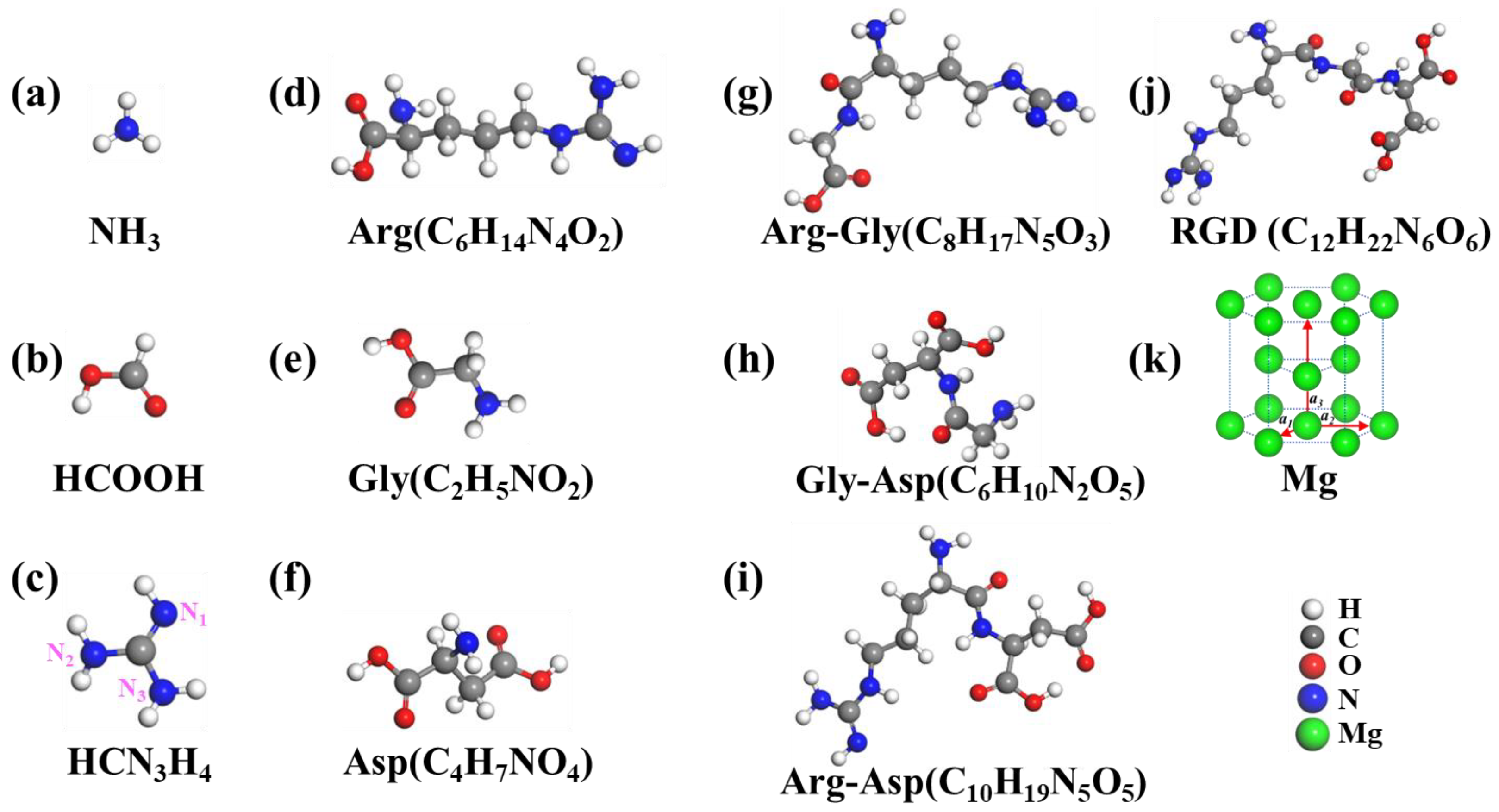
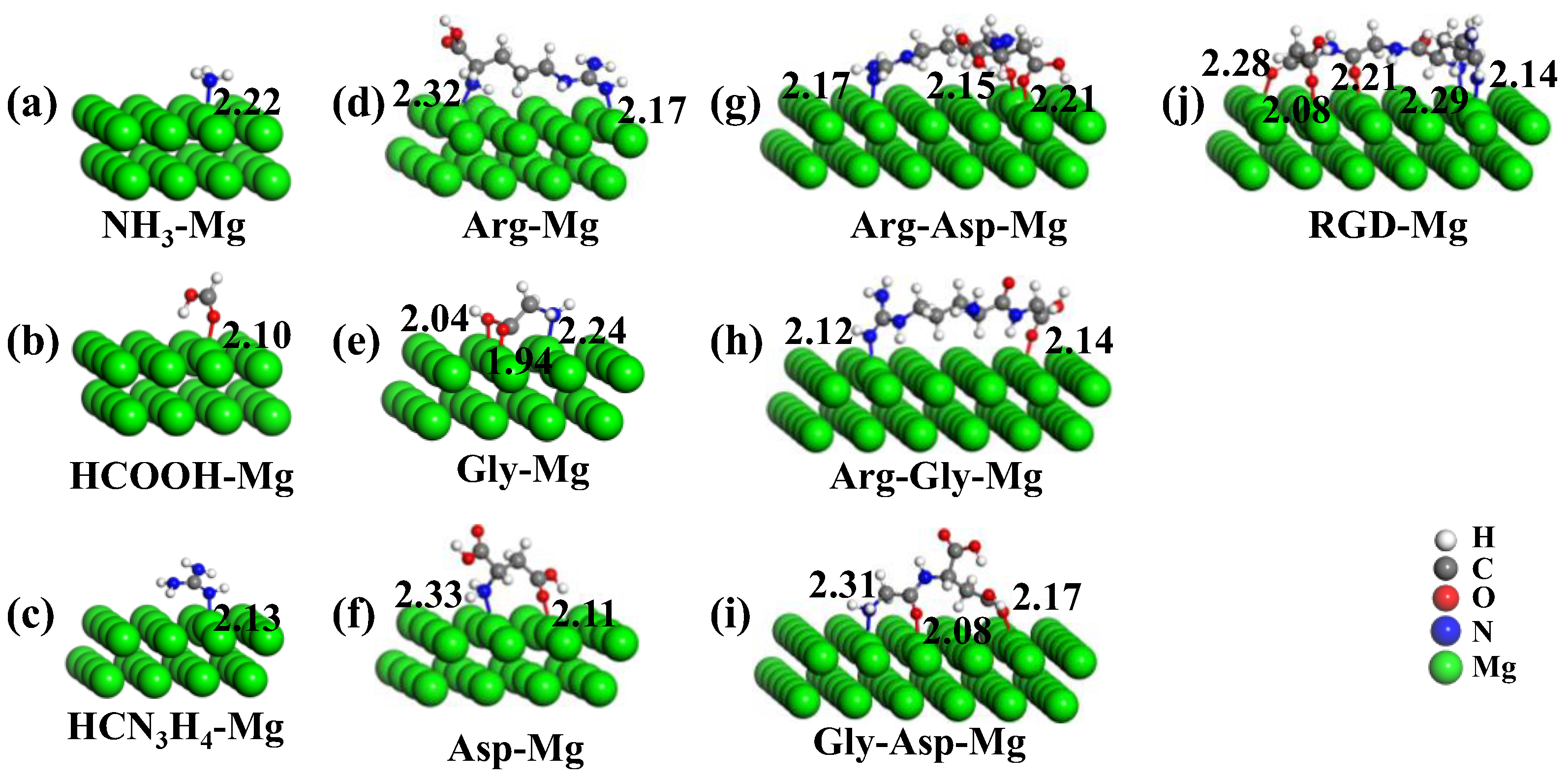
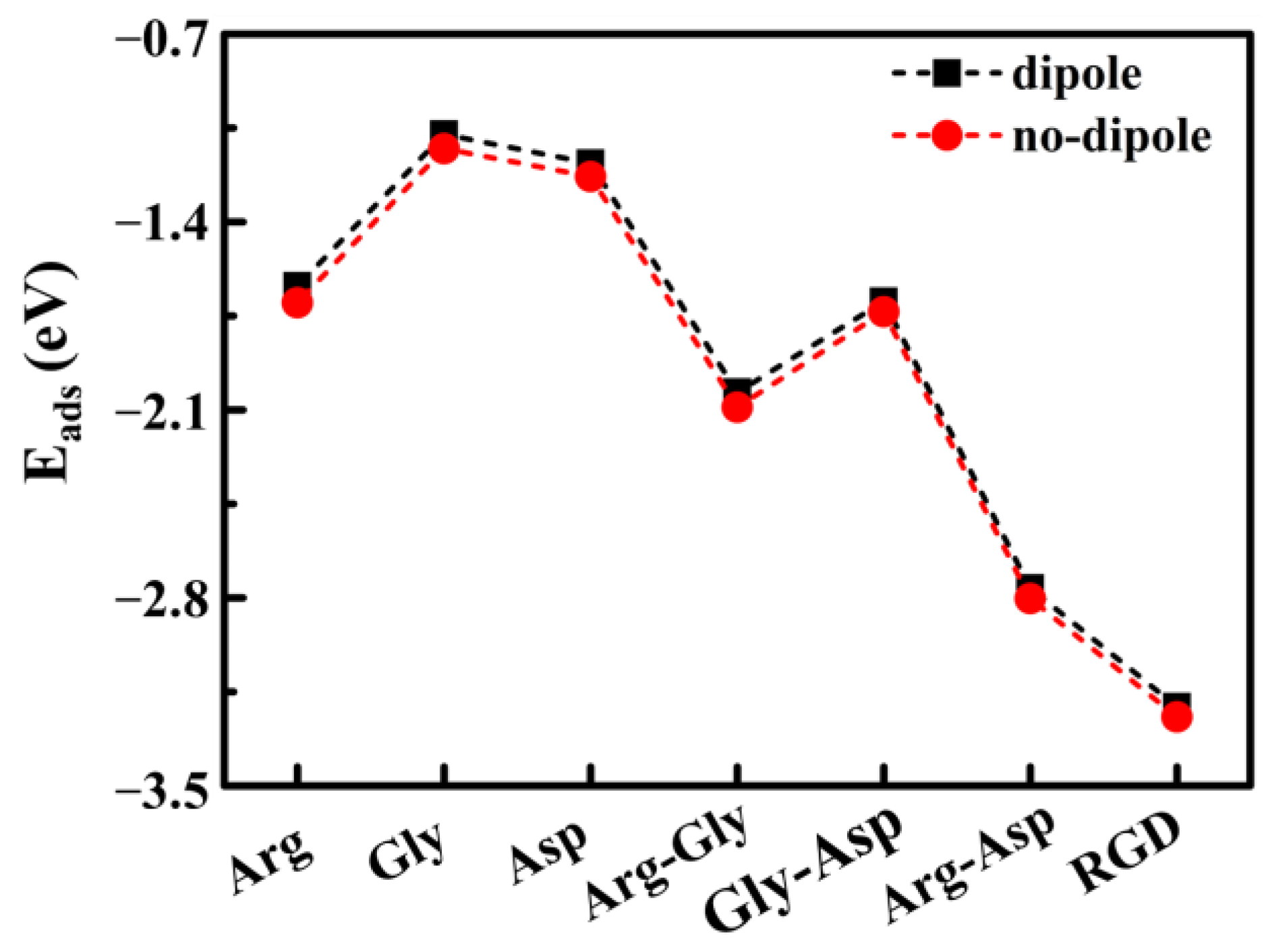
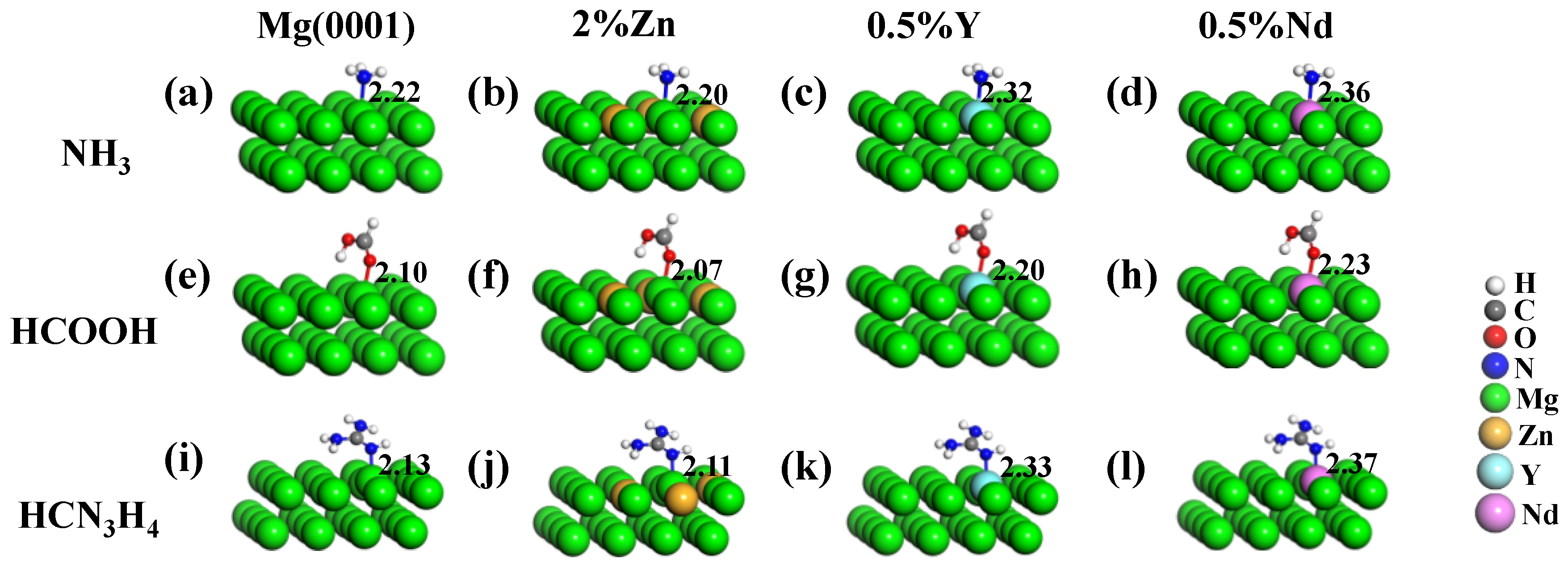


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Adsorption of Functional Groups | |||
|---|---|---|---|
| NH3 | HCOOH | HCN3H4 | |
| Eads (eV) | −0.71 | −0.64 | −1.08 |
| Adsorption of Amino Acids | |||
|---|---|---|---|
| B | Gly | Asp | |
| Eads (eV) | −1.67 | −1.16 | −1.23 |
| Adsorption of Dipeptides and RGD Tripeptide | ||||
|---|---|---|---|---|
| Arg-Asp | Arg-Gly | Gly-Asp | RGD | |
