
Oxford Nanopore MinION Direct RNA-Seq for Systems Biology

 , , and
, , and
Abstract
:Simple Summary
Abstract
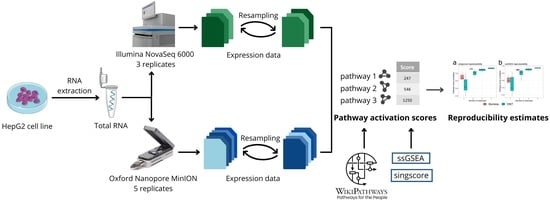
1. Introduction
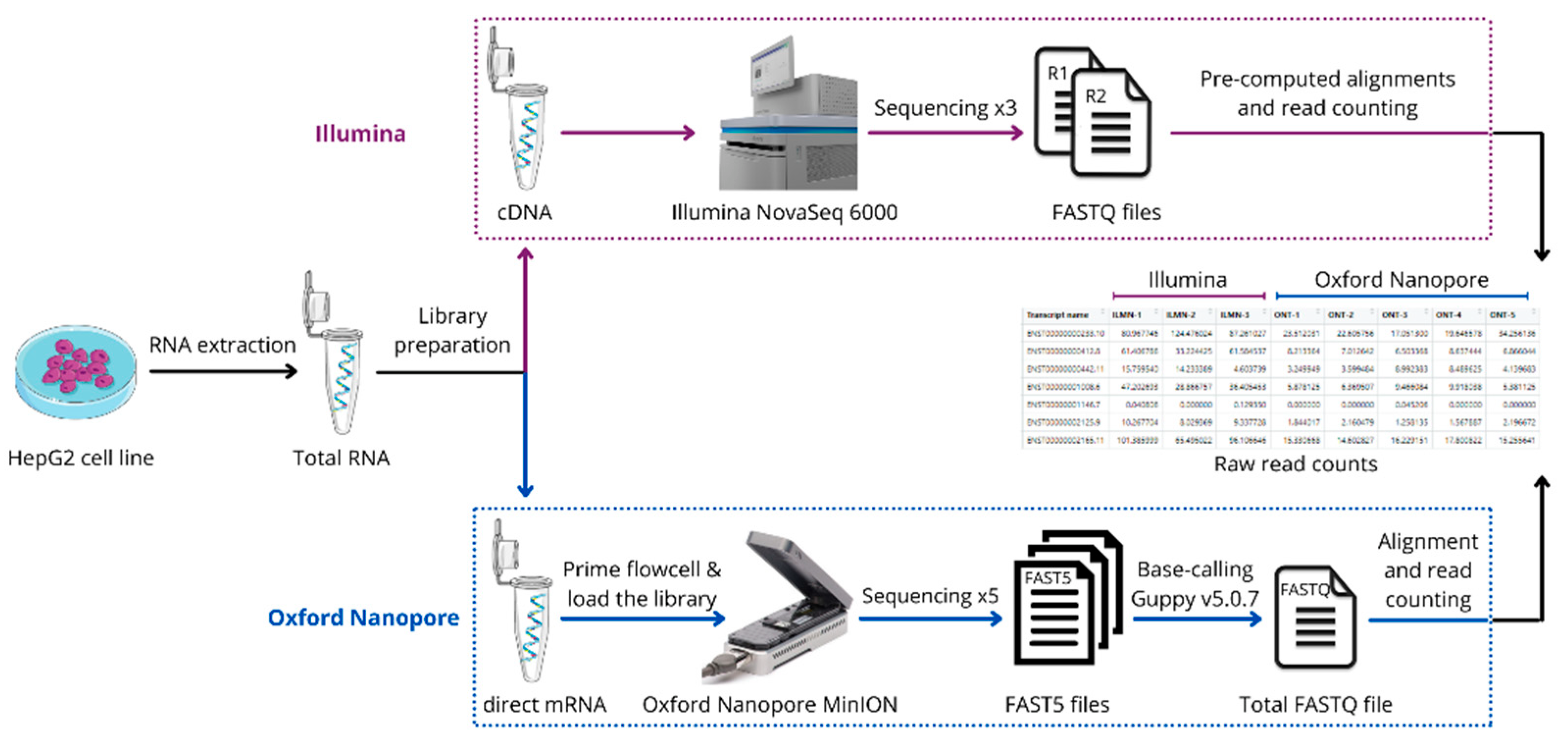
2. Materials and Methods
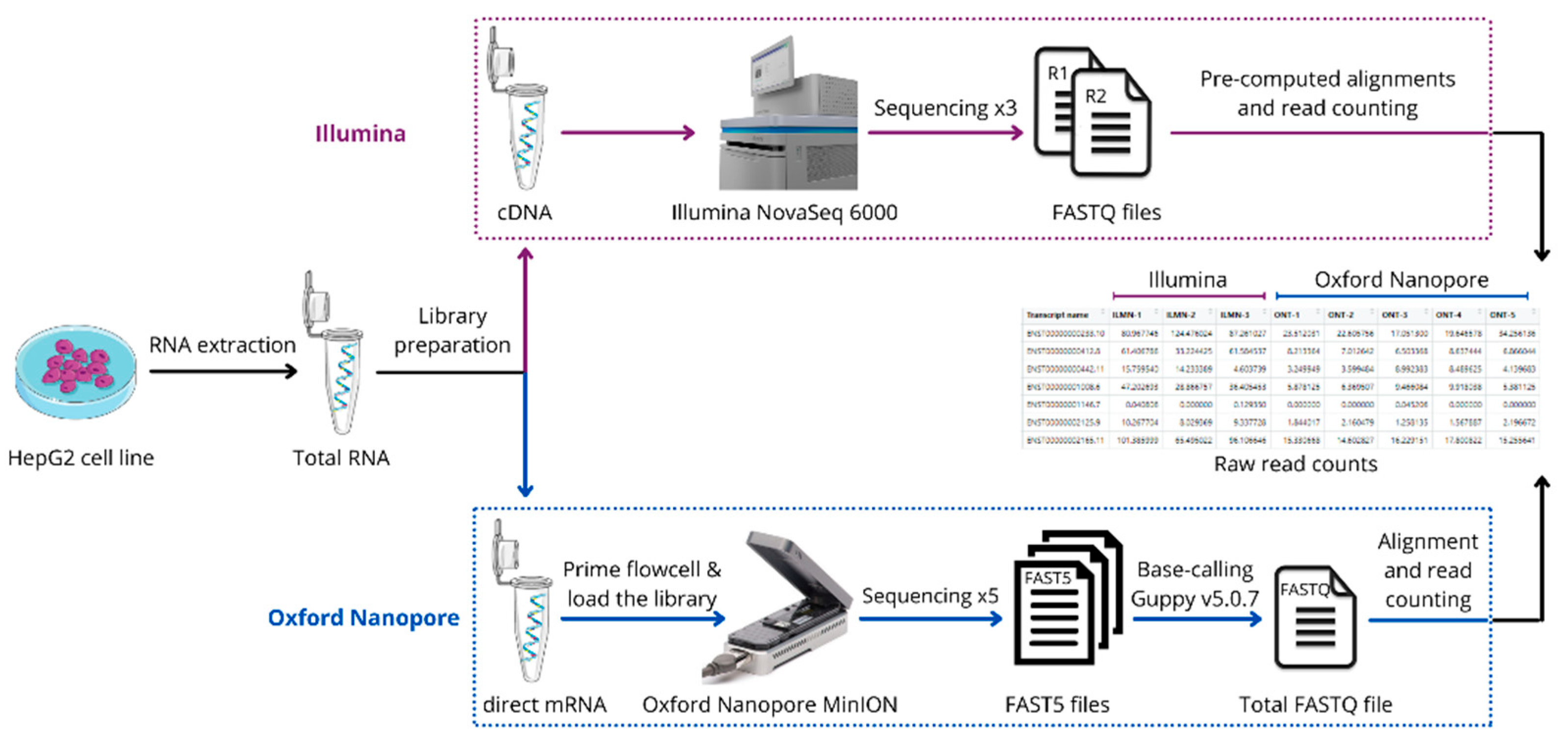
2.1. Cells Preparation
2.2. Sample and Library Preparation
2.3. Transcriptome Profiling
2.4. Data Analysis
3. Results
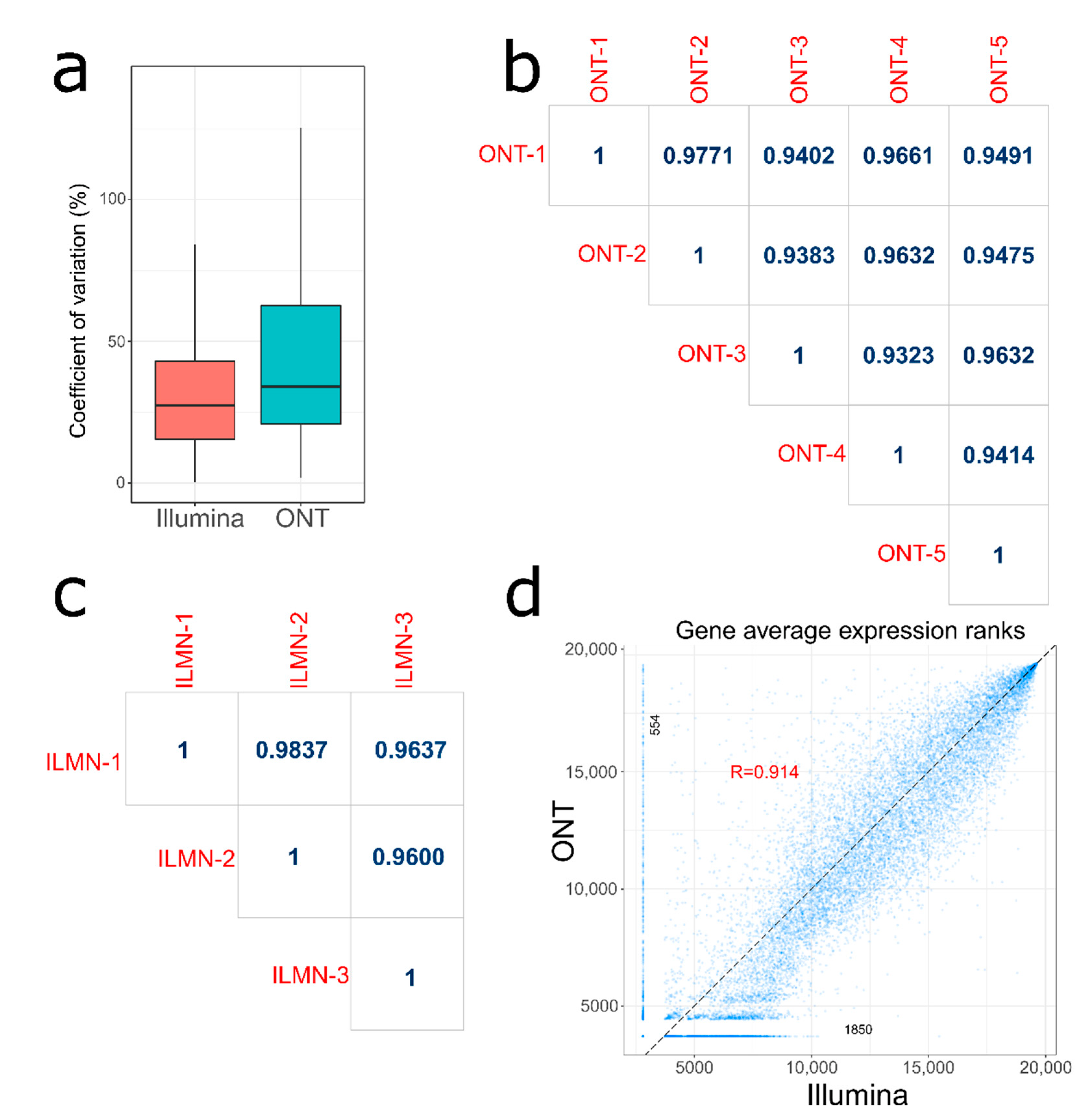
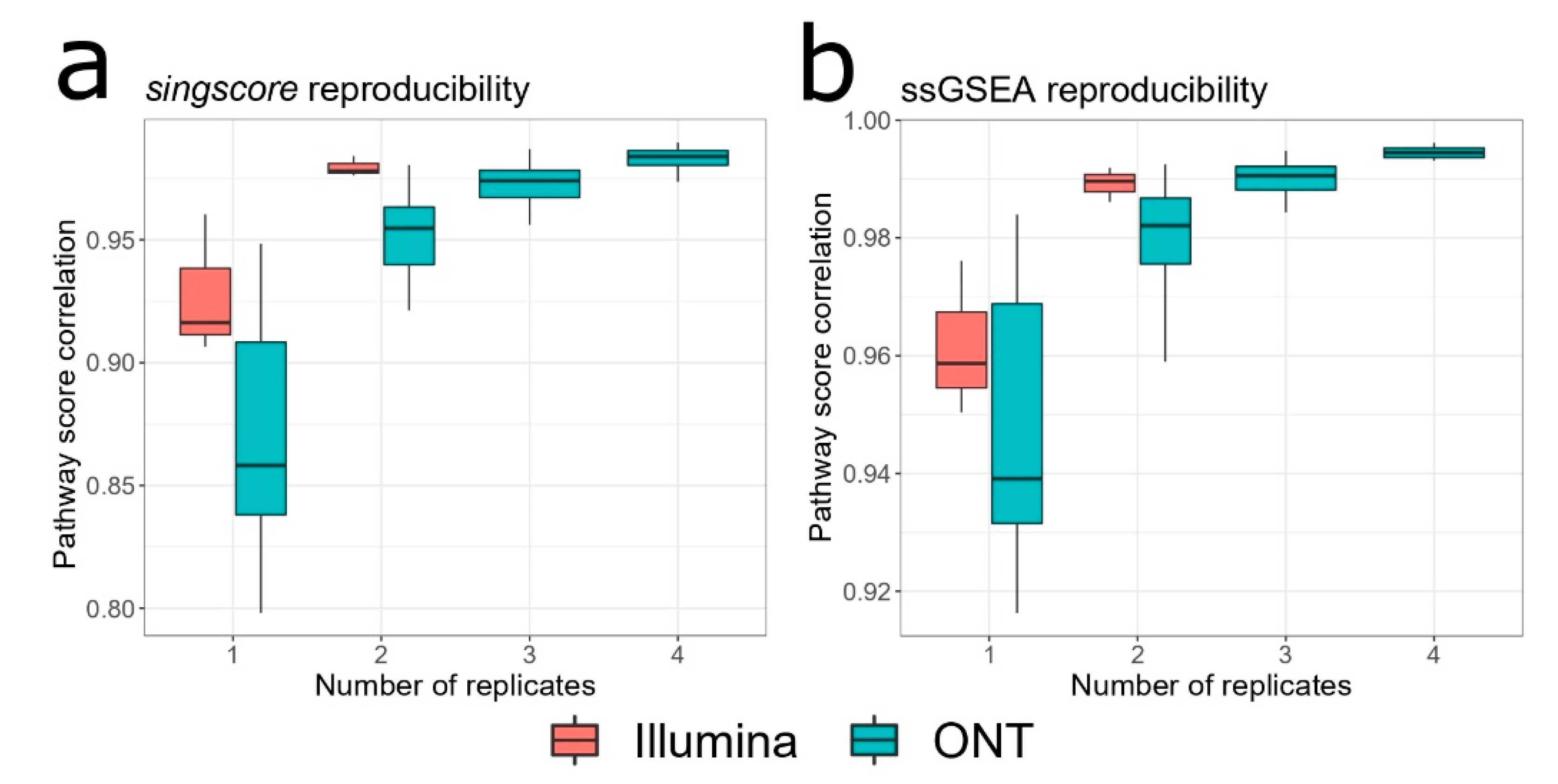
3.1. Intra- and Inter-Platform Reproducibility
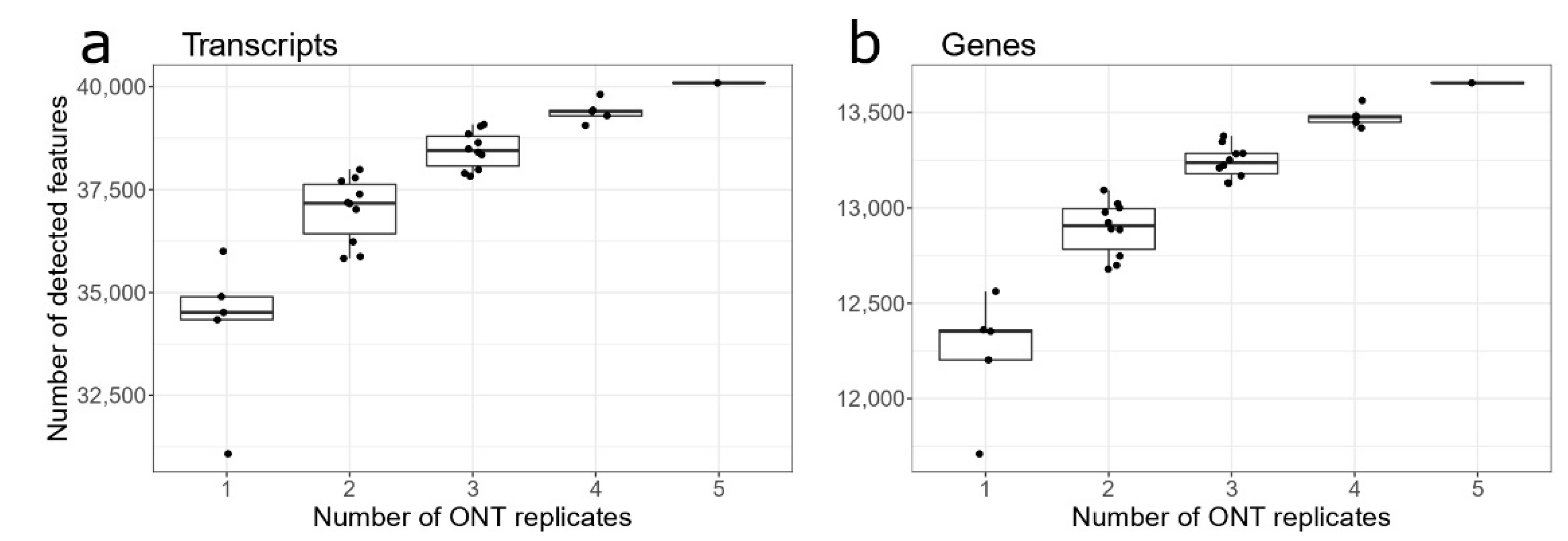
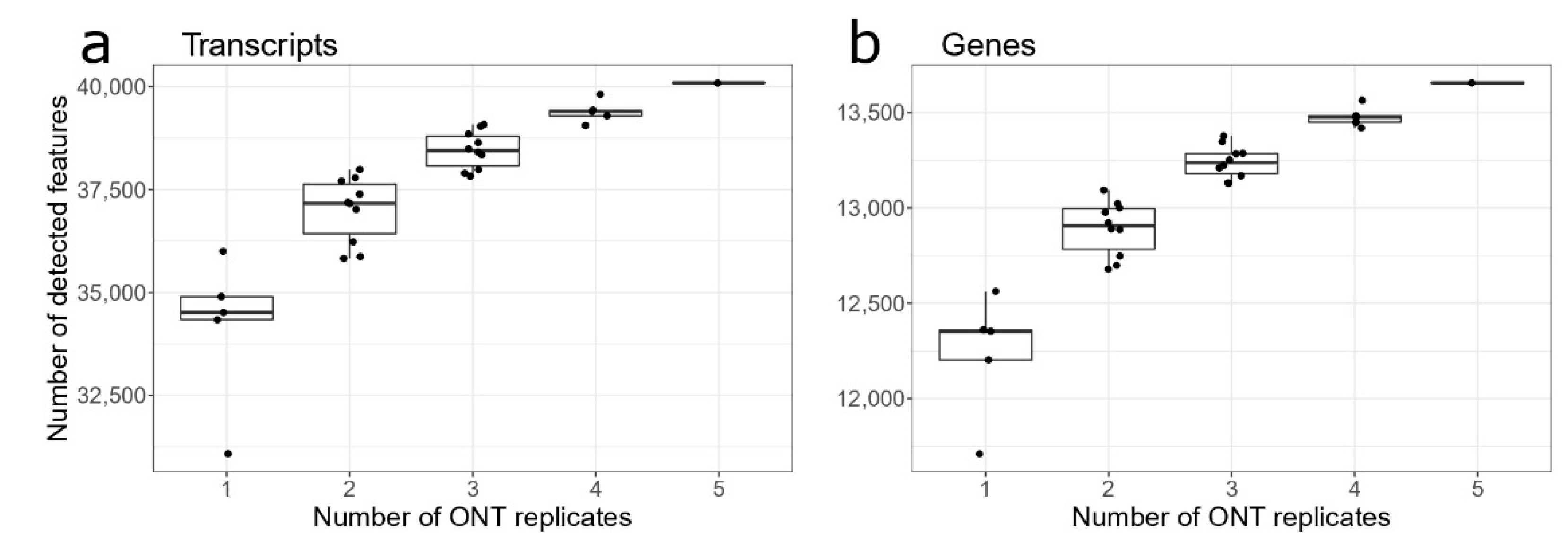
3.2. ONT Replicates for Gene/Transcripts Identification
3.3. ONT Replicates for Quantifying Pathway Activation
3.4. Confirmation of Results Using Published Data
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Ethical Statement
References
- Guo, Y.; Sheng, Q.; Li, J.; Ye, F.; Samuels, D.C.; Shyr, Y. Large Scale Comparison of Gene Expression Levels by Microarrays and RNAseq Using TCGA Data. PLoS ONE 2013, 8, e71462. [Google Scholar] [CrossRef] [Green Version]
- Stark, R.; Grzelak, M.; Hadfield, J. RNA Sequencing: The Teenage Years. Nat. Rev. Genet. 2019, 20, 631–656. [Google Scholar] [CrossRef]
- Schuierer, S.; Carbone, W.; Knehr, J.; Petitjean, V.; Fernandez, A.; Sultan, M.; Roma, G. A Comprehensive Assessment of RNA-Seq Protocols for Degraded and Low-Quantity Samples. BMC Genom. 2017, 18, 442. [Google Scholar] [CrossRef] [Green Version]
- Amarasinghe, S.L.; Su, S.; Dong, X.; Zappia, L.; Ritchie, M.E.; Gouil, Q. Opportunities and Challenges in Long-Read Sequencing Data Analysis. Genome Biol. 2020, 21, 30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kono, N.; Arakawa, K. Nanopore Sequencing: Review of Potential Applications in Functional Genomics. Dev. Growth Differ. 2019, 61, 316–326. [Google Scholar] [CrossRef] [Green Version]
