Full-Length Transcriptome Sequencing of the Scleractinian Coral Montipora foliosa Reveals the Gene Expression Profile of Coral–Zooxanthellae Holobiont

 , , and
, , and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Specimen Collection and Coral Culture System
2.2. Sampling and RNA Extraction
2.3. Library Preparation and Sequencing
2.3.1. PacBio Library Preparation and Sequencing
2.3.2. Illumina Library Preparation and Sequencing
2.4. Bioinformatics
2.4.1. Data Processing
2.4.2. Error Correction Using Illumina Reads
2.4.3. Gene Functional Annotation
2.4.4. Coral and Symbiodinium Gene Identification
2.4.5. Gene Expression Quantification
2.4.6. GO, KEGG, KOG/COG, and Pfam Enrichment Analysis
3. Results
3.1. Polymerase Read Statistics
3.2. Correction of Transcripts and Removal of Redundant Transcripts
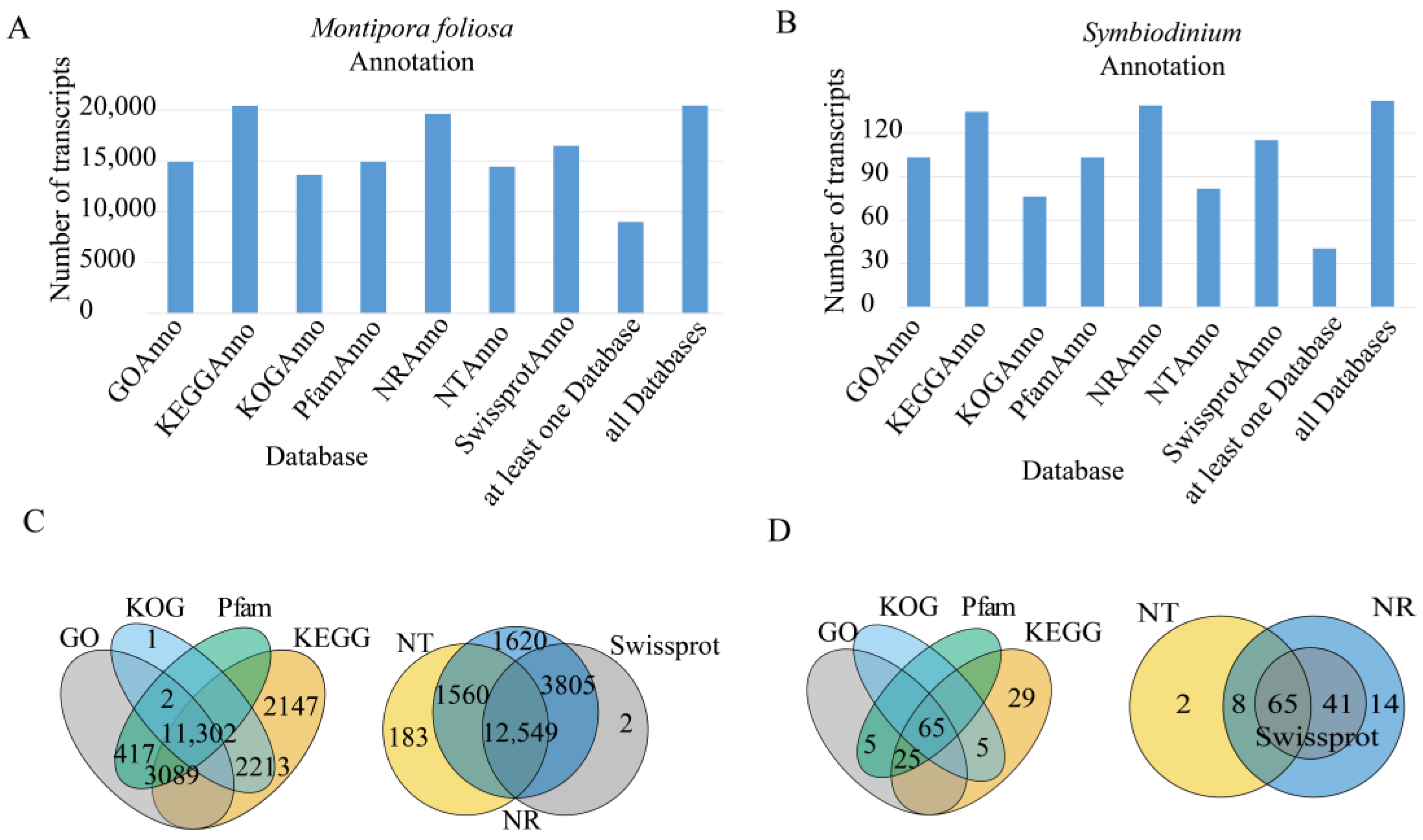
3.3. Gene Function Annotation
3.3.1. NR Database Annotation Results
3.3.2. GO Classification
3.3.3. KOG Classification
3.3.4. KEGG Classification
3.3.5. Pfam Database Annotations
3.3.6. Swiss-Prot Database Annotations
3.4. Gene Structure Analysis
3.4.1. CDS Prediction
3.4.2. Transcription Factor Analysis
3.4.3. LncRNA Prediction
3.5. Gene Expression Analysis
3.5.1. Gene Enrichment Analysis of All Zooxanthellae
3.5.2. Enrichment Analysis of M. foliosa Highly Expressed Genes
3.5.3. Enrichment Analysis of M. foliosa Metabolism-Related Genes Located on the Cell Membrane
4. Discussion
4.1. Gene Function of All Zooxanthellae Related to Symbiosis
4.2. Functions of Highly Expressed M. foliosa Genes Related to Symbiosis
4.2.1. GO Entries Related to Symbiosis
4.2.2. KEGG, KOG, and Pfam Entries Related to Symbiosis
4.3. Molecular Biological Mechanisms of Intracellular Symbiosis
4.4. Symbiosis Recognition-Related Genes
4.5. M. foliosa May Convert the Monosaccharides Transported by Zooxanthellae into Glycans, Thereby Avoiding the Fluctuation of the Sugar Concentration in the Cell
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type | Before Correcting | After Correcting |
|---|---|---|
| Total nucleotides | 1.31 × 108 | 1.31 × 108 |
| Total number | 38,365 | 38,365 |
| Mean length | 3414 | 3411 |
| Min length | 57 | 57 |
| Max length | 12,445 | 12,442 |
| N50 | 4501 | 4501 |
| N90 | 1988 | 1987 |
| Transcript’s Length Interval | Number of Transcripts | Number of Genes |
|---|---|---|
| <500 bp | 326 | 160 |
| 500–1k bp | 1362 | 842 |
| 1k–2k bp | 7511 | 4089 |
| 2k–3k bp | 12,753 | 6882 |
| >3k bp | 16,413 | 8487 |
| Total | 38,365 | 20,460 |
| Metrics | Count |
|---|---|
| Total nucleotides | 67,436,488 |
| Total number | 20,460 |
| Mean length | 3297 |
| Min length | 99 |
| Max length | 12,442 |
| N50 | 3844 |
| N90 | 1959 |
| FPKM Interval | MF_1 | MF_2 | MF_3 |
