
A Review of Capacity Fade Mechanism and Promotion Strategies for Lithium Iron Phosphate Batteries

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
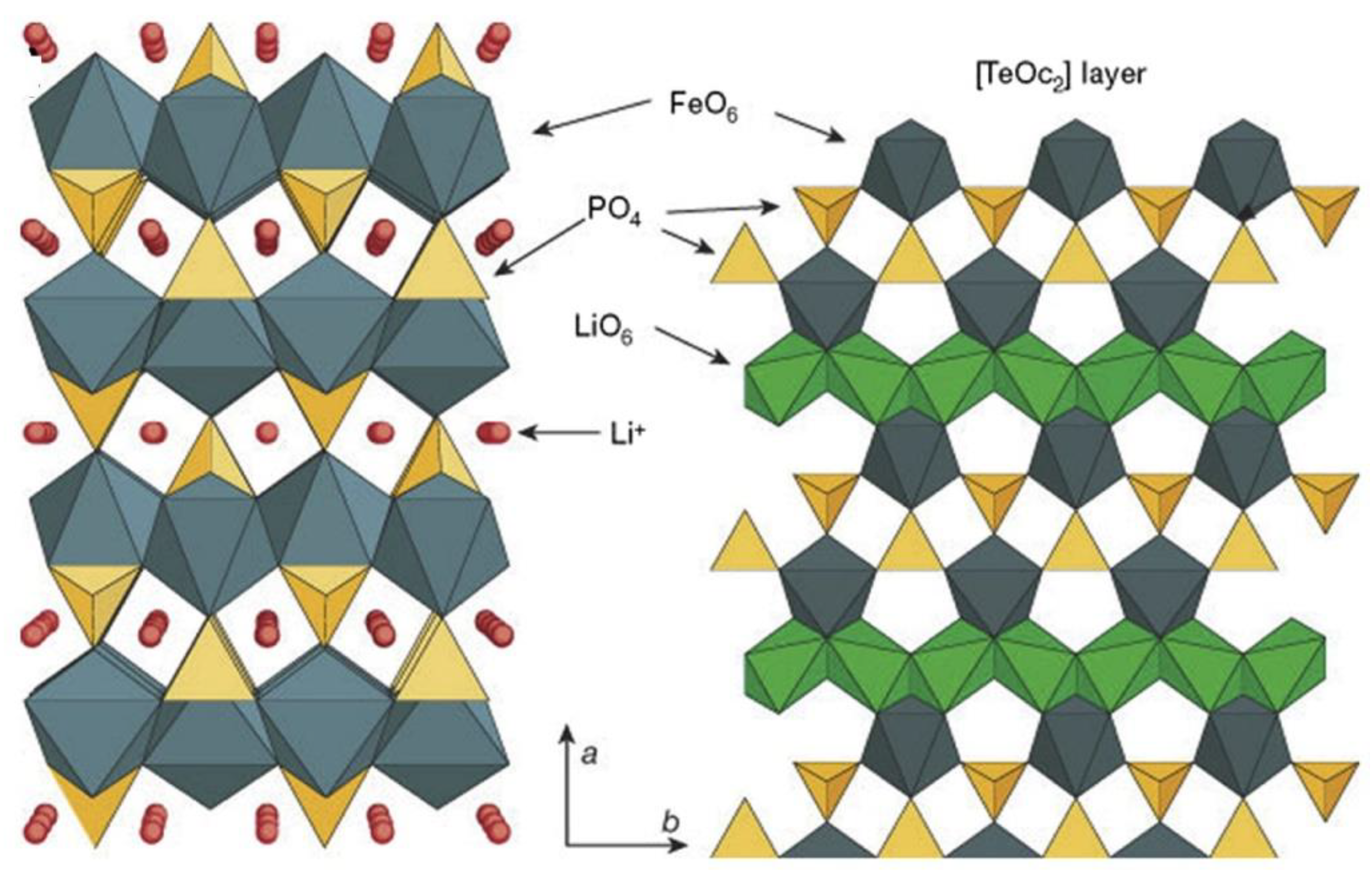
2. Crystal Structure and Electrochemical Performance of LiFePO4
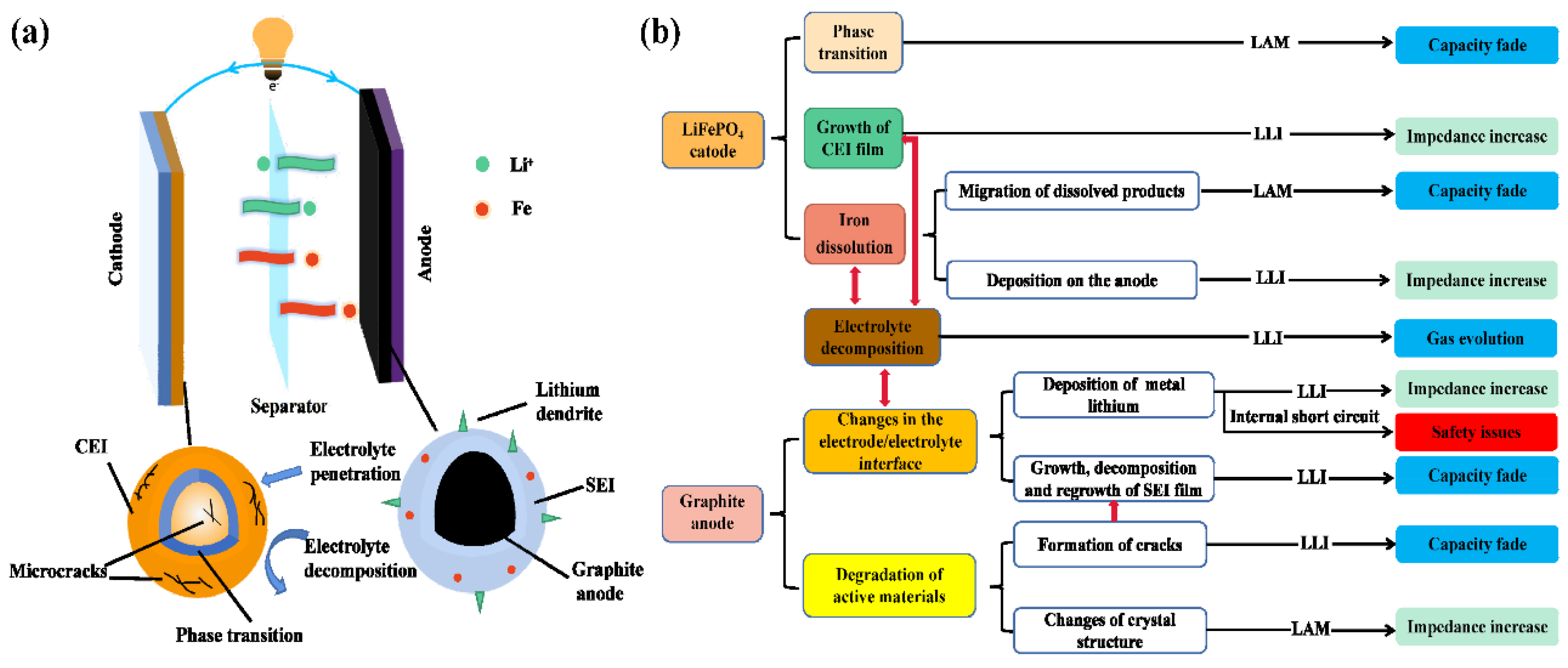
3. Capacity Fade Mechanism
3.1. Degradations at the Cathode
3.1.1. Irreversible Phase Transition

3.1.2. Formation of Cathode–Electrolyte Interface (CEI) Layer
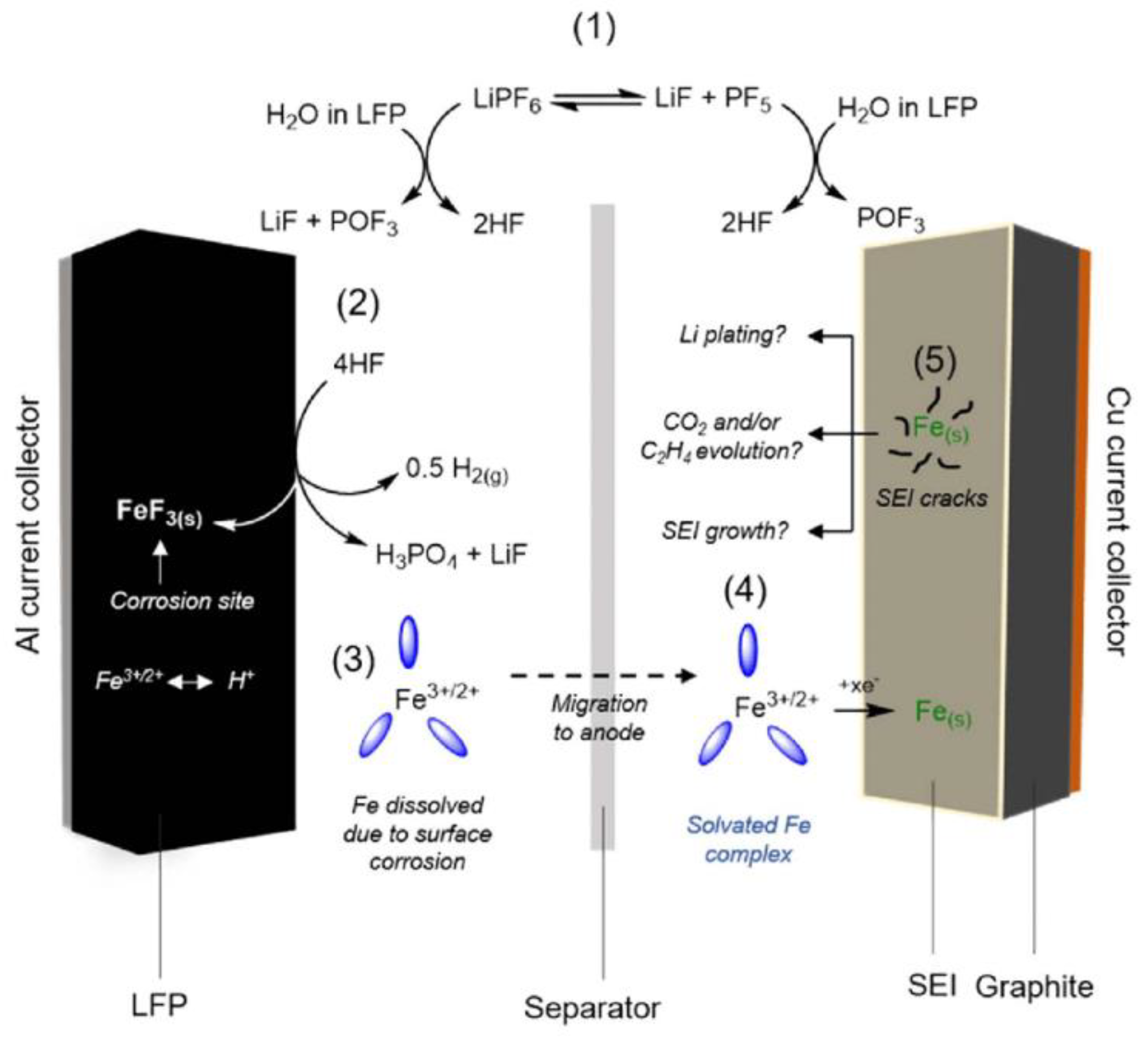
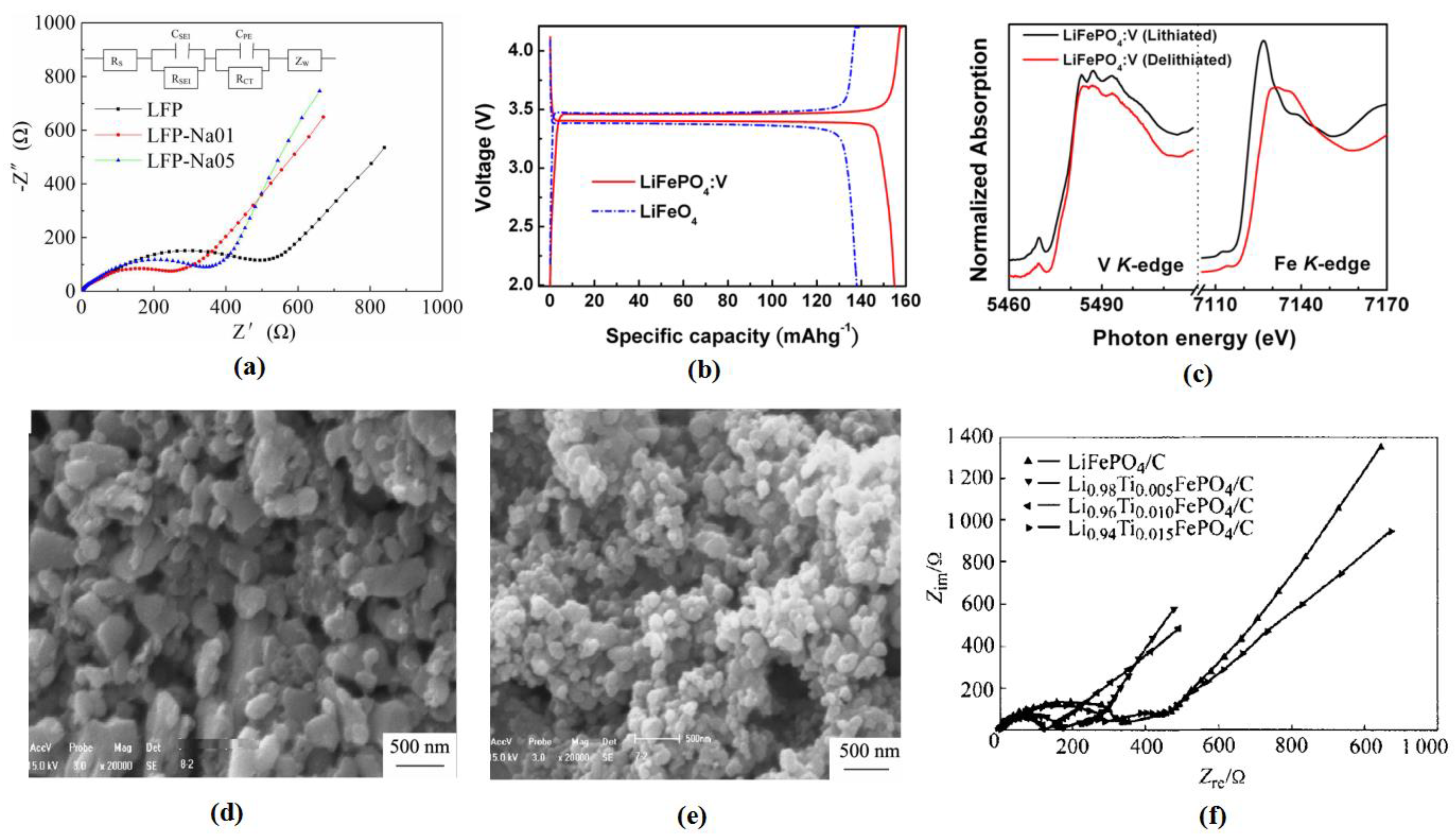
3.1.3. Dissolution and Deposition of Iron
3.2. Degradations at the Graphite Anode
3.2.1. Formation of Irreversible SEI Layer
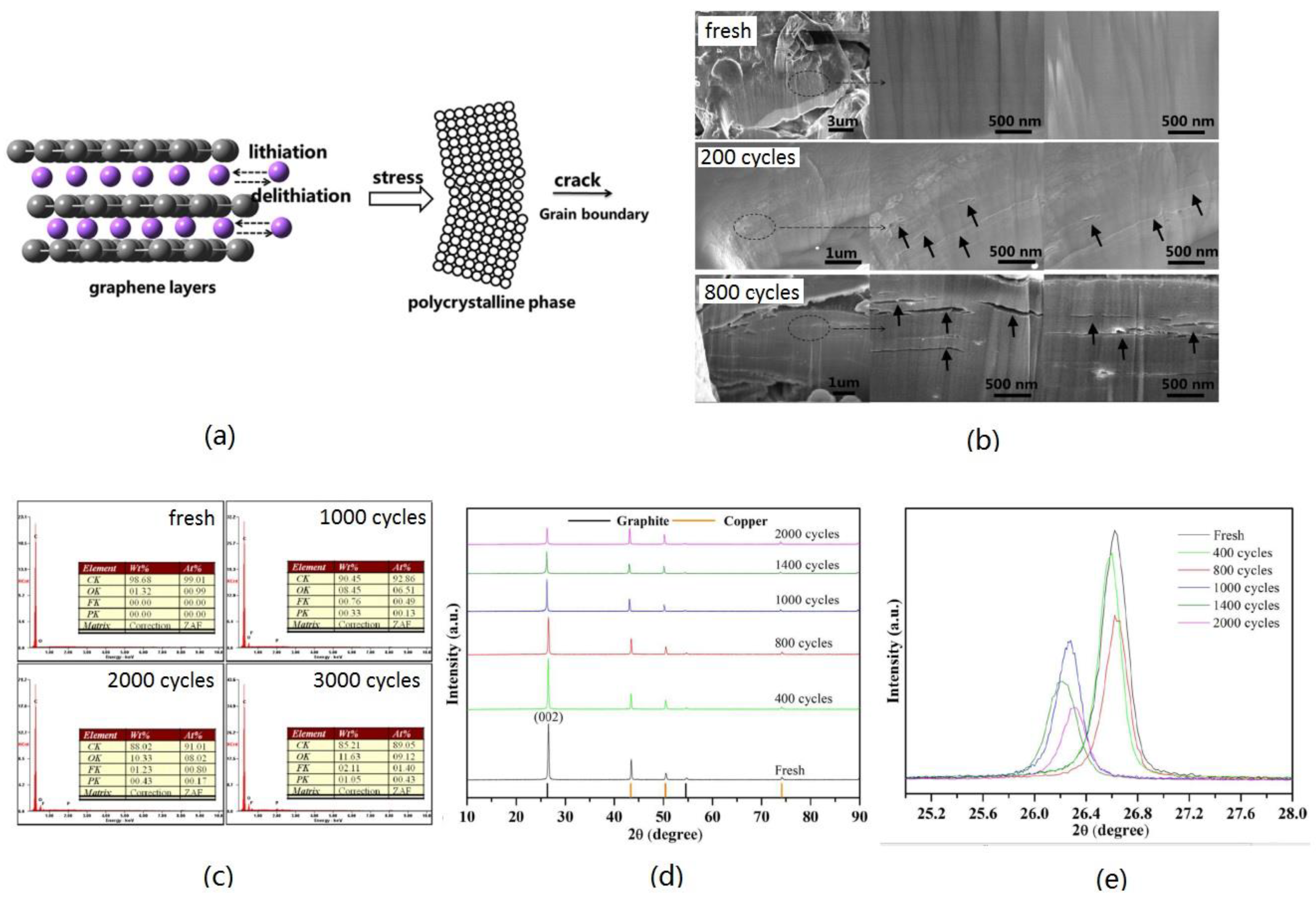
3.2.2. Structure Deterioration
3.2.3. Growth of Lithium Dendrites
4. Enhancement Strategies
4.1. Doping
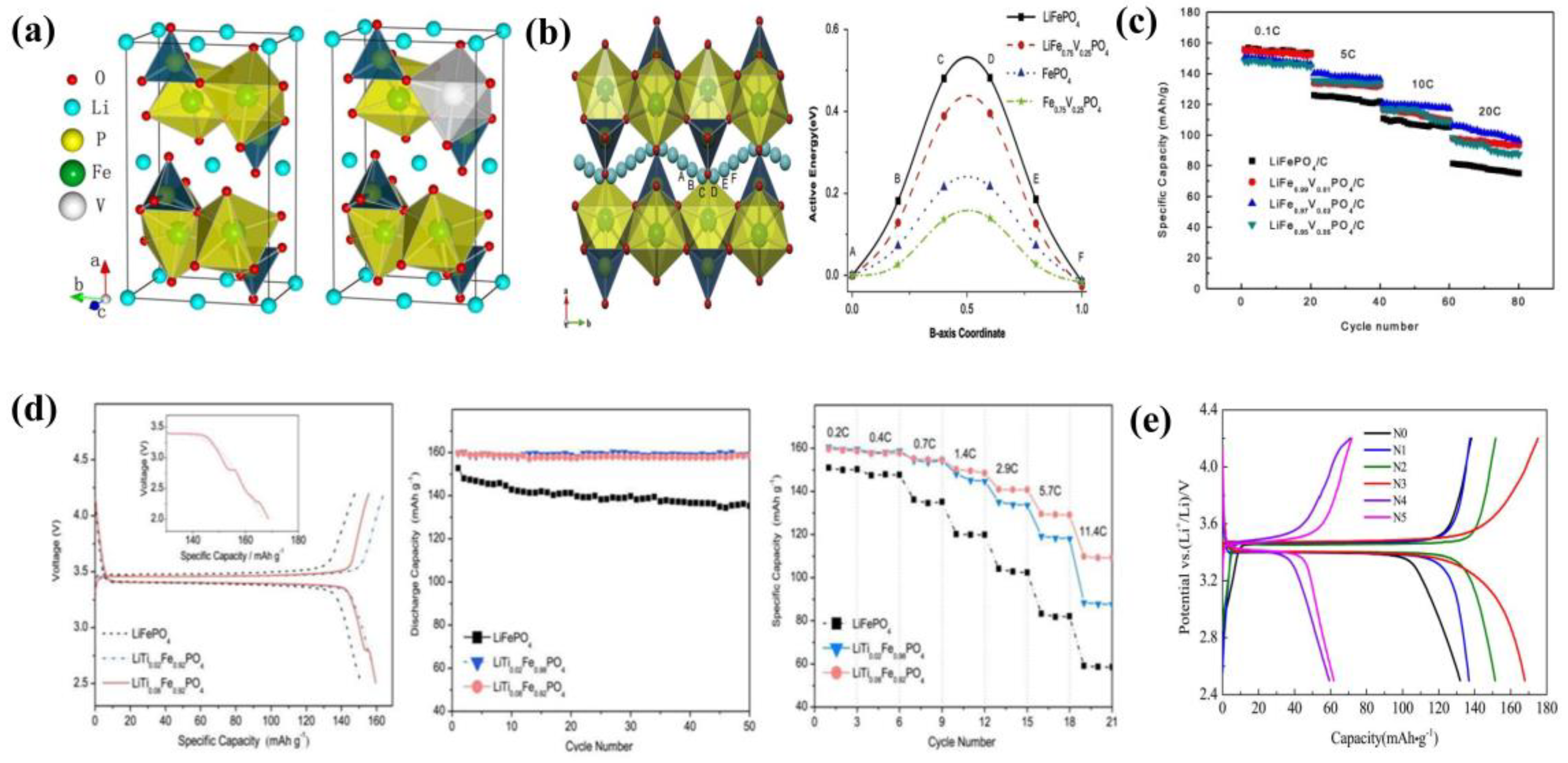
4.1.1. Lithium Site Substitution
4.1.2. Iron Site Substitution
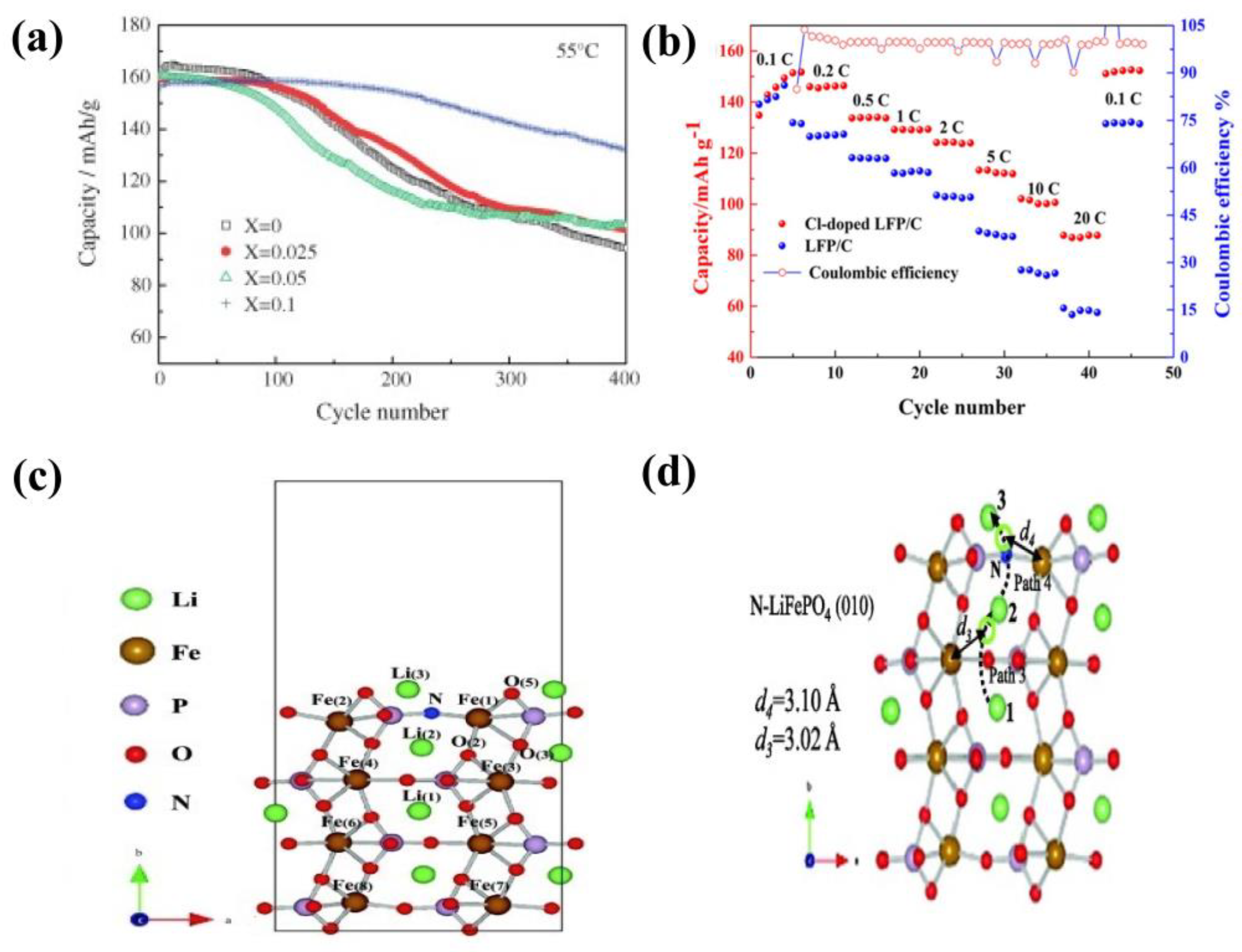
4.1.3. Polyanion Doping
4.2. Coating
4.2.1. Carbon Coating
4.2.2. Other Layers
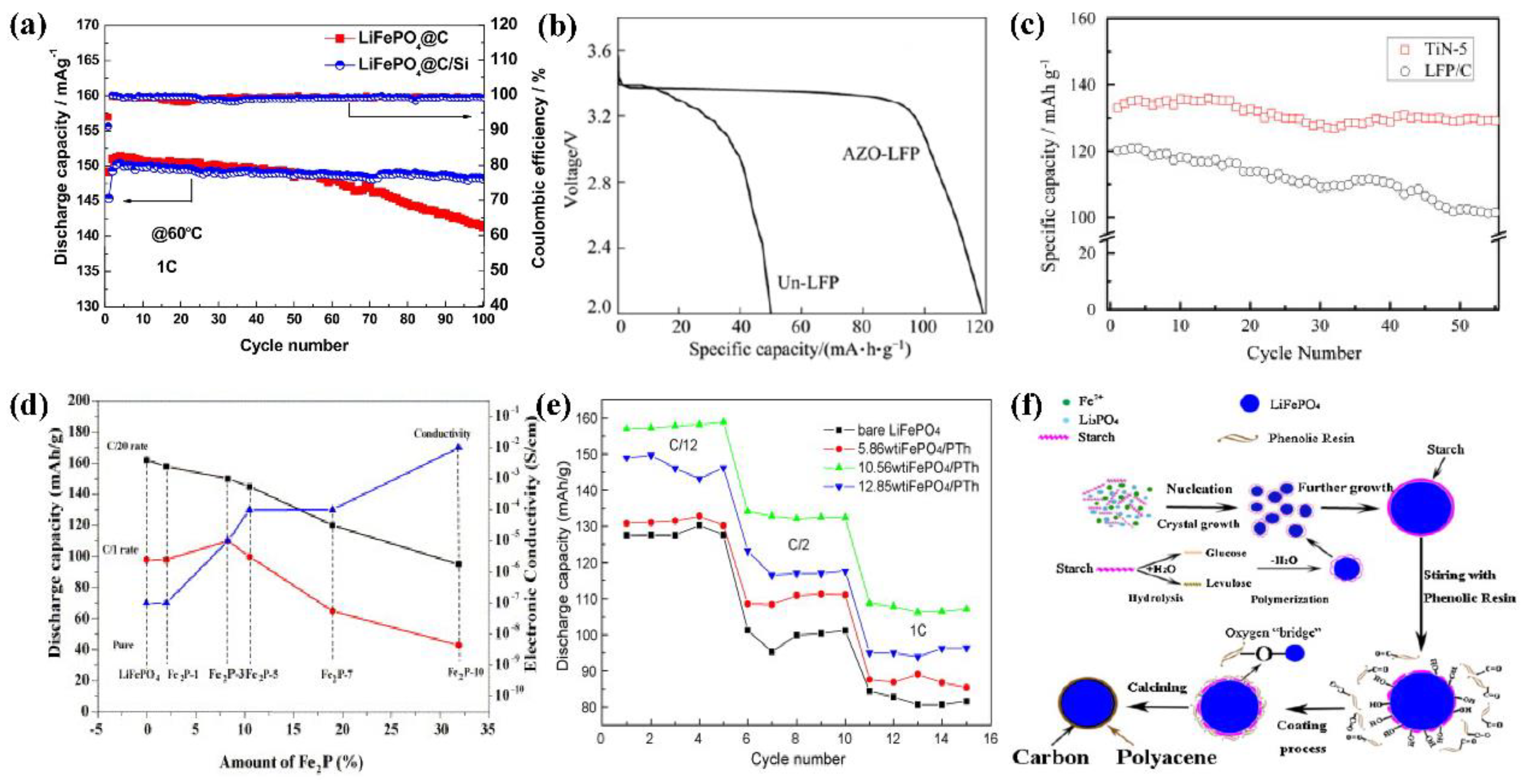
4.3. Microstructure Control
4.4. Electrolyte

4.5. Anode
5. Conclusion and Outlook
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Armand, M.; Tarascon, J.M. Building better batteries. Nature 2008, 451, 652–657. [Google Scholar] [CrossRef] [PubMed]
- Goodenough, J.B. Energy storage materials: A perspective. Energy Storage Mater. 2015, 1, 158–161. [Google Scholar] [CrossRef]
- Dunn, B.; Kamath, H.; Tarascon, J.M. Electrical Energy Storage for the Grid: A Battery of Choices. Science 2011, 334, 928–935. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.-Q.; Zhu, C.-X.; Yu, S.-C. Status and challenges facing representative anode materials for rechargeable lithium batteries. J. Energy Chem. 2022, 66, 260–294. [Google Scholar] [CrossRef]
- Gao, J.-Y.; Jin, B.-W.; Shao, M.-F. Layered double hydroxides functionalization toward rechargeable batteries. Particuology 2024, 91, 138–154. [Google Scholar] [CrossRef]
- Peled, E.; Menkin, S. Review-SEI: Past, present and future. J. Electrochem. Soc. 2017, 164, 1703–1719. [Google Scholar] [CrossRef]
- Li, R.; Bao, L.-Y.; Chen, L. Accelerated aging of lithium-ion batteries: Bridging battery aging analysis and operational lifetime prediction. Sci. Bull. 2023, 68, 3055–3079. [Google Scholar] [CrossRef]
- Chen, Y.; Xiao, R.; Cui, Y.-L. Research Review on Early Warning and Suppression Technology of Lithium-ion Battery Fire in Energy Storage Power Station. J. Electr. Eng. 2022, 17, 72–87. [Google Scholar]
- Li, Z.-L.; Li, X.-H.; Wang, J. Research Progress in Thermal Runaway Warning and Protection of Lithium-ion Battery Energy Storage Power Station. High Volt. Appar. 2024, 60, 87–99. [Google Scholar]
- Yuan, Q.-F.; Zhao, F.-G.; Wang, W.-D. Overcharge failure investigation of lithium-ion batteries. Electrochim. Acta 2015, 178, 682–6880. [Google Scholar] [CrossRef]
- Wang, J.; Purewal, J.; Liu, P. Degradation of lithium ion batteries employing graphite negatives and nickel-cobalt-manganese oxide + spinel manganese oxide positives: Part 1, aging mechanisms and life estimation. J. Power Sources 2014, 269, 937–948. [Google Scholar] [CrossRef]
- Atalay, S.; Sheikh, M.; Mariani, A.; Merla, Y.; Bower, E.; Widanage, W.D. Theory of battery ageing in a lithium-ion battery: Capacity fade, nonlinear ageing and lifetime prediction. J. Power Sources 2020, 478, 229026–229034. [Google Scholar] [CrossRef]
- Mussa, A.S.; Klett, M.; Lindbergh, G.; Lindström, R.W. Effects of external pressure on the performance and ageing of single-layer lithium-ion pouch cells. J. Power Sources 2018, 385, 18–26. [Google Scholar] [CrossRef]
- Sauerteig, D.; Hanselmann, N.; Arzberger, A. Electrochemical-mechanical coupled modeling and parameterization of swelling and ionic transport in lithium-ion batteries. J. Power Sources 2018, 378, 235–247. [Google Scholar] [CrossRef]
- Liu, Y.-K.; Zhao, C.-Z.; Du, J. Research Progresses of Liquid Electrolytes in Lithium-Ion Batteries. Small 2023, 22, 2205315. [Google Scholar] [CrossRef]
- Gong, C.; Xue, Z.; Wen, S.; Ye, Y.; Xie, X. Advanced carbon materials/olivine LiFePO4 composites cathode for lithium ion batteries. J. Power Sources 2016, 318, 93–112. [Google Scholar] [CrossRef]
- Zhang, W.J. Structure and performance of LiFePO4 cathode materials: A review. J. Power Sources 2011, 196, 2962–2970. [Google Scholar] [CrossRef]
- Yu, F.; Ge, S.; Li, B.; Sun, G.; Mei, R.; Zheng, L. Three-Dimensional Porous LiFePO4: Design, Architectures and High Performance for Lithium Ion Batteries, Curr. Inorg. Chem. 2012, 2, 194–212. [Google Scholar] [CrossRef]
- Padhi, A.K.; Nanjundaswamy, K.S.; Goodenough, J. Phospho-olivines and positive-electrode materials for rechargeable lithium batteries. J. Electrochem. Soc. 1997, 144, 1188–1194. [Google Scholar]
- Ren, X.-G.; Li, Y.-J.; He, Z.-J.; Xi, X.-M.; Shen, X.-J. In-situ growth of LiFePO4 with interconnected pores supported on carbon nanotubes via tavorite-olivine phase transition, Ceram. Inter. 2023, 49, 40131–40139. [Google Scholar]
- Zhao, Y.-S.; Li, W.-L.; Wu, J.; Zhou, X.-F.; Liu, Z.-P. A Scalable and Controllable Li-Powder-Coating Prelithiation Method Toward Ultralong-life and High-Energy-Density LiFePO4 Battery. Energy Technol. 2023, 11, 23003. [Google Scholar] [CrossRef]
- Islam, M.S.; Driscoll, D.J.; Fisher, C.A.J.; Slater, P.R. Atomic scale investigation of defects, dopants, and lithium transport in a LiFePO4 olivine-type battery material. Chem. Mater. 2005, 17, 5085. [Google Scholar] [CrossRef]
- Siddique, N.A.; Allen, A.M.; Mukherjee, P.P.; Liu, F.-Q. Simulation of effect of interfacial lithium flux on miscibility gap in non-equilibrium phase transformation of LiFePO4 particles. J. Power Sources 2014, 245, 83–88. [Google Scholar] [CrossRef]
- Wang, H.; Li, J.-B.; Miao, Z.-Y.; Huang, K.; Liao, Y.-Q.; Xu, X.-J.; Meng, J.-T.; Li, Z.; Huang, Y.-H. Hierarchical Electrode Architecture Enabling Ultrahigh-Capacity LiFePO4 Cathodes with Low Tortuosity. ACS Appl. Mater. Interfaces 2023, 15, 26824–26833. [Google Scholar] [CrossRef]