- Garalde, D.R.; Snell, E.A.; Jachimowicz, D.; Sipos, B.; Lloyd, J.H.; Bruce, M.; Pantic, N.; Admassu, T.; James, P.; Warland, A.; et al. Highly Parallel Direct RNA Sequencing on an Array of Nanopores. Nat. Methods 2018, 15, 201–206. [Google Scholar] [CrossRef]
- Workman, R.E.; Tang, A.D.; Tang, P.S.; Jain, M.; Tyson, J.R.; Razaghi, R.; Zuzarte, P.C.; Gilpatrick, T.; Payne, A.; Quick, J.; et al. Nanopore Native RNA Sequencing of a Human Poly(A) Transcriptome. Nat. Methods 2019, 16, 1297–1305. [Google Scholar] [CrossRef] [PubMed]
- Leger, A.; Amaral, P.P.; Pandolfini, L.; Capitanchik, C.; Capraro, F.; Barbieri, I.; Migliori, V.; Luscombe, N.M.; Enright, A.J.; Tzelepis, K.; et al. RNA Modifications Detection by Comparative Nanopore Direct RNA Sequencing. BioRxiv 2019. [Google Scholar] [CrossRef] [Green Version]
- Sessegolo, C.; Cruaud, C.; Da Silva, C.; Cologne, A.; Dubarry, M.; Derrien, T.; Lacroix, V.; Aury, J.-M. Transcriptome Profiling of Mouse Samples Using Nanopore Sequencing of CDNA and RNA Molecules. Sci. Rep. 2019, 9, 14908. [Google Scholar] [CrossRef] [Green Version]
- Sahlin, K.; Medvedev, P. Error Correction Enables Use of Oxford Nanopore Technology for Reference-Free Transcriptome Analysis. Nat. Commun. 2021, 12, 2. [Google Scholar] [CrossRef]
- Shi, L.; Jones, W.D.; Jensen, R.V.; Harris, S.C.; Perkins, R.G.; Goodsaid, F.M.; Guo, L.; Croner, L.J.; Boysen, C.; Fang, H.; et al. The Balance of Reproducibility, Sensitivity, and Specificity of Lists of Differentially Expressed Genes in Microarray Studies. BMC Bioinform. 2008, 9, S10. [Google Scholar] [CrossRef] [Green Version]