|---|---|---|---|
| 0–0.1 | 50,757 (62.88%) | 50,735 (62.85%) | 50,604 (62.69%) |
| 0.1–1 | 5930 (7.35%) | 5904 (7.31%) | 6014 (7.45%) |
| 1–5 | 9405 (11.65%) | 9309 (11.53%) | 9409 (11.66%) |
| 5–15 | 7673 (9.51%) | 7819 (9.69%) | 7776 (9.63%) |
| 15–60 | 5047 (6.25%) | 5044 (6.25%) | 5044 (6.25%) |
| >60 | 1911 (2.37%) | 1912 (2.37%) | 1876 (2.32%) |

References
- Cohen, A.L.; McConnaughey, T.A. Geochemical perspectives on coral mineralization. In Biomineralization; Dove, P.M., DeYoreo, J.J., Weiner, S., Eds.; Elsevier: Amsterdam, The Netherlands, 2003; Volume 54, pp. 151–187. ISBN 0-93995-066-9. [Google Scholar]
- Tambutté, S.; Holcomb, M.; Ferrier-Pagès, C.; Reynaud, S.; Tambutté, É.; Zoccola, D.; Allemand, D. Coral biomineralization: From the gene to the environment. J. Exp. Mar. Biol. Ecol. 2011, 408, 58–78. [Google Scholar] [CrossRef]
- Weis, V.M. Cell Biology of Coral Symbiosis: Foundational Study Can Inform Solutions to the Coral Reef Crisis. Integr. Comp. Biol. 2019, 59, 845–855. [Google Scholar] [CrossRef]
- Davy, S.K.; Allemand, D.; Weis, V.M. Cell Biology of Cnidarian-Dinoflagellate Symbiosis. Microbiol. Mol. Biol. Rev. 2012, 76, 229–261. [Google Scholar] [CrossRef] [Green Version]
- Stat, M.; Morris, E.; Gates, R.D. Functional diversity in coral-dinoflagellate symbiosis. Proc. Natl. Acad. Sci. USA 2008, 105, 9256–9261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nielsen, D.A.; Petrou, K.; Gates, R.D. Coral bleaching from a single cell perspective. ISME J. 2018, 12, 1558–1567. [Google Scholar] [CrossRef] [PubMed]
- Cunning, R.; Silverstein, R.N.; Baker, A.C. Investigating the causes and consequences of symbiont shuffling in a multi-partner reef coral symbiosis under environmental change. Proc. R. Soc. B Boil. Sci. 2015, 282, 20141725. [Google Scholar] [CrossRef] [PubMed]
- Silverstein, R.N.; Correa, A.M.S.; Baker, A.C. Specificity is rarely absolute in coral–algal symbiosis: Implications for coral response to climate change. Proc. R. Soc. B Boil. Sci. 2012, 279, 2609–2618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacobovitz, M.R.; Rupp, S.; Voss, P.A.; Maegele, I.; Gornik, S.G.; Guse, A. Dinoflagellate symbionts escape vomocytosis by host cell immune suppression. Nat. Microbiol. 2021, 6, 769–782. [Google Scholar] [CrossRef]
- Xiang, T.; Lehnert, E.; Jinkerson, R.E.; Clowez, S.; Kim, R.G.; Denofrio, J.C.; Pringle, J.R.; Grossman, A.R. Symbiont population control by host-symbiont metabolic interaction in Symbiodiniaceae-cnidarian associations. Nat. Commun. 2020, 11, 108. [Google Scholar] [CrossRef] [Green Version]