- Huang, H.Y.S.; Wang, Y.X. Dislocation Based Stress Developments in Lithium-Ion Batteries. J. Electrochem. Soc. 2012, 159, A815–A821. [Google Scholar] [CrossRef]
- Channagiri, S.A.; Nagpure, S.C.; Babu, S.S.; Noble, G.J.; Hart, R.T. Porosity and phase fraction evolution with aging in lithium iron phosphate battery cathodes. J. Power Sources 2013, 243, 750–757. [Google Scholar] [CrossRef]
- Scipion, R.; Jørgensen, P.S.; Ngo, D.; Simonsen, S.; Liu, Z.; Yakal-Kremski, K.; Wang, H.Q.; Hjelm, J.; Norby, P.; Barnett, S.; et al. Electron microscopy investigations of changes in morphology and conductivity of LiFePO4/C electrodes. J. Power Sources 2016, 307, 259–269. [Google Scholar] [CrossRef]
- Weichert, K.; Sigle, W.; Aken, P.; Jamnik, J.; Zhu, C.B.; Amin, R.; Acarturk, T.; Starke, U.; Maier, J. Phase Boundary Propagation in Large LiFePO4 Single Crystals on Delithiation. J. Am. Chem. Soc. 2012, 134, 2988–2992. [Google Scholar] [CrossRef]
- Konda, K.; Jacob, M.S.; Seth, J.; Juvekar, V.; Gopalan, R.; Moodakare, S. Capacity degradation of lithium-ion cell: The role of free carbon black content in the slurry and drying induced cracks in LiFePO4 electrode. J. Energy Storage 2023, 74, 109477. [Google Scholar] [CrossRef]
- ChiuHuang, C.K.; Huangz, H.Y.S. Stress Evolution on the Phase Boundary in LiFePO4 Particles. J. Electrochem. Soc. 2013, 160, 2184–2188. [Google Scholar] [CrossRef]
- Zhang, B.-Q.; Wang, S.-Z.; Li, Y.-H.; Sun, P.-P.; Yang, C.; Wang, D.; Liu, L. Review: Phase transition mechanism and supercritical hydrothermal synthesis of nano lithium iron phosphate. Ceram. Inter. 2020, 46, 27922–27939. [Google Scholar] [CrossRef]
- Takahashi, I.; Mori, T.; Yoshinari, T.; Orikasa, Y.; Koyama, Y.; Murayama, H.; Fukuda, K.; Hatano, M.; Arai, H.; Uchimoto, Y.; et al. Irreversible phase transition between LiFePO4 and FePO4 during highrate charge-discharge reaction by operando X-ray diffraction. J. Power Sources 2016, 309, 122–126. [Google Scholar] [CrossRef]
- Scipioni, R.; Jørgensen, P.S.; Stroe, D.I.; Younesi, R.; Simonsen, S.B.; Norby, P.; Hjelm, J.; Jensen, S.H. Complementary analyses of aging in a commercial LiFePO4/Graphite 26650 cell. Electrochim. Acta 2018, 284, 454–468. [Google Scholar] [CrossRef]
- Kobayashi, Y.; Miyashiro, H.; Mita, Y. Capacity fading of a LiFePO4/graphite cell during shallow cycling due to planar inhomogeneity in LiFePO4. J. Power Sources 2020, 451, 227782. [Google Scholar] [CrossRef]
- Cuisinier, M.; Dupré, N.; Martin, J.; Kanno, R.; Guyomard, D. Evolution of the LiFePO4 positive electrode interface along cycling monitored by MAS NMR. J. Power Sources 2013, 224, 50–58. [Google Scholar] [CrossRef]
- Zhang, J.-W.; Yu, H.-X.; Zhang, X.-K.; Xia, M.-T.; Zhang, X.Q.; Zhang, L.Y.; Shui, M.; Cui, Y.H.; Shu, J. Surface chemistry of LiFePO4 cathode material as unraveled by HRTEM and XPS. Ionics 2021, 27, 31–37. [Google Scholar] [CrossRef]
- Liu, Y.-D.; Xie, J. Failure Study of Commercial LiFePO4 Cells in Overcharge Conditions Using Electrochemical Impedance Spectroscopy. J. Electrochem. Soc. 2015, 162, A2208–A2217. [Google Scholar] [CrossRef]
- Dupré, N.; Martin, J.F.; Degryse, J.; Fernandez, V.; Soudan, P.; Guyomard, D. Aging of the LiFePO4 positive electrode interface in electrolyte. J. Power Sources 2010, 195, 7415–7425. [Google Scholar] [CrossRef]
- Edstr¨om, K.; Gustafsson, T.; Thomas, J.O. The cathode–electrolyte interface in the Li-ion battery. Electrochim. Acta 2004, 50, 397–403. [Google Scholar] [CrossRef]
- Simolka, M.; Heger, J.-F.; Traub, N.; Kaess, H.; Friedrich, K.A. Influence of Cycling Profile, Depth of Discharge and Temperature on Commercial LFP/C Cell Ageing: Cell Level Analysis with ICA, DVA and OCV Measurements. J. Electrochem. Soc. 2020, 167, 110502. [Google Scholar] [CrossRef]
- Eldesoky, A.; Logan, E.R.; Johnson, M.B.; McFarlane, C.R.M.; Dahn, J.R. Scanning Micro X-ray Fluorescence (μXRF) as an Effective Tool in Quantifying Fe Dissolution in LiFePO4 Cells: Towards a Mechanistic Understanding of Fe Dissolution. J. Electrochem. Soc. 2020, 167, 130539. [Google Scholar] [CrossRef]
- Li, D.J.; Danilov, D.; Xie, J.; Raijmakers, L.; Gao, L.; Yang, Y.; Notten, P. Degradation Mechanisms of C6/LiFePO4 Batteries: Experimental Analyses of Calendar Aging. Electrochim. Acta 2016, 190, 1124–1133. [Google Scholar] [CrossRef]
- Lai, Y.-Q.; Cao, Z.; Song, H.-S.; Zhang, Z.; Chen, X.; Lu, H.; Jia, M.; Li, J. Influence of Fe (II) Species in Electrolyte on Performance of Graphite Anode for Lithium-Ion Batteries. J. Electrochem. Soc. 2012, 159, 1961–1966. [Google Scholar] [CrossRef]
- Joshi, T.; Eom, K.S.; Yushin, G.; Fuller, T. Effects of Dissolved Transition Metals on the Electrochemical Performance and SEI Growth in Lithium-Ion Batteries. J. Electrochem. Soc. 2014, 161, 1915. [Google Scholar] [CrossRef]
- Guo, Z.-J.; Chen, Z.-L. High-temperature capacity fading mechanism for LiFePO4/graphite soft-packed cell without Fe dissolution. J. Electroan. Chem. 2015, 754, 148–153. [Google Scholar] [CrossRef]
- Zhi, X.-K.; Liang, G.-C.; Wang, L.; Ou, X.-Q.; Zhang, J.-P.; Cui, J.-Y. The cycling performance of LiFePO4/C cathode materials. J. Power Sources 2009, 189, 779–782. [Google Scholar] [CrossRef]
- Agubra, V.; Fergus, J. The formation and stability of the solid electrolyte interface on the graphite anode. J. Power Sources 2014, 268, 153–162. [Google Scholar] [CrossRef]
- Meda, U.S.; Lal, L.; Garg, P. Solid Electrolyte Interphase (SEI), a boon or a bane for lithium batteries: A review on the recent advances. J. Energy Storage 2021, 47, 103564. [Google Scholar] [CrossRef]
- Sharifi, H.; Mosallanejad, B.; Mohammadzad, M.; Hosseini-Hosseinabad, S.M.; Ramakrishna, S. Cycling performance of LiFePO4/graphite batteries and their degradation mechanism analysis via electrochemical and microscopic techniques. Ionics 2022, 28, 213–228. [Google Scholar] [CrossRef]