- Haury, A.-C.; Gestraud, P.; Vert, J.-P. The Influence of Feature Selection Methods on Accuracy, Stability and Interpretability of Molecular Signatures. PLoS ONE 2011, 6, e28210. [Google Scholar] [CrossRef] [PubMed]
- Venet, D.; Dumont, J.E.; Detours, V. Most Random Gene Expression Signatures Are Significantly Associated with Breast Cancer Outcome. PLoS Comput. Biol. 2011, 7, e1002240. [Google Scholar] [CrossRef] [PubMed]
- Khatri, P.; Sirota, M.; Butte, A.J. Ten Years of Pathway Analysis: Current Approaches and Outstanding Challenges. PLoS Comput. Biol. 2012, 8, e1002375. [Google Scholar] [CrossRef] [PubMed]
- García-Campos, M.A.; Espinal-Enríquez, J.; Hernández-Lemus, E. Pathway Analysis: State of the Art. Front. Physiol. 2015, 6, 383. [Google Scholar] [CrossRef] [Green Version]
- Pyatnitskiy, M.; Mazo, I.; Shkrob, M.; Schwartz, E.; Kotelnikova, E. Clustering Gene Expression Regulators: New Approach to Disease Subtyping. PLoS ONE 2014, 9, e84955. [Google Scholar] [CrossRef] [Green Version]
- Vitali, F.; Li, Q.; Schissler, A.G.; Berghout, J.; Kenost, C.; Lussier, Y.A. Developing a ‘Personalome’ for Precision Medicine: Emerging Methods That Compute Interpretable Effect Sizes from Single-Subject Transcriptomes. Brief. Bioinform. 2019, 20, 789–805. [Google Scholar] [CrossRef] [Green Version]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene Set Enrichment Analysis: A Knowledge-Based Approach for Interpreting Genome-Wide Expression Profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [Green Version]