- Morris, L.A.; Voolstra, C.R.; Quigley, K.M.; Bourne, D.G.; Bay, L.K. Nutrient Availability and Metabolism Affect the Stability of Coral–Symbiodiniaceae Symbioses. Trends Microbiol. 2019, 27, 678–689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shinzato, C.; Shoguchi, E.; Kawashima, T.; Hamada, M.; Hisata, K.; Tanaka, M.; Fujie, M.; Fujiwara, M.; Koyanagi, R.; Ikuta, T.; et al. Using the Acropora digitifera genome to understand coral responses to environmental change. Nature 2011, 476, 320–323. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.; Cheng, S.; Song, B.; Zhong, X.; Lin, X.; Li, W.; Li, L.; Zhang, Y.; Zhang, H.; Ji, Z.; et al. The Symbiodinium kawagutii genome illuminates dinoflagellate gene expression and coral symbiosis. Science 2015, 350, 691–694. [Google Scholar] [CrossRef] [Green Version]
- Shinzato, C.; Mungpakdee, S.; Satoh, N.; Shoguchi, E. A genomic approach to coral-dinoflagellate symbiosis: Studies of Acropora digitifera and Symbiodinium minutum. Front. Microbiol. 2014, 5, 336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, H.; Stephens, T.G.; González-Pech, R.A.; Beltran, V.H.; Lapeyre, B.; Bongaerts, P.; Cooke, I.; Aranda, M.; Bourne, D.G.; Forêt, S.; et al. Symbiodinium genomes reveal adaptive evolution of functions related to coral-dinoflagellate symbiosis. Commun. Biol. 2018, 1, 95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baumgarten, S.; Simakov, O.; Esherick, L.Y.; Liew, Y.J.; Lehnert, E.M.; Michell, C.T.; Li, Y.; Hambleton, E.A.; Guse, A.; Oates, M.E.; et al. The genome of Aiptasia, a sea anemone model for coral symbiosis. Proc. Natl. Acad. Sci. USA 2015, 112, 11893–11898. [Google Scholar] [CrossRef] [Green Version]
- Yuyama, I.; Ishikawa, M.; Nozawa, M.; Yoshida, M.-A.; Ikeo, K. Transcriptomic changes with increasing algal symbiont reveal the detailed process underlying establishment of coral-algal symbiosis. Sci. Rep. 2018, 8, 16802. [Google Scholar] [CrossRef]
- Chin, C.-S.; Peluso, P.; Sedlazeck, F.J.; Nattestad, M.; Concepcion, G.T.; Clum, A.; Dunn, C.; O’malley, R.; Figueroa-Balderas, R.; Morales-Cruz, A.; et al. Phased diploid genome assembly with single-molecule real-time sequencing. Nat. Methods 2016, 13, 1050–1054. [Google Scholar] [CrossRef] [Green Version]
- Salmela, L.; Rivals, E. LoRDEC: Accurate and efficient long read error correction. Bioinformatics 2014, 30, 3506–3514. [Google Scholar] [CrossRef]
- Fu, L.; Niu, B.; Zhu, Z.; Wu, S.; Li, W. CD-HIT: Accelerated for clustering the next-generation sequencing data. Bioinformatics 2012, 28, 3150–3152. [Google Scholar] [CrossRef]
- Li, W.; Jaroszewski, L.; Godzik, A. Tolerating some redundancy significantly speeds up clustering of large protein databases. Bioinformatics 2002, 18, 77–82. [Google Scholar] [CrossRef]
- Tatusov, R.L.; Fedorova, N.D.; Jackson, J.D.; Jacobs, A.R.; Kiryutin, B.; Koonin, E.V.; Krylov, D.M.; Mazumder, R.; Mekhedov, S.L.; Nikolskaya, A.N.; et al. The COG database: An updated version includes eukaryotes. BMC Bioinform. 2003, 4, 41. [Google Scholar] [CrossRef] [Green Version]