- Zheng, Y.; He, Y.-B.; Qian, K.; Li, B.-H.; Wang, X.-D.; Li, J.-L.; Chiang, S.-W.; Miao, C.; Kang, F.-Y.; Zhang, J.-B. Deterioration of lithium iron phosphate/graphite power batteries under high-rate discharge cycling. Electrochim. Acta 2015, 176, 270–279. [Google Scholar] [CrossRef]
- Chang, C.-C.; Huang, S.-Y.; Chen, W.-H. Thermal and solid electrolyte interphase characterization of lithiumion battery. Energy 2019, 174, 999–1011. [Google Scholar] [CrossRef]
- Tasaki, K.; Goldberg, A.; Lian, J.J.; Walker, M.; Timmons, A.; Harris, S. Solubility of Lithium Salts Formed on the Lithium-Ion Battery Negative Electrode Surface in Organic Solvents. J. Electrochem. Soc. 2009, 156, 1019–1027. [Google Scholar] [CrossRef]
- Ding, L.; Bagul, P.; Cui, L.Y.; Oswald, S.; Pohle, B.; Leones, R.; Mikhailova, D. Graphite Anode Functionalized with a Gel Biopolymer Binder for Li-Ion Batteries Operating in a Broad Temperature Range. ACS Appl. Energy Mater. 2023, 6, 4404–4412. [Google Scholar] [CrossRef]
- Kim, J.; Woo, S.C.; Park, M.S.; Kim, K.J.; Yim, T.; Kim, J.S.; Kim, Y.J. Capacity fading mechanism of LiFePO4-based lithium secondary batteries for stationary energy storage. J. Power Sources 2013, 229, 190–197. [Google Scholar] [CrossRef]
- Zhang, Y.-C.; Wang, C.-Y.; Tang, X.-D. Cycling degradation of an automotive LiFePO4 lithium-ion battery. J. Power Sources 2011, 196, 1513–1520. [Google Scholar] [CrossRef]
- Tan, L.; Zhang, L.; Sun, Q.-N.; Shen, M.; Qu, Q.-T.; Zheng, H.-H. Capacity loss induced by lithium deposition at graphite anode for LiFePO4/graphite cell cycling at different temperatures. Electrochim. Acta. 2013, 111, 802–808. [Google Scholar] [CrossRef]
- Dubarry, M.; Liaw, B.Y. Identify capacity fading mechanism in a commercial LiFePO4 cell. J. Power Sources 2009, 194, 541–549. [Google Scholar] [CrossRef]
- Li, N.; Jia, Z.; Wang, Z.-H.; Zhao, H.; Ai, G.; Son, X.Y.; Bai, Y.; Battaglia, V.; Sun, C.D.; Qiao, J.; et al. Understanding the crack formation of graphite particles in cycled commercial lithium-ion batteries by focused ion beam—Scanning electron microscopy. J. Power Sources 2017, 365, 235–239. [Google Scholar]
- Da, K.H.; Wang, Z.-H.; Ai, G.; Zha, H.; Yuan, W.; Song, X.-Y.; Battaglia, V.; Sun, C.D.; Wu, K.; Liu, G. The transformation of graphite electrode materials in lithium-ion batteries after cycling. J. Power Sources 2015, 298, 349–354. [Google Scholar] [CrossRef]
- Zinth, V.; Lüders, C.; Wilhelm, J.; Erhard, S.V.; Hofmann, M.; Seidlmayer, S.; Rebelo-Kornmeier, J.; Gan, W.; Jossen, A.; Gilles, R. Inhomogeneity and relaxation phenomena in the graphite anode of a lithium-ion battery probed by in situ neutron diffraction. J. Power Sources 2017, 361, 54–60. [Google Scholar] [CrossRef]
- Sethuraman, V.A.; Hardwick, L.J.; Srinivasan, V.; Kostecki, R. Surface structural disordering in graphite upon lithium intercalation/deintercalation. J. Power Sources 2010, 195, 3655–3660. [Google Scholar] [CrossRef]
- Rao, M.; Zhang, L.-F.; Li, L.W.; Rong, L.-B.; Ye, C.-C.; Zhou, G.-M.; Xu, H.; Qiu, Y.C. Investigation of lithium content changes to understand the capacity fading mechanism in LiFePO4/graphite battery. J. Electroan. Chem. 2019, 853, 113544. [Google Scholar] [CrossRef]
- Liu, P.; Wang, J.; Hicks-Garner, J.; Sherman, E.; Soukiazian, S.; Verbrugge, M.; Tataria, H.; Musser, J.; Finamore, P. Aging Mechanisms of LiFePO4 Batteries Deduced by Electrochemical and Structural Analyses. J. Electrochem. Soc. 2010, 157, 499–507. [Google Scholar] [CrossRef]
- Zhang, P.-C.; Yuan, T.; Pang, Y.-P.; Peng, C.-X.; Yang, J.-H.; Ma, Z.-F.; Zheng, S.-Y. Influence of Current Density on Graphite Anode Failure in Lithium-Ion Batteries. J. Electrochem. Soc. 2019, 166, 5489–5495. [Google Scholar] [CrossRef]
- Ouyang, M.-G.; Chu, Z.-Y.; Lu, L.-G.; Li, J.-Q.; Han, X.-B.; Fen, X.-N.; Liu, G.-M. Low temperature aging mechanism identification and lithium deposition in a large format lithium iron phosphate battery for different charge profiles. J. Power Sources 2015, 286, 309–320. [Google Scholar] [CrossRef]
- Rauhala, T.; Jalkanen, K.; Romann, T.; Lust, E.; Omar, N.; Kallio, T. Low-temperature aging mechanisms of commercial graphite/LiFePO4 cells cycled with a simulated electric vehicle load profile—A post-mortem study. J. Energy Storage 2018, 20, 344–356. [Google Scholar] [CrossRef]
- Wu, X.-G.; Wang, W.-B.; Du, J.-Y. Effect of charge rate on capacity degradation of LiFePO4 power battery at low temperature. Int. J. Energy Res. 2020, 44, 1775–1788. [Google Scholar] [CrossRef]
- Petzl, M.; Kaspe, M.; Danzer, M. Lithium plating in a commercial lithium-ion battery—A low temperature aging study. J. Power Sources 2015, 275, 799–807. [Google Scholar] [CrossRef]
- Ansena, D.; Dubarr, M.; Devie, A.; Liaw, B.Y.; García, V.M.; Viera, J.C.; Gonzalez, M. Operando lithium plating quantification and early detection of a commercial LiFePO4 cell cycled under dynamic driving schedule. J. Power Sources 2017, 356, 36–46. [Google Scholar] [CrossRef]
- Wang, Z.-W.; Kong, X.-P.; Fan, Z.-W.; Ding, S.-J.; Rong, Q.; Su, Y.-Q. A First-Principles Study of Anion Doping in LiFePO4 Cathode Materials for Li-Ion Batterie. ChemPhysChem 2024, 25, e202300756. [Google Scholar] [CrossRef]
- Harrison, K.L.; Bridges, C.A.; Paranthaman, M.P.; Segre, C.U.; Katsoudas, J.; Maroni, V.A.; Idrobo, J.C.; Goodenough, J.B.; Manthiram, A. Temperature Dependence of Aliovalent-Vanadium Doping in LiFePO4 Cathodes. Chem. Mater. 2013, 25, 768–781. [Google Scholar] [CrossRef]
- Liu, Q.-S.; Wen, D.-H.; Yu, X.-Y.; Jiang, H. Effect of Na-Si co-doping on the performance of LiFePO4. J. Electroanal. Chem. 2023, 950, 117891. [Google Scholar] [CrossRef]
- Wang, S.-C.; Wang, F.-Z.; Zhang, J.-C.; Chen, Z.; Zhao, X.-N.; Wu, H.; Cui, J.-Q. First-principles investigation of the electronic and Li-ion diffusion properties of C and B doped LiFePO4 (010) surface. Comput. Theor. Chem. 2023, 1227, 114254. [Google Scholar] [CrossRef]