- Barbie, D.A.; Tamayo, P.; Boehm, J.S.; Kim, S.Y.; Moody, S.E.; Dunn, I.F.; Schinzel, A.C.; Sandy, P.; Meylan, E.; Scholl, C.; et al. Systematic RNA Interference Reveals That Oncogenic KRAS-Driven Cancers Require TBK1. Nature 2009, 462, 108–112. [Google Scholar] [CrossRef] [PubMed]
- Glazko, G.; Rahmatallah, Y.; Zybailov, B.; Emmert-Streib, F. Extracting the Strongest Signals from Omics Data: Differentially Expressed Pathways and Beyond. In Biological Networks and Pathway Analysis; Tatarinova, T.V., Nikolsky, Y., Eds.; Methods in Molecular Biology; Springer: New York, NY, USA, 2017; Volume 1613, pp. 125–159. ISBN 978-1-4939-7025-4. [Google Scholar]
- Shen, S.; Wang, G.; Zhang, R.; Zhao, Y.; Yu, H.; Wei, Y.; Chen, F. Development and Validation of an Immune Gene-Set Based Prognostic Signature in Ovarian Cancer. EBioMedicine 2019, 40, 318–326. [Google Scholar] [CrossRef] [Green Version]
- Xie, Z.; Li, X.; He, Y.; Wu, S.; Wang, S.; Sun, J.; He, Y.; Lun, Y.; Zhang, J. Immune Cell Confrontation in the Papillary Thyroid Carcinoma Microenvironment. Front. Endocrinol. 2020, 11, 570604. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Jin, Y.; Gong, L.; He, D.; Cheng, Y.; Xiao, M.; Zhu, Y.; Wang, Z.; Cao, K. Bioinformatics Analysis Finds Immune Gene Markers Related to the Prognosis of Bladder Cancer. Front. Genet. 2020, 11, 607. [Google Scholar] [CrossRef]
- Foroutan, M.; Bhuva, D.D.; Lyu, R.; Horan, K.; Cursons, J.; Davis, M.J. Single Sample Scoring of Molecular Phenotypes. BMC Bioinform. 2018, 19, 404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhuva, D.D.; Foroutan, M.; Xie, Y.; Lyu, R.; Cursons, J.; Davis, M.J. Using Singscore to Predict Mutation Status in Acute Myeloid Leukemia from Transcriptomic Signatures. F1000Research 2019, 8, 776. [Google Scholar] [CrossRef]