- Bairoch, A.; Apweiler, R. The SWISS-PROT protein sequence database and its supplement TrEMBL in 2000. Nucleic Acids Res. 2000, 28, 45–48. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S.; Kawashima, S.; Okuno, Y.; Hattori, M. The KEGG resource for deciphering the genome. Nucleic Acids Res. 2004, 32, D277–D280. [Google Scholar] [CrossRef] [Green Version]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.-M.; Liu, T.; Liu, C.-J.; Song, S.; Zhang, X.; Liu, W.; Jia, H.; Xue, Y.; Guo, A.-Y. AnimalTFDB 2.0: A resource for expression, prediction and functional study of animal transcription factors. Nucleic Acids Res. 2014, 43, D76–D81. [Google Scholar] [CrossRef]
- Zheng, Y.; Jiao, C.; Sun, H.; Rosli, H.G.; Pombo, M.A.; Zhang, P.; Banf, M.; Dai, X.; Martin, G.; Giovannoni, J.J.; et al. iTAK: A Program for Genome-wide Prediction and Classification of Plant Transcription Factors, Transcriptional Regulators, and Protein Kinases. Mol. Plant 2016, 9, 1667–1670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, L.; Luo, H.; Bu, D.; Zhao, G.; Yu, K.; Zhang, C.; Liu, Y.; Chen, R.; Zhao, Y. Utilizing sequence intrinsic composition to classify protein-coding and long non-coding transcripts. Nucleic Acids Res. 2013, 41, e166. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.-J.; Yang, D.-C.; Kong, L.; Hou, M.; Meng, Y.-Q.; Wei, L.; Gao, G. CPC2: A fast and accurate coding potential calculator based on sequence intrinsic features. Nucleic Acids Res. 2017, 45, W12–W16. [Google Scholar] [CrossRef] [Green Version]
- Finn, R.D.; Coggill, P.; Eberhardt, R.Y.; Eddy, S.R.; Mistry, J.; Mitchell, A.L.; Potter, S.C.; Punta, M.; Qureshi, M.; Sangrador-Vegas, A.; et al. The Pfam protein families database: Towards a more sustainable future. Nucleic Acids Res. 2016, 44, D279–D285. [Google Scholar] [CrossRef] [PubMed]
- Li, A.; Zhang, J.; Zhou, Z. PLEK: A tool for predicting long non-coding RNAs and messenger RNAs based on an improved k-mer scheme. BMC Bioinform. 2014, 15, 311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Helmkampf, M.; Bellinger, M.R.; Geib, S.M.; Sim, S.B.; Takabayashi, M. Draft Genome of the Rice Coral Montipora capitata Obtained from Linked-Read Sequencing. Genome Biol. Evol. 2019, 11, 2045–2054. [Google Scholar] [CrossRef] [Green Version]
- Shinzato, C.; Khalturin, K.; Inoue, J.; Zayasu, Y.; Kanda, M.; Kawamitsu, M.; Yoshioka, Y.; Yamashita, H.; Suzuki, G.; Satoh, N. Eighteen Coral Genomes Reveal the Evolutionary Origin of Acropora Strategies to Accommodate Environmental Changes. Mol. Biol. Evol. 2020, 38, 16–30. [Google Scholar] [CrossRef]