- Cong, J.; Luo, S.; Li, K.; Wang, J.C.; Wang, Y.F.; Yan, S.X.; Li, P.W. High-performance Na-doped LiFePO4 cathode material derived from acid- washed iron red for the simultaneous immobilization of multi-metals. Ionics 2023, 29, 3915–3926. [Google Scholar] [CrossRef]
- Yin, X.-G.; Huang, K.-L.; Liu, S.-Q.; Wang, H.-Y.; Wang, H. Preparation and characterization of Na-doped LiFePO4/C composites as cathode materials for lithium-ion batteries. J. Power Sources 2010, 195, 4308–4312. [Google Scholar] [CrossRef]
- Liu, Y.; Qin, W.-C.; Zhang, D.-K.; Feng, L.-W.; Wu, L. Effect of Na+ in situ doping on LiFePO4/C cathode material for lithium-ion batteries. Prog. Nat. Sci. Mater. Int. 2021, 31, 14–18. [Google Scholar] [CrossRef]
- Ma, Z.-P.; Shao, G.-J.; Wang, G.-L.; Zhang, Y.; Du, J.-P. Effects of Nb-doped on the structure and electrochemical performance of LiFePO4/C composites. J. Solid State Chem. 2014, 210, 232–237. [Google Scholar] [CrossRef]
- Chiang, C.-Y.; Su, H.-C.; Wu, P.-J.; Liu, H.-J.; Hu, C.-W.; Sharma, N.; Peterson, V.; Hsieh, H.W.; Lin, Y.-F.; Chou, W.-C.; et al. Vanadium Substitution of LiFePO4 Cathode Materials to Enhance the Capacity of LiFePO4-Based Lithium-Ion Batteries. J. Phys. Chem. C, 2012; 116, 24424–24429. [Google Scholar]
- Kulka, A.; Braun, A.; Huang, T.W.; Wolska, A.; Klepka, M.; Szewczyk, A.; Baster, D.; Zając, W.; Świerczek, K.; Molenda, J. Evidence for Al doping in lithium sublattice of LiFePO4. Solid State Ion. 2015, 270, 33–38. [Google Scholar] [CrossRef]
- Tian, Y.-W.; Kang, X.-X.; Liu, L.-Y.; Xu, C.-Q.; Qu, T. Research on cathode material of Li-ion battery by yttrium doping. J. Rare Earths 2008, 26, 2. [Google Scholar] [CrossRef]
- Luo, S.-H.; Tian, Y.; Li, H.; Shi, K.-J.; Tang, Z.-L.; Zhang, Z.-T. Influence of lanthanum doping on performance of LiFePO4 cathode materials for lithium-ion batteries. J. Rare Earths 2010, 28, 439. [Google Scholar] [CrossRef]
- Yang, R.; Song, X.-P.; Zhao, M.-S.; Wang, F. Characteristics of Li0.98Cu0.01FePO4 prepared from improved co-precipitation. J. Alloys Comp. 2009, 468, 365–369. [Google Scholar] [CrossRef]
- Hu, G.-R.; Gao, X.-G.; Peng, Z.-D.; Du, K.; Tan, X.-Y.; Liu, Y.-J. Influence of Ti4+ doping on electrochemical properties of LiFePO4/C cathode material for lithium-ion batteries.Trans. Nonferrous Met. SOC. China 2007, 17, 296–300. [Google Scholar] [CrossRef]
- Zheng, Z.-H.; Bei, F.-L.; Zhou, L.; Xia, W.-C.; Sun, J.-T.; Qian, H. Efficient Structural Regulation Plat form for the Controlled Synthesis of LiFePO4 Cathodes with Shorter Li-Ion Diffusion Paths. Langmuir 2024, 40, 2396–2404. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.-H.; Yuan, L.-X.; Wu, M.; Sun, D.; Huang, Y.-H. Effects of Na+ and Cl− co-doping on electrochemical performance in LiFePO4/C. Electrochim. Acta 2011, 56, 8477–8483. [Google Scholar] [CrossRef]
- Yiming, W.; Giuli, G.; Moretti, A.; Nobili, F.; Fehr, K.T.; Paris, E.; Marassi, R. Synthesis and characterization of Zn-doped LiFePO4 cathode materials for Li-ion battery. Mater. Chem. Phys. 2015, 155, 191–204. [Google Scholar] [CrossRef]
- Wu, J.Y.; Tsai, C.J. Discovered Intermediate Phase and Improved Electrochemical Performance of Zn-Doped LiCoPO4 for High-Energy Li-Ion Batteries. ACS Appl. Energy Mater. 2021, 7, 6408–6413. [Google Scholar] [CrossRef]
- Lama, B.; Smirnova, A.; Paudel, T. Enhanced Li-Ion Diffusivity of LiFePO4 by Ru Doping: Ab Initio and Machine Learning Force Field Results. ACS Appl. Energy Mater. 2023, 6, 10424–10431. [Google Scholar] [CrossRef]
- Lin, H.; Wen, Y.-W.; Zhang, C.-X.; Zhang, L.-L.; Huang, Y.-H.; Shan, B.; Chen, R. A GGA+U study of lithium diffusion in vanadium doped LiFePO4. Solid State Commun. 2012, 152, 999–1003. [Google Scholar] [CrossRef]
- Chen, M.; Shao, L.-L.; Yang, H.-B.; Ren, T.-Z.; Du, G.-H.; Yuan, Z.-Y. Vanadium-doping of LiFePO4/carbon composite cathode materials synthesized with organophosphorus source. Electrochim. Acta 2015, 167, 278–286. [Google Scholar] [CrossRef]
- Gao, H.-Y.; Jiao, L.-F.; Yang, J.-Q.; Qi, Z.; Wang, Y.-J.; Yuan, H.-T. High rate capability of Co-doped LiFePO4/C. Electrochim. Acta 2013, 97, 143–149. [Google Scholar] [CrossRef]
- Latif, C.; Firdausi, A.; Nihlatunnur, N.; Saiyasombat, C.; Klysubun, W.; Subhan, A.; Zainuri, M.; Pratapa, S. Synchrotron crystal and local structures, microstructure, and electrical characterization of Cu-doped LiFePO4/C via dissolution method with ironstone as Fe source. J. Mater. Sci. Mater. Electron. 2022, 33, 17722–17732. [Google Scholar] [CrossRef]
- Kim, S.; Mathew, V.; Kang, J.; Gim, J.; Song, J.; Jo, J.; Kim, J. High ratecapability of LiFePO4 cathodes doped with a high amount of Ti. Ceram. Int. 2016, 42, 7230–7236. [Google Scholar] [CrossRef]
- Wen, L.-Z.; Guan, Z.-W.; Wang, L.; Liu, X.-M.; Wen, G.-Q.; Zhao, Y.; Pang, D.-F.; Dou, R.-Z. Synthesis and Electrochemical Properties of Molybdenum-Doped LiMn0.6Fe0.4PO4 Cathode Materials. J. Mater. Eng. Perform. 2024, 42, 7230–7236. [Google Scholar] [CrossRef]
- Ren, X.-G.; Li, Y.-J.; He, Z.-J.; Xi, X.-M.; Yang, J.-C.; Hao, S.-P.; Shen, X.-J.; Wu, Q. Preparation of LiFe0.99Mn0.01PO4 Cathode Material with Lower Fe-Li Antisite via Wet-Lithiation Following by Tavorite-Olivine Phase Transition. J. Electrochem. Soc. 2023, 170, 100526. [Google Scholar] [CrossRef]
- Yang, H.-T.; Pan, Z.-W.; Wang, L.; Liu, C.; Wang, Z.-C.; Zhang, C.-X.; Lu, W.-Q. Enhancing electrochemical performance of Mn doped LiFePO4 cathode materials for lithium-ion batteries. Int. J. Electrochem. Sci. 2023, 18, 100079. [Google Scholar] [CrossRef]
- Mi, Y.-Y.; Yang, C.-K.; Zuo, Z.-C.; Qi, L.-Y.; Tang, C.-X.; Zhang, W.-D.; Zhou, H.-H. Positive Effect of Minor Manganese Doping on the Electrochemical Performance of LiFePO4/C under Extreme Conditions. Electrochim. Acta 2015, 176, 642–648. [Google Scholar] [CrossRef]