- Cursons, J.; Souza-Fonseca-Guimaraes, F.; Foroutan, M.; Anderson, A.; Hollande, F.; Hediyeh-Zadeh, S.; Behren, A.; Huntington, N.D.; Davis, M.J. A Gene Signature Predicting Natural Killer Cell Infiltration and Improved Survival in Melanoma Patients. Cancer Immunol. Res. 2019, 7, 1162–1174. [Google Scholar] [CrossRef] [Green Version]
- Bhuva, D.D.; Cursons, J.; Davis, M.J. Stable Gene Expression for Normalisation and Single-Sample Scoring. Nucleic Acids Res. 2020, 48, e113. [Google Scholar] [CrossRef]
- Trivedi, U.H.; Cezard, T.; Bridgett, S.; Montazam, A.; Nichols, J.; Blaxter, M.; Gharbi, K. Quality Control of Next-Generation Sequencing Data without a Reference. Front. Genet. 2014, 5, 111. [Google Scholar] [CrossRef] [Green Version]
- Li, H. Minimap2: Pairwise Alignment for Nucleotide Sequences. Bioinformatics 2018, 34, 3094–3100. [Google Scholar] [CrossRef] [PubMed]
- Patro, R.; Duggal, G.; Love, M.I.; Irizarry, R.A.; Kingsford, C. Salmon Provides Fast and Bias-Aware Quantification of Transcript Expression. Nat. Methods 2017, 14, 417–419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hänzelmann, S.; Castelo, R.; Guinney, J. GSVA: Gene Set Variation Analysis for Microarray and RNA-Seq Data. BMC Bioinform. 2013, 14, 7. [Google Scholar] [CrossRef] [Green Version]
- Slenter, D.N.; Kutmon, M.; Hanspers, K.; Riutta, A.; Windsor, J.; Nunes, N.; Mélius, J.; Cirillo, E.; Coort, S.L.; Digles, D.; et al. WikiPathways: A Multifaceted Pathway Database Bridging Metabolomics to Other Omics Research. Nucleic Acids Res. 2018, 46, D661–D667. [Google Scholar] [CrossRef]