- Yoshioka, Y.; Yamashita, H.; Suzuki, G.; Zayasu, Y.; Tada, I.; Kanda, M.; Satoh, N.; Shoguchi, E.; Shinzato, C. Whole-Genome Transcriptome Analyses of Native Symbionts Reveal Host Coral Genomic Novelties for Establishing Coral–Algae Symbioses. Genome Biol. Evol. 2020, 13, evaa240. [Google Scholar] [CrossRef] [PubMed]
- González-Pech, R.A.; Stephens, T.G.; Chen, Y.; Mohamed, A.R.; Cheng, Y.; Shah, S.; Dougan, K.E.; Fortuin, M.D.A.; Lagorce, R.; Burt, D.W.; et al. Comparison of 15 dinoflagellate genomes reveals extensive sequence and structural divergence in family Symbiodiniaceae and genus Symbiodinium. BMC Biol. 2021, 19, 73. [Google Scholar] [CrossRef] [PubMed]
- Shoguchi, E.; Shinzato, C.; Hisata, K.; Satoh, N.; Mungpakdee, S. The Large Mitochondrial Genome of Symbiodinium minutum Reveals Conserved Noncoding Sequences between Dinoflagellates and Apicomplexans. Genome Biol. Evol. 2015, 7, 2237–2244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beedessee, G.; Hisata, K.; Roy, M.C.; Satoh, N.; Shoguchi, E. Multifunctional polyketide synthase genes identified by genomic survey of the symbiotic dinoflagellate, Symbiodinium minutum. BMC Genom. 2015, 16, 941. [Google Scholar] [CrossRef] [Green Version]
- Shoguchi, E.; Shinzato, C.; Kawashima, T.; Gyoja, F.; Mungpakdee, S.; Koyanagi, R.; Takeuchi, T.; Hisata, K.; Tanaka, M.; Fujiwara, M.; et al. Draft Assembly of the Symbiodinium minutum Nuclear Genome Reveals Dinoflagellate Gene Structure. Curr. Biol. 2013, 23, 1399–1408. [Google Scholar] [CrossRef] [Green Version]
- Li, H. Minimap2: Pairwise alignment for nucleotide sequences. Bioinformatics 2018, 34, 3094–3100. [Google Scholar] [CrossRef]
- Ruffalo, M.; Koyutürk, M.; Ray, S.; LaFramboise, T. Accurate estimation of short read mapping quality for next-generation genome sequencing. Bioinformatics 2012, 28, i349–i355. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.; Li, M.-C.; Konaté, M.M.; Chen, L.; Das, B.; Karlovich, C.; Williams, P.M.; Evrard, Y.A.; Doroshow, J.H.; McShane, L.M. TPM, FPKM, or Normalized Counts? A Comparative Study of Quantification Measures for the Analysis of RNA-seq Data from the NCI Patient-Derived Models Repository. J. Transl. Med. 2021, 19, 269. [Google Scholar] [CrossRef]
- Liberzon, A.; Birger, C.; Thorvaldsdóttir, H.; Ghandi, M.; Mesirov, J.P.; Tamayo, P. The Molecular Signatures Database Hallmark Gene Set Collection. Cell Syst. 2015, 1, 417–425. [Google Scholar] [CrossRef] [Green Version]
- Lichtenberg, M.; Larkum, A.W.D.; Kühl, M. Photosynthetic acclimation of Symbiodinium in hospite depends on vertical position in the tissue of the scleractinian coral Montastrea curta. Front. Microbiol. 2016, 7, 230. [Google Scholar] [CrossRef] [Green Version]