- Liu, Y.; Gu, Y.-J.; Luo, G.-Y.; Chen, Z.-L.; Wu, F.-Z.; Dai, X.-Y.; Mai, Y.; Li, J.-Q. Ni-doped LiFePO4/C as high-performance cathode composites for Li-ion batteries. Ceram. Inter. 2020, 46, 14857–14863. [Google Scholar] [CrossRef]
- Tong, D.-G.; Luo, F.-L.; Chu, W.; Li, Y.-L.; Wu, P. Effect of rhodium substitution on the electrochemical performance of LiFePO4/C. Mater. Chem. Phys. 2010, 124, 1–5. [Google Scholar] [CrossRef]
- Zhao, N.-N.; Li, Y.-S.; Zhi, X.-K.; Wang, L.; Zhao, X.-X.; Wang, Y.-M.; Liang, G.-C. Effect of Ce3+ doping on the properties of LiFePO4 cathode material. J. Rare Earths 2016, 34, 174. [Google Scholar] [CrossRef]
- Meng, X.-D.; Han, B.; Wang, Y.-F. Effects of samarium doping on the electrochemical performance of LiFePO4/C cathode material or lithium-ion batteries. Ceram. Int. 2016, 42, 2599–2604. [Google Scholar] [CrossRef]
- Yuan, H.; Wang, X.-Y.; Wu, Q. Effects of Ni and Mn doping on physicochemical and electrochemical performances of LiFePO4/C. J. Alloys Compd. 2016, 675, 187–194. [Google Scholar] [CrossRef]
- Long, Y.-F.; Su, J.; Cui, X.-R.; Lv, X.-Y.; Wen, Y.-X. Enhanced rate performance of LiFePO4/C by co-doping titanium and vanadium. Solid State Sci. 2015, 48, 104–111. [Google Scholar] [CrossRef]
- Jugović, D.; Mitrić, M.; Milović, M.; Cvjetićanin, N.; Jokić, B.; Umićević, A.; Uskoković, D. The influence of fluorine doping on the structural and electrical properties of the LiFePO4 powder. Ceram. Int. 2017, 43, 3224–3230. [Google Scholar] [CrossRef]
- Li, X.-T.; Shao, Z.-B.; Liu, K.-R.; Zhao, Q.; Liu, G.-F.; Xu, B.-S. Effect of F-doping on the properties of LiFePO4-x/3Fx/C cathode materials via wet mechanical agitation-assisted high-temperature ball milling method. J. Solid State Electrochem. 2018, 22, 2837–2843. [Google Scholar] [CrossRef]
- Gao, C.; Zhou, J.; Liu, G.-Z.; Wang, L. Synthesis of F-doped LiFePO4/C cathode materials for high performance lithium-ion batteries using co-precipitation method with hydrofluoric acid source. J. Alloys Compd. 2017, 727, 501–513. [Google Scholar] [CrossRef]
- Liao, X.-Z.; He, Y.-S.; Ma, Z.-F.; Zhang, X.-M.; Wang, L. Effects of fluorine-substitution on the electrochemical behavior of LiFePO4/C cathode materials. J. Power Sources 2007, 174, 720–725. [Google Scholar] [CrossRef]
- Zaki, N.H.M.; Ahmad, S.I.; Sazman, F.N.; Badrudin, F.W.; Abdullah, A.L.A.; Taib, M.F.M.; Hassan, O.H.; Yahya, M.Z.A. The influence of Cl doping on the structural, electronic properties and Li-ion migration of LiFePO4: A DFT study. Comput. Theor. Chem. 2023, 1221, 114029. [Google Scholar] [CrossRef]
- Sun, C.S.; Zhang, Y.; Zhang, X.-J.; Zhou, Z. Structural and electrochemical properties of Cl-doped LiFePO4/C. J. Power Sources 2010, 195, 3680–3683. [Google Scholar] [CrossRef]
- Liu, H.; Luo, S.-h.; Yan, S.-X.; Wang, Y.-F.; Wang, Q.; Li, M.-Q.; Zhang, Y.-H. A novel and low-cost iron source for synthesizing Cl-doped LiFePO4/C cathode materials for lithium-ion batteries. J. Electroanal. Chem. 2019, 850, 113434. [Google Scholar] [CrossRef]
- Okada, K.; Kimura, I.; Machida, K. High rate capability by sulfur-doping into LiFePO4 matrix. RSC Adv. 2018, 8, 5848–5853. [Google Scholar] [CrossRef]
- Lee, S.-B.; Cho, S.-H.; Aravindan, V.; Kim, H.-S.; Lee, Y.-S. Improved Cycle Performance of Sulfur-Doped LiFePO4 Material at High Temperatures. Bull. Korean Chem. Soc. 2009, 30, 10. [Google Scholar]
- Li, Y.; Wang, L.; Liang, F.; Yao, Y.-C.; Zhang, K.Y. Enhancing high rate performance and cyclability of LiFePO4 cathode materials for lithium ion batteries by boron doping. J. Alloys Compd. 2021, 880, 160560. [Google Scholar] [CrossRef]
- Trócoli, R.; Franger, S.; Cruz, M.; Morales, J.; Jesús, S.-P. Improving the electrochemical properties of nanosized LiFePO4-basedelectrode by boron doping. Electrochim. Acta 2014, 135, 558–567. [Google Scholar] [CrossRef]
- Liu, Z.-J.; Huang, X.-J.; Wang, D.-S. First-principle investigations of N doping in LiFePO4. Solid State Commun. 2008, 147, 505–509. [Google Scholar] [CrossRef]
- Xu, G.; Zhong, K.; Zhang, J.-M.; Huang, Z. First-principles study of structural, electronic and Li-ion diffusion properties of N-doped LiFePO4 (010) surface. Solid State Ion. 2015, 281, 1–5. [Google Scholar] [CrossRef]
- Tian, Z.; Zhou, Z.-F.; Liu, S.-S.; Ye, F.; Yao, S.-J. Enhanced properties of olivine LiFePO4/graphene co-doped with Nb5+ and Ti4+ by a sol–gel method. Solid State Ion. 2015, 278, 186–191. [Google Scholar] [CrossRef]
- Zhang, B.-F.; Ma, X.-N.; Hou, W.-Q.; Yuan, W.; He, L.-X.; Yang, O.; Liu, Y.-B.; Wang, J.; Xu, Y.L. Revealing the Ultrahigh Rate Performance of the La and Ce Co-doping LiFePO4 Composite. ACS Appl. Energy Mater. 2022, 5, 14712–14719. [Google Scholar] [CrossRef]
- Yang, X.; Hu, Z.; Liang, J. Effects of sodium and vanadium co-doping on the structure and electrochemical performance of LiFePO4/C cathode material for lithium-ion batteries. Ceram. Int. 2015, 41, 2863–2868. [Google Scholar] [CrossRef]
- Wang, Y.-Z.; Cui, C.; Cheng, R.-F.; Wang, J.-C.; Wang, X.-H. Tannic Acid-Derived Carbon Coating on LiFePO4 Nanocrystals Enables High-Rate Cathode Materials for Lithium-Ion Batteries. ACS Appl. Nano Mater. 2023, 6, 9124–9129. [Google Scholar] [CrossRef]
- Wen, L.-Z.; Guan, Z.-W.; Wang, L.; Hu, S.-T.; Lv, D.-H.; Liu, X.-M.; Duan, T.-T.; Liang, G.-C. Effect of Carbon-Coating on Internal Resistance and Performance of Lithium Iron Phosphate Batteries. J. Electrochem. Soc. 2022, 169, 050536. [Google Scholar] [CrossRef]
- Huang, C.-Y.; Kuo, T.-R.; Yougbaré, S.; Lin, L.-Y. Design of LiFePO4 and porous carbon composites with excellent High-Rate charging performance for Lithium-Ion secondary battery. J. Colloid Interface Sci. 2022, 607, 1457–1465. [Google Scholar] [CrossRef]
- Wu, G.; Liu, N.; Gao, X.G.; Tian, X.-H.; Zhu, Y.-B.; Zhou, Y.-K.; Zhu, Q.-Y. A hydrothermally synthesized LiFePO4/C composite with superior low-temperature performance and cycle life. Appl. Surf. Sci. 2018, 435, 1329–1336. [Google Scholar] [CrossRef]