- R: The R Project for Statistical Computing. Available online: https://www.r-project.org/ (accessed on 8 October 2021).
- Wick, R.R.; Judd, L.M.; Holt, K.E. Performance of Neural Network Basecalling Tools for Oxford Nanopore Sequencing. Genome Biol. 2019, 20, 129. [Google Scholar] [CrossRef] [Green Version]
- Parker, M.T.; Knop, K.; Sherwood, A.V.; Schurch, N.J.; Mackinnon, K.; Gould, P.D.; Hall, A.J.; Barton, G.J.; Simpson, G.G. Nanopore Direct RNA Sequencing Maps the Complexity of Arabidopsis MRNA Processing and M6A Modification. eLife 2020, 9, e49658. [Google Scholar] [CrossRef]
- Massaiu, I.; Songia, P.; Chiesa, M.; Valerio, V.; Moschetta, D.; Alfieri, V.; Myasoedova, V.A.; Schmid, M.; Cassetta, L.; Colombo, G.I.; et al. Evaluation of Oxford Nanopore MinION RNA-Seq Performance for Human Primary Cells. Int. J. Mol. Sci. 2021, 22, 6317. [Google Scholar] [CrossRef] [PubMed]
- Sheng, Q.; Vickers, K.; Zhao, S.; Wang, J.; Samuels, D.C.; Koues, O.; Shyr, Y.; Guo, Y. Multi-Perspective Quality Control of Illumina RNA Sequencing Data Analysis. Brief. Funct. Genom. 2016, 16, 194–204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- SEQC/MAQC-III Consortium A Comprehensive Assessment of RNA-Seq Accuracy, Reproducibility and Information Content by the Sequencing Quality Control Consortium. Nat. Biotechnol. 2014, 32, 903–914. [CrossRef] [PubMed]
- Duan, W.; Kong, X.; Li, J.; Li, P.; Zhao, Y.; Liu, T.; Binang, H.B.; Wang, Y.; Du, L.; Wang, C. LncRNA AC010789.1 Promotes Colorectal Cancer Progression by Targeting MicroRNA-432-3p/ZEB1 Axis and the Wnt/β-Catenin Signaling Pathway. Front. Cell Dev. Biol. 2020, 8, 565355. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Davidson, N.M.; Wan, Y.K.; Patel, H.; Yao, F.; Low, H.M.; Hendra, C.; Watten, L.; Sim, A.; Sawyer, C.; et al. A Systematic Benchmark of Nanopore Long Read RNA Sequencing for Transcript Level Analysis in Human Cell Lines. BioRxiv 2021. [Google Scholar] [CrossRef]
- Lin, B.; Hui, J.; Mao, H. Nanopore Technology and Its Applications in Gene Sequencing. Biosensors 2021, 11, 214. [Google Scholar] [CrossRef]
- Ilgisonis, E.; Vavilov, N.; Ponomarenko, E.; Lisitsa, A.; Poverennaya, E.; Zgoda, V.; Radko, S.; Archakov, A. Genome of the Single Human Chromosome 18 as a “Gold Standard” for Its Transcriptome. Front. Genet. 2021, 12, 674534. [Google Scholar] [CrossRef]
- Soneson, C.; Yao, Y.; Bratus-Neuenschwander, A.; Patrignani, A.; Robinson, M.D.; Hussain, S. A Comprehensive Examination of Nanopore Native RNA Sequencing for Characterization of Complex Transcriptomes. Nat. Commun. 2019, 10, 3359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, X.; Regan, K.; Huang, Y.; Zhang, Q.; Li, J.; Seiwert, T.Y.; Cohen, E.E.W.; Xing, H.R.; Lussier, Y.A. Single Sample Expression-Anchored Mechanisms Predict Survival in Head and Neck Cancer. PLoS Comput. Biol. 2012, 8, e1002350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schurch, N.J.; Schofield, P.; Gierliński, M.; Cole, C.; Sherstnev, A.; Singh, V.; Wrobel, N.; Gharbi, K.; Simpson, G.G.; Owen-Hughes, T.; et al. How Many Biological Replicates Are Needed in an RNA-Seq Experiment and Which Differential Expression Tool Should You Use? RNA 2016, 22, 839–851. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Num Total Reads | Average Read Length | Num Mapped Reads | Num Expressed Protein-Coding Transcripts | Num Expressed Protein-Coding Genes |