- Rädecker, N.; Pogoreutz, C.; Voolstra, C.R.; Wiedenmann, J.; Wild, C. Nitrogen cycling in corals: The key to understanding holobiont functioning? Trends Microbiol. 2015, 23, 490–497. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosic, N.N.; Ling, E.Y.S.; Chan, C.-K.K.; Lee, H.C.; Kaniewska, P.; Edwards, D.; Dove, S.; Hoegh-Guldberg, O. Unfolding the secrets of coral–algal symbiosis. ISME J. 2015, 9, 844–856. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bellantuono, A.J.; Granados-Cifuentes, C.; Miller, D.J.; Hoegh-Guldberg, O.; Rodriguez-Lanetty, M. Coral Thermal Tolerance: Tuning Gene Expression to Resist Thermal Stress. PLoS ONE 2012, 7, e50685. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, Y.; Suzuki, T.; Awai, K.; Shioi, Y. Isolation and characterization of a tandem-repeated cysteine protease from the symbiotic dinoflagellate Symbiodinium sp. KB8. PLoS ONE 2019, 14, e0211534. [Google Scholar] [CrossRef]
- Graham, D.; Smillie, R. Carbonate Dehydratase in Marine Organisms of the Great Barrier Reef. Funct. Plant Biol. 1976, 3, 113–119. [Google Scholar] [CrossRef]
- Levy, S.; Elek, A.; Grau-Bové, X.; Menéndez-Bravo, S.; Iglesias, M.; Tanay, A.; Mass, T.; Sebé-Pedrós, A. A stony coral cell atlas illuminates the molecular and cellular basis of coral symbiosis, calcification, and immunity. Cell 2021, 184, 2973–2987.e18. [Google Scholar] [CrossRef]
- Bessho-Uehara, M.; Francis, W.R.; Haddock, S.H.D. Biochemical characterization of diverse deep-sea anthozoan bioluminescence systems. Mar. Biol. 2020, 167, 114. [Google Scholar] [CrossRef]
- Bollati, E.; D’Angelo, C.; Alderdice, R.; Pratchett, M.; Ziegler, M.; Wiedenmann, J. Optical Feedback Loop Involving Dinoflagellate Symbiont and Scleractinian Host Drives Colorful Coral Bleaching. Curr. Biol. 2020, 30, 2433–2445.e3. [Google Scholar] [CrossRef]
- Császár, N.B.M.; Seneca, F.O.; van Oppen, M.J.H. Variation in antioxidant gene expression in the scleractinian coral Acropora millepora under laboratory thermal stress. Mar. Ecol. Prog. Ser. 2009, 392, 93–102. [Google Scholar] [CrossRef] [Green Version]
- Weston, A.J.; Dunlap, W.C.; Shick, J.M.; Klueter, A.; Iglic, K.; Vukelic, A.; Starcevic, A.; Ward, M.; Wells, M.L.; Trick, C.G.; et al. A Profile of an Endosymbiont-enriched Fraction of the Coral Stylophora pistillata Reveals Proteins Relevant to Microbial-Host Interactions. Mol. Cell. Proteom. 2012, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pernice, M.; Meibom, A.; Van Den Heuvel, A.; Kopp, C.; Domart-Coulon, I.; Hoegh-Guldberg, O.; Dove, S. A single-cell view of ammonium assimilation in coral–dinoflagellate symbiosis. ISME J. 2012, 6, 1314–1324. [Google Scholar] [CrossRef] [Green Version]