- Wang, X.-F.; Feng, Z.-J.; Hou, X.-L.; Liu, L.-L.; He, M.; He, X.-S.; Huang, J.-T.; Wen, Z.-H. Fluorine doped carbon coating of LiFePO4 as a cathode material for lithiumion batteries. Chem. Eng. J. 2020, 379, 122371. [Google Scholar] [CrossRef]
- Liu, Y.-Y.; Gu, J.-J.; Zhang, J.-L.; Yu, F.; Dong, L.-T.; Nie, N.; Li, W. Metal organic frameworks derived porous lithium iron phosphate with continuous nitrogen-doped carbon networks for lithium ion batteries. J. Power Sources 2016, 304, 42–50. [Google Scholar] [CrossRef]
- Feng, J.-P.; Wang, Y.-L. High-rate and ultralong cycle-life LiFePO4 nanocrystals coated byboron-doped carbon as positive electrode for lithium-ion batteries. Appl. Surf. Sci. 2016, 390, 481–488. [Google Scholar] [CrossRef]
- Wang, X.-F.; Feng, Z.-J.; Huang, J.-T.; Deng, W.; Li, X.-B.; Zhang, H.-S.; Wen, Z.-H. Graphene-decorated carbon-coated LiFePO4 nanospheres as a high- performance cathode material for lithium-ion batteries. Carbon 2018, 127, 149–157. [Google Scholar] [CrossRef]
- Wu, G.; Zhou, Y.-K.; Shao, Z.-P. Carbon nanotube and graphene nanosheet co-modified LiFePO4 nanoplate composite cathode material by a facile polyol process. Appl. Surf. Sci. 2013, 283, 999–1005. [Google Scholar] [CrossRef]
- Liu, J.-Y.; Lin, X.-R.; Han, T.-L.; Li, X.-X.; Gu, C.-P.; Li, J.-J. A novel litchi-like LiFePO4 sphere/reduced graphene oxide composite Li-ion battery cathode with high capacity, good rate-performance and low temperature. Appl. Surf. Sci. 2018, 459, 233–241. [Google Scholar] [CrossRef]
- Wang, J.; Gu, Y.-J.; Kong, W.-L.; Liu, H.-Q.; Chen, Y.-B.; Liu, W. Effect of carbon coating on the crystal orientation and electrochemical performance of nanocrystalline LiFePO4. Solid State Ion. 2018, 327, 11–17. [Google Scholar] [CrossRef]
- Lin, Y.-B.; Lin, Y.; Zhou, T.; Zhao, G.-Y.; Huang, Y.-D.; Huang, Z.-G. Enhanced electrochemical performances of LiFePO4/C by surface modification with Sn nanoparticles. J. Power Sources 2013, 226, 20–26. [Google Scholar] [CrossRef]
- Wen, L.; Liu, J.-W.; Hou, J.-J.; Zhao, S.-N.; Liu, J.-L.; Zheng, Z.-H.; Bei, F.-L. A High Performance LiFePO4 Cathode Material with 3D Conduction Network Connected by 1D helix-like Ag Nano-chains. Int. J. Electrochem. Sci. 2021, 16, 151002. [Google Scholar] [CrossRef]
- Yang, W.-Y.; Zhuang, Z.-Y.; Chen, X.; Zou, M.-Z.; Zhao, G.-Y.; Feng, Q.; Li, J.-X.; Lin, Y.-B.; Huang, Z.-G. A simple and novel Si surface modification on LiFePO4@C electrode and its suppression of degradation of lithium ion batteries. Appl. Surf. Sci. 2015, 359, 875–882. [Google Scholar] [CrossRef]
- Li, Y.-D.; Zhao, S.-X.; Nan, C.-W.; Li, B.-H. Electrochemical performance of SiO2-coated LiFePO4 cathode materials forlithium ion battery. J. Alloys Compd. 2011, 509, 957–960. [Google Scholar] [CrossRef]
- Yao, J.-W.; Wu, F.; Qiu, X.-P.; Ning, L.; Su, Y.-F. Effect of CeO2-coating on the electrochemical performances of LiFePO4/C cathode material. Electrochim. Acta 2011, 56, 5587–5592. [Google Scholar] [CrossRef]
- Jin, Y.; Yang, C.-P.; Rui, X.-H.; Cheng, T.; Chen, C.H. V2O3 modified LiFePO4/C composite with improved electrochemical performance. J. Power Sources 2011, 196, 5623–5630. [Google Scholar] [CrossRef]
- Ziolkowska, D.; Korona, K.P.; Hamankiewicz, B.; Wu, S.-H.; Chen, M.-S.; Jasinski, J.B.; Kaminska, M.; Czerwinski, A. The role of SnO2 surface coating on the electrochemical performance of LiFePO4 cathode materials. Electrochim. Acta 2013, 108, 532–539. [Google Scholar] [CrossRef]
- Cui, Y.; Zhao, X.-L.; Guo, R.-S. Enhanced electrochemical properties of LiFePO4 cathode material by CuO and carbon co-coating, J. Alloys Compd. 2010, 490, 236–240. [Google Scholar] [CrossRef]
- Moustafaa, M.G.; Sanad, M.S. Green fabrication of ZnAl2O4-coated LiFePO4 nanoparticles for enhanced electrochemical performance in Li-ion batteries. J. Alloys Compd. 2022, 903, 163910. [Google Scholar] [CrossRef]
- Tang, H.; Tan, L.; Xu, J. Synthesis and characterization of LiFePO4 coating with aluminum doped zinc oxide. Trans. Nonferrous Met. Soc. China 2013, 23, 451–455. [Google Scholar] [CrossRef]
- Liu, C.-W.; An, J.; Guo, R.-S.; Li, Y.; Liu, L. Enhanced electrochemical performance of LiFePO4/C cathode material modified with highly conductive TiN. J. Alloys Compd. 2013, 563, 33–38. [Google Scholar] [CrossRef]
- Cai, G.L.; Guo, R.-S.; Liu, L.; Yang, Y.-X.; Zhang, C.; Wu, C.; Guo, W.-N.; Jiang, H. Enhanced low temperature electrochemical performances of LiFePO4/C by surface modification with Ti3SiC2. J. Power Sources 2015, 288, 136–144. [Google Scholar] [CrossRef]
- Yuan, G.; Zhang, X.-J.; Lu, S.-G.; Jiang, D.-P.; Wu, A.-D. High rate performance of LiF modified LiFePO4/C cathode material. Solid State Ion. 2015, 269, 30–36. [Google Scholar]
- Kim, C.W.; Park, J.S.; Lee, K.S. Effect of Fe2P on the electron conductivity and electrochemical performance of LiFePO4 synthesized by mechanical alloying using Fe3+ raw material. J. Power Sources 2006, 163, 144–150. [Google Scholar] [CrossRef]
- Li, Y.; Wang, J.; Huang, H.-X.; Wang, J.; Zhang, M.; Liang, M.-M. Co-coating effect of GdPO4 and carbon on LiFePO4 cathode surface for lithium ion batteries. Adv. Powder Technol. 2019, 30, 1442–1449. [Google Scholar] [CrossRef]
- Hu, G.-R.; Chen, P.-E.; Cao, Y.-B.; Peng, Z.-D.; Ke, D.; Zhang, Z.-J. Enhanced electrochemical performance of LiFePO4 cathode with synergistic modification of Na3V2(PO4)3 and carbon network. Solid State Ion. 2017, 301, 138–145. [Google Scholar] [CrossRef]
- Zhao, S.-X.; Ding, H.; Wang, Y.-C.; Li, B.-H.; Nan, C.-W. Improving rate performance of LiFePO4 cathode materials by hybrid coating of nano-Li3PO4 and carbon. J. Alloys Compd. 2013, 566, 206–211. [Google Scholar] [CrossRef]
- Fedorková, A.; Nacher-Alejos, A.; Gómez-Romero, P.; Orináková, R.; Kaniansky, D. Structural and electrochemical studies of PPy/PEG-LiFePO4 cathode material for Li-ion batteries. Electrochim. Acta 2010, 55, 943–947. [Google Scholar] [CrossRef]