|---|---|---|---|---|---|
| ONT-1 | 2,416,117 | 1174 | 2,329,641 | 36,001 | 12,562 |
| ONT-2 | 1,813,263 | 1212 | 1,175,355 | 34,898 | 12,361 |
| ONT-3 | 956,446 | 1381 | 914,186 | 31,078 | 11,710 |
| ONT-4 | 1,807,655 | 1176 | 1,630,381 | 34,341 | 12,203 |
| ONT-5 | 1,875,286 | 1333 | 1,799,768 | 34,517 | 12,353 |
| ILMN-1 | 59,789,684 | 100 | 53,499,563 | 30,552 | 13,961 |
| ILMN-2 | 95,048,978 | 100 | 86,520,672 | 31,954 | 14,104 |
| ILMN-3 | 54,075,176 | 100 | 46,702,470 | 28,993 | 13,972 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pyatnitskiy, M.A.; Arzumanian, V.A.; Radko, S.P.; Ptitsyn, K.G.; Vakhrushev, I.V.; Poverennaya, E.V.; Ponomarenko, E.A. Oxford Nanopore MinION Direct RNA-Seq for Systems Biology. Biology 2021, 10, 1131. https://doi.org/10.3390/biology10111131
Pyatnitskiy MA, Arzumanian VA, Radko SP, Ptitsyn KG, Vakhrushev IV, Poverennaya EV, Ponomarenko EA. Oxford Nanopore MinION Direct RNA-Seq for Systems Biology. Biology. 2021; 10(11):1131. https://doi.org/10.3390/biology10111131
Chicago/Turabian StylePyatnitskiy, Mikhail A., Viktoriia A. Arzumanian, Sergey P. Radko, Konstantin G. Ptitsyn, Igor V. Vakhrushev, Ekaterina V. Poverennaya, and Elena A. Ponomarenko. 2021. "Oxford Nanopore MinION Direct RNA-Seq for Systems Biology" Biology 10, no. 11: 1131. https://doi.org/10.3390/biology10111131
APA StylePyatnitskiy, M. A., Arzumanian, V. A., Radko, S. P., Ptitsyn, K. G., Vakhrushev, I. V., Poverennaya, E. V., & Ponomarenko, E. A. (2021). Oxford Nanopore MinION Direct RNA-Seq for Systems Biology. Biology, 10(11), 1131. https://doi.org/10.3390/biology10111131