| Eads (eV) | −2.80 | −2.09 | −1.73 | −3.24 |
| Eads (eV) | ||||
|---|---|---|---|---|
| Mg(0001) | 2% Zn | 0.5% Y | 0.5% Nd | |
| NH3 | −0.71 | −0.83 | −1.19 | −1.22 |
| HCOOH | −0.64 | 0.72 | −1.21 | −1.17 |
| HCN3H4 | −1.08 | −1.19 | −1.70 | −1.76 |
| Eads (eV) | ||||
|---|---|---|---|---|
| Mg (0001) | 2% Zn | 0.5% Y | 0.5% Nd | |
| Arg | −1.67 | −1.99 | −2.39 | −2.37 |
| Gly | −1.16 | −1.35 | −2.30 | −1.77 |
| Asp | −1.23 | −1.39 | −1.72 | −2.02 |
| Eads (eV) | ||||
|---|---|---|---|---|
| Mg(0001) | 2% Zn | 0.5% Y | 0.5% Nd | |
| Arg-Asp | −2.80 | −3.06 | −3.41 | −3.53 |
| Arg-Gly | −2.09 | −2.29 | −2.73 | −2.85 |
| Gly-Asp | −1.73 | −2.13 | −2.27 | −2.45 |
| RGD | −3.24 | −3.73 | −3.89 | −4.03 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fang, Z.; Ma, B.; Liang, E.; Jia, Y.; Guan, S. Interaction Regularity of Biomolecules on Mg and Mg-Based Alloy Surfaces: A First-Principles Study. Coatings 2024, 14, 25. https://doi.org/10.3390/coatings14010025
Fang Z, Ma B, Liang E, Jia Y, Guan S. Interaction Regularity of Biomolecules on Mg and Mg-Based Alloy Surfaces: A First-Principles Study. Coatings. 2024; 14(1):25. https://doi.org/10.3390/coatings14010025
Chicago/Turabian StyleFang, Zhe, Baiwei Ma, Erjun Liang, Yu Jia, and Shaokang Guan. 2024. "Interaction Regularity of Biomolecules on Mg and Mg-Based Alloy Surfaces: A First-Principles Study" Coatings 14, no. 1: 25. https://doi.org/10.3390/coatings14010025
APA StyleFang, Z., Ma, B., Liang, E., Jia, Y., & Guan, S. (2024). Interaction Regularity of Biomolecules on Mg and Mg-Based Alloy Surfaces: A First-Principles Study. Coatings, 14(1), 25. https://doi.org/10.3390/coatings14010025