- Bai, Y.-M.; Qiu, P.; Wen, Z.-L.; Han, S.-C. Improvement of electrochemical performances of LiFePO4 cathode materials by coating of polythiophene. J. Alloys Compd. 2010, 508, 1–4. [Google Scholar] [CrossRef]
- Zhou, J.-P.; Tu, J.-P.; Cheng, L.-J.; Shi, S.-J.; Qiao, Y.-Q.; Liu, W.-L.; Wang, X.-L.; Gu, C.-D. LiFePO4/Polyacene Nanocomposite Synthesized from a Pretreatment of Iron Phosphate: In-situ Polymerization with Phenolic-Formaldehyde Resin. J. Electrochem. Soc. 2011, 158, 1237–1242. [Google Scholar] [CrossRef]
- Chen, Z.-Y.; Du, B.-L.; Xu, M.; Zhu, H.-l.; Li, L.-J.; Wang, W.-H. Polyacene coated carbon/LiFePO4 cathode for Li ion batteries: Understanding the stabilized double coating structure and enhanced lithium ion diffusion kinetics. Electrochim. Acta 2013, 109, 262–268. [Google Scholar] [CrossRef]
- Trinh, N.D.; Saulnier, M.; Lepage, D.; Schougaard, S.B. Conductive polymer film supporting LiFePO4 as composite cathode for lithium ion batteries. J. Power Sources 2013, 221, 284–289. [Google Scholar] [CrossRef]
- Delacourt, C.; Poizot, P.; Levasseur, S.; Masquelier, C. Size Effects on Carbon-Free LiFePO4 Powders The Key to Superior Energy Density. Electrochem. Solid-State Lett. 2006, 9, 352–355. [Google Scholar] [CrossRef]
- Kim, D.H.; Kim, J. Synthesis of LiFePO4 nanoparticles and their electrochemical properties. J. Phys. Chem. Solids 2007, 68, 734–737. [Google Scholar] [CrossRef]
- Konarova, M.; Taniguchi, I. Synthesis of carbon-coated LiFePO4 nanoparticles with high rate performance in lithium secondary batterie. J. Power Sources 2010, 195, 3661–3667. [Google Scholar] [CrossRef]
- Zheng, Z.-M.; Pan, W.-K.; Tang, X.-C.; Jia, D.-Z.; Huang, Y.-D.; Guo, Z.-P. Solvothermal synthesis and electrochemical performance of hollow LiFePO4 nanoparticles. J. Alloys Compd. 2015, 640, 95–100. [Google Scholar] [CrossRef]
- Sarmadi, A.; Masoudpanah, S.M.; Alamolhoda, S. L-Lysine-assisted solvothermal synthesis of hollow-like structure LiFePO4/C powders as cathode materials for Li-ion batteries. J. Mater. Res. Technol. 2021, 15, 5405–5413. [Google Scholar] [CrossRef]
- Du, J.; Kong, L.-B.; Liu, H.; Liu, J.-B.; Liu, M.-C.; Zhang, P.; Luo, Y.-C.; Kang, L. Template-free synthesis of porous–LiFePO4/C nanocomposite for high power lithium-ion batteries. Electrochim. Acta 2014, 123, 1–6. [Google Scholar] [CrossRef]
- Du, G.-D.; Zhou, Y.-K.; Tian, X.-H.; Wu, G.; Xi, Y.-K.; Zhao, S.-Y. High-performance 3D directional porous LiFePO4/C materials synthesized by freeze casting. Appl. Surf. Sci. 2018, 453, 493–501. [Google Scholar] [CrossRef]
- Tang, X.; Lv, S.-Y.; Jiang, K.; Zhou, G.-H.; Liu, X.-M. Recent development of ionic liquid-based electrolytes in lithium-ion batteries. J. Power Sources 2022, 542, 231792. [Google Scholar] [CrossRef]
- Kaina, S.; Answer, J.; Hamid, A.; Gull, N.; Khan, S.M. Electrolytes in Lithium-Ion Batteries: Advancements in the Era of Twenties (2020’s). Mater. Chem. Phys. 2024, 313, 128796. [Google Scholar] [CrossRef]
- Kautz, S.; Cao, X.; Gao, P.-Y.; Matthews, B.E.; Xu, Y.-B.; Han, K.S. Designing Electrolytes With Controlled Solvation Structurefor Fast-Charging Lithium-Ion Batteries. Adv. Energy Mater. 2023, 13, 2301199. [Google Scholar] [CrossRef]
- Lima, J.-E.; Ohb, M.-S.; Ahnc, J.-H.; Kim, J.-K. Electrochemical properties of LiMn0.4Fe0.6PO4 with polyimide-based gel polymer electrolyte for high safety and improvement of rate capability. Electrochim. Acta 2017, 238, 107–111. [Google Scholar] [CrossRef]
- Li, F.-Q.; Yan, G.; Jia, G.-F.; Wang, Q.-L.; Peng, Z.-J.; Fan, W.; Bai, B. A novel dual-salts of LiTFSI and LiODFB in LiFePO4-based batteries for suppressing aluminum corrosion and improving cycling stability. J. Power Sources 2015, 295, 47–54. [Google Scholar] [CrossRef]
- Zhang, L.-F.; Chai, L.-L.; Zhang, L.; Shen, M.; Zhang, X.-L.; Battaglia, V.S.; Stephenson, T.; Zheng, H.-H. Synergistic effect between lithium bis (fluorosulfonyl) imide (LiFSI) and lithium bis-oxalato borate (LiBOB) salts in LiPF6-based electrolyte for high-performance Li-ion batteries. Electrochim. Acta 2014, 127, 39–44. [Google Scholar] [CrossRef]
- Qin, B.-S.; Liu, Z.-H.; Zheng, J.; Hu, P.; Ding, G.-L.; Zhang, C.-J.; Zhao, J.-H.; Kong, D.-S.; Cui, G.-L. Single-ion dominantly conducting polyborates towards high performance electrolytes in lithium batteries. J. Mater. Chem. A. 2015, 3, 7773. [Google Scholar] [CrossRef]
- Shi, J.-L.; Ehteshami, N.; Ma, J.-L.; Zhang, H.; Liu, H.-D.; Zhang, X.; Li, J.; Elie, P. Improving the graphite/electrolyte interface in lithium-ion battery for fast charging and low temperature operation: Fluorosulfonyl isocyanate as electrolyte additive. J. Power Sources 2019, 429, 67–74. [Google Scholar] [CrossRef]
- Moreno-Fernández, G.; Boulanger, N.; Nordenström, A.; Iakunkov, A.; Talyzin, A.; Carriazo, D.; Mysyk, R. Ball-milling-enhanced capacitive charge storage of activated graphene in aqueous, organic and ionic liquid electrolytes. Electrochim. Acta 2021, 370, 137738. [Google Scholar] [CrossRef]
- Chan, C.-C.; Lee, K.-Y.; Lee, H.-Y.; Su, Y.-H.; Her, L.-J. Trimethyl borate and triphenyl borate as electrolyte additives for LiFePO4 cathode with enhanced high temperature performance. J. Power Sources 2012, 217, 524–529. [Google Scholar] [CrossRef]
- Cai, Z.-J.; Liu, Y.-B.; Zhao, J.-H.; Li, L.; Zhang, Y.-M.; Zhang, J. Tris(trimethylsilyl) borate as electrolyte additive to improve performance of lithium-ion batteries. J. Power Sources 2012, 202, 341–346. [Google Scholar] [CrossRef]
- Liao, L.-X.; Chen, X.-Q.; Ma, Y.-L.; Zuo, P.-J.; Fang, W.; Yin, G.-P.; Gao, Y.-Z. Fluoroethylene carbonate as electrolyte additive to improve low temperature performance of LiFePO4 electrode. Electrochim. Acta 2013, 87, 466–472. [Google Scholar] [CrossRef]
- Wu, B.-R.; Ren, Y.-H.; Mu, D.-B.; Liu, X.-J.; Yang, G.-C.; Sun, Z. Lithium insertion/desertion properties of LiFePO4 cathode in a low temperature electrolyte modified with sodium chloride additive. Solid State Ion. 2014, 260, 8–14. [Google Scholar] [CrossRef]