- Chalker, B.E.; Taylor, D.L. Light-enhanced calcification, and the role of oxidative phosphorylation in calcification of the coral Acropora Cervicornis. Proc. R. Soc. London. Ser. B Boil. Sci. 1975, 190, 323–331. [Google Scholar] [CrossRef]
- He, C.; Han, T.; Liao, X.; Zhou, Y.; Wang, X.; Guan, R.; Tian, T.; Li, Y.; Bi, C.; Lu, N.; et al. Phagocytic intracellular digestion in amphioxus (Branchiostoma). Proc. R. Soc. B Boil. Sci. 2018, 285, 20180438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fitt, W.K.; Trench, R.K. Endocytosis of the symbiotic dinoflagellate Symbiodinium microadriaticum Freudenthal by endo-dermal cells of the scyphistomae of Cassiopeia xamachana and resistance of the algae to host digestion. J. Cell Sci. 1983, 64, 195–212. [Google Scholar] [CrossRef]
- Aihara, Y.; Maruyama, S.; Baird, A.H.; Iguchi, A.; Takahashi, S.; Minagawa, J. Green fluorescence from cnidarian hosts attracts symbiotic algae. Proc. Natl. Acad. Sci. USA 2019, 116, 2118–2123. [Google Scholar] [CrossRef] [Green Version]
- Fransolet, D.; Roberty, S.; Plumier, J.-C. Establishment of endosymbiosis: The case of cnidarians and Symbiodinium. J. Exp. Mar. Biol. Ecol. 2012, 420–421, 1–7. [Google Scholar] [CrossRef]
- Zhu, Y.; Thangamani, S.; Ho, B.; Ding, J.L. The ancient origin of the complement system. EMBO J. 2004, 24, 382–394. [Google Scholar] [CrossRef] [Green Version]
- Poole, A.Z.; Kitchen, S.A.; Weis, V.M. The Role of Complement in Cnidarian-Dinoflagellate Symbiosis and Immune Challenge in the Sea Anemone Aiptasia pallida. Front. Microbiol. 2016, 7, 519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rus, H.; Cudrici, C.; Niculescu, F. The Role of the Complement System in Innate Immunity. Immunol. Res. 2005, 33, 103–112. [Google Scholar] [CrossRef]
- Nesargikar, P.; Spiller, B.; Chavez, R. The complement system: History, pathways, cascade and inhibitors. Eur. J. Microbiol. Immunol. 2012, 2, 103–111. [Google Scholar] [CrossRef]
- Janeway, C.A., Jr.; Travers, P.; Walport, M.; Shlomchik, M.J. Immunobiology: The Immune System in Health and Disease, 5th ed.; T-cell receptor gene rearrangement; Garland Science: New York, NY, USA, 2001. [Google Scholar]
- Li, Y.; Liew, Y.J.; Cui, G.; Cziesielski, M.J.; Zahran, N.; Michell, C.T.; Voolstra, C.R.; Aranda, M. DNA methylation regulates transcriptional homeostasis of algal endosymbiosis in the coral model Aiptasia. Sci. Adv. 2018, 4, eaat2142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Endo, Y.; Takahashi, M.; Fujita, T. Lectin complement system and pattern recognition. Immunobiology 2006, 211, 283–293. [Google Scholar] [CrossRef]
- Neubauer, E.-F.; Poole, A.Z.; Neubauer, P.; Detournay, O.; Tan, K.; Davy, S.K.; Weis, V.M. A diverse host thrombospondin-type-1 repeat protein repertoire promotes symbiont colonization during establishment of cnidarian-dinoflagellate symbiosis. eLife 2017, 6, e24494. [Google Scholar] [CrossRef]