- Chen, M.-Y.; Mei, J.; Wang, S.-J.; Chen, Q.-P.; Zhao, L.-Y.; Kong, Q.-H.; Wu, X.-Y. Comparative studies on the combustion characters of the lithium-ion battery electrolytes with composite flame-retardant additives. J. Energy Storage 2022, 47, 103642. [Google Scholar] [CrossRef]
- Wu, B.-B.; Pei, F.; Wu, Y.; Mao, R.-J.; Ai, X.-P.; Yang, H.-X.; Cao, Y.-L. An electrochemically compatible and flame-retardant electrolyte additive for safe lithium ion batteries. J. Power Sources 2013, 227, 106–110. [Google Scholar] [CrossRef]
- Madram, A.R.; Zarandi, M.; Beni, A.S.; Bayat, Y. Designed Novel Carbazole Based Electrolyte Additive for Overcharge Protection of Lithium-Ion Batteries. Russ. J. Electrochem. 2019, 55, 637–642. [Google Scholar] [CrossRef]
- Ahmed, F.; Choi, I.; Ryu, T.; Yoon, S.-J.; Rahman, M.M.; Zhang, W.; Jang, H.; Kim, W. Highly conductive divalent fluorosulfonyl imide based electrolytes improving Li-ion battery performance: Additive potentiating electrolytes action. J. Power Sources 2020, 455, 227980. [Google Scholar] [CrossRef]
- Xu, F.; Liu, C.-Y.; Feng, W.-F.; Nie, J.; Li, H.; Huang, X.-J.; Zhou, Z.-B. Molten salt of lithium bis(fluorosulfonyl)imide (LiFSI)-potassiumbis (fluorosulfonyl)imide (KFSI) as electrolyte for the natural graphite/LiFePO4 lithium-ion cell. Electrochim. Acta 2014, 135, 217–223. [Google Scholar] [CrossRef]
- Pohl, B.; Grünebaum, M.; Drews, M.; Passerini, S.; Winte, M.; Wiemhöfer, H.-D. Nitrile functionalized silyl ether with dissolved LiTFSI as new electrolyte solvent for lithium-ion batteries. Electrochim. Acta 2015, 180, 795–800. [Google Scholar] [CrossRef]
- Seki, S.; Takei, K.; Miyashiro, H.; Watanabe, M. Physicochemical and Electrochemical Properties of Glyme-LiN(SO2F)2 Complex for Safe Lithium-ion Secondary Battery Electrolyte. J. Electrochem. Soc. 2011, 158, 769–774. [Google Scholar] [CrossRef]
- Long, M.-C.; Wu, G.; Wang, X.-L.; Wang, Y.-Z. Self-adaptable gel polymer electrolytes enable high-performance and all-round safety lithium ion batteries. Energy Storage Mater. 2022, 53, 62–71. [Google Scholar] [CrossRef]
- Kim, D.S.; Kim, Y.E.; Kim, H. Improved fast charging capability of graphite anodes via amorphous Al2O3 coating for high power lithium ion batteries. J. Power Sources 2019, 422, 18–24. [Google Scholar] [CrossRef]
- Yang, N.-H.; Wu, Y.-S.; Chou, J.; Wu, H.-C.; Wu, N.-L. Silicon oxide-on-graphite planar composite synthesized using a microwave-assisted coating method for use as a fast-charging lithiumion battery anode. J. Power Sources 2015, 296, 314–317. [Google Scholar] [CrossRef]
- Yang, X.-Y.; Zhan, C.-Z.; Ren, X.-L.; Wang, C.; Wei, L.; Yu, Q.-T.; Xu, D.-P.; Nan, D.; Lv, R.-T.; Shen, W.-C.; et al. Nitrogen-doped hollow graphite granule as anode materials for high-performance lithium-ion batteries. J. Solid State Chem. 2021, 303, 122500. [Google Scholar] [CrossRef]
- Li, Y.-X.; Song, J.; Tian, Q.-H. Li4Ti5O12 nanowires intertwined with carbon nanotubes for ultra-long life and conductive additive-free anodes of lithium-ion batteries. Mater. Lett. 2023, 348, 134698. [Google Scholar] [CrossRef]
- Wang, P.-C.; Wang, Y.; Zhang, W.-L.; Xing, Y.-F.; Huang, B.; Zhang, H.-G. One-step synthesis of Cl-doped Li4Ti5O12 during electrode fabrication process with improved specific capacity. J Solid State Electrochem. 2024, 348, 134698. [Google Scholar] [CrossRef]
- Zhang, Z.-H.; Ying, H.-J.; Huang, P.-F.; Zhang, S.-L.; Zhang, Z.; Yang, T.-T.; Han, W.-Q. Porous Si decorated on MXene as free-standing anodes for lithium-ion batteries with enhanced diffusion properties and mechanical stability. Chem. Eng. J. 2023, 451, 138785. [Google Scholar] [CrossRef]
- Yang, C.-B.; Yan, X.; Gao, W.-W.; Xia, Y.; Liu, H.-J.; Hu, L.-Y.; Song, W.-L.; Zhang, W.-K.; Xia, X.-H.; Huang, H. Facile Synthesis of Laminated Si@C–Fe3O4 Composite from Exfoliation of Zintl Phase: A Promising Lithium Battery Anode with Superior Rate Capability and Cycling Stability. ACS Appl. Energy Mater. 2024, 7, 1313–1319. [Google Scholar] [CrossRef]
- Kang, H.E.; Ko, J.; Song, S.G.; Yoon, Y.S. Recent progress in utilizing carbon nanotubes and graphene to relieve volume expansion and increase electrical conductivity of Si-based composite anodes for lithium-ion batteries. Carbon 2024, 219, 118800. [Google Scholar] [CrossRef]
- Bandyopadhyay, P.; Senthamaraikannan, T.G.; Lim, D.-H.; Sahoo, G.; Baasanjav, E.; Kim, J.-K.; Jeong, S.M. Hierarchically nanofibers embedded with NiMnS nanocrystals as anode for high-performance lithium-ion batteries: Experimental and theoretical studies. Chem. Eng. J. 2024, 481, 148578. [Google Scholar] [CrossRef]
- Zhang, M.; Deng, Z.-P.; Zhang, X.-F.; Huo, L.-H.; Gao, S. Co9S8/Biocarbon Tubular Nanocomposite with Low-Temperature Performance for Lithium Storage. ACS Appl. Nano Mater. 2024, 7, 5396–5404. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hu, C.; Geng, M.; Yang, H.; Fan, M.; Sun, Z.; Yu, R.; Wei, B. A Review of Capacity Fade Mechanism and Promotion Strategies for Lithium Iron Phosphate Batteries. Coatings 2024, 14, 832. https://doi.org/10.3390/coatings14070832
Hu C, Geng M, Yang H, Fan M, Sun Z, Yu R, Wei B. A Review of Capacity Fade Mechanism and Promotion Strategies for Lithium Iron Phosphate Batteries. Coatings. 2024; 14(7):832. https://doi.org/10.3390/coatings14070832
Chicago/Turabian StyleHu, Chen, Mengmeng Geng, Haomiao Yang, Maosong Fan, Zhaoqin Sun, Ran Yu, and Bin Wei. 2024. "A Review of Capacity Fade Mechanism and Promotion Strategies for Lithium Iron Phosphate Batteries" Coatings 14, no. 7: 832. https://doi.org/10.3390/coatings14070832
APA StyleHu, C., Geng, M., Yang, H., Fan, M., Sun, Z., Yu, R., & Wei, B. (2024). A Review of Capacity Fade Mechanism and Promotion Strategies for Lithium Iron Phosphate Batteries. Coatings, 14(7), 832. https://doi.org/10.3390/coatings14070832