- Adams, J.C.; Tucker, R.P. The thrombospondin type 1 repeat (TSR) superfamily: Diverse proteins with related roles in neu-ronal development. Dev. Dyn. 2000, 218, 280–299. [Google Scholar] [CrossRef]
- Tucker, R.P. The thrombospondin type 1 repeat superfamily. Int. J. Biochem. Cell Biol. 2004, 36, 969–974. [Google Scholar] [CrossRef]
- Lopez-Dee, Z.P.; Chittur, S.V.; Patel, B.; Stanton, R.; Wakeley, M.; Lippert, B.; Menaker, A.; Eiche, B.; Terry, R.; Gutierrez, L.S. Thrombospondin-1 Type 1 Repeats in a Model of Inflammatory Bowel Disease: Transcript Profile and Therapeutic Effects. PLoS ONE 2012, 7, e34590. [Google Scholar] [CrossRef] [Green Version]
- Hourcade, D.E. The Role of Properdin in the Assembly of the Alternative Pathway C3 Convertases of Complement. J. Biol. Chem. 2006, 281, 2128–2132. [Google Scholar] [CrossRef] [Green Version]
- Wolfowicz, I.; Baumgarten, S.; Voss, P.A.; Hambleton, E.A.; Voolstra, C.R.; Hatta, M.; Guse, A. Aiptasia sp. larvae as a model to reveal mechanisms of symbiont selection in cnidarians. Sci. Rep. 2016, 6, 32366. [Google Scholar] [CrossRef] [Green Version]
- Schwarz, R.S.; Hodes-Villamar, L.; Fitzpatrick, K.A.; Fain, M.G.; Hughes, A.L.; Cadavid, L.F. A gene family of putative immune recognition molecules in the hydroid Hydractinia. Immunogenetics 2007, 59, 233–246. [Google Scholar] [CrossRef]
- Detournay, O.; Schnitzler, C.E.; Poole, A.; Weis, V.M. Regulation of cnidarian–dinoflagellate mutualisms: Evidence that activation of a host TGFβ innate immune pathway promotes tolerance of the symbiont. Dev. Comp. Immunol. 2012, 38, 525–537. [Google Scholar] [CrossRef] [Green Version]
- Berthelier, J.; Schnitzler, C.E.; Wood-Charlson, E.M.; Poole, A.Z.; Weis, V.M.; Detournay, O. Implication of the host TGFβ pathway in the onset of symbiosis between larvae of the coral Fungia scutaria and the dinoflagellate Symbiodinium sp. (clade C1f). Coral Reefs 2017, 36, 1263–1268. [Google Scholar] [CrossRef]
- Spiegel, S.; Milstien, S. Sphingosine-1-phosphate: An enigmatic signalling lipid. Nat. Rev. Mol. Cell Biol. 2003, 4, 397–407. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Lanetty, M.; Phillips, W.S.; Weis, V. Transcriptome analysis of a cnidarian–dinoflagellate mutualism reveals complex modulation of host gene expression. BMC Genom. 2006, 7, 23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kitchen, S.A.; Weis, V.M. The sphingosine rheostat is involved in the cnidarian heat stress response but not necessarily in bleaching. J. Exp. Biol. 2017, 220, 1709–1720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Detournay, O.; Weis, V.M. Role of the Sphingosine Rheostat in the Regulation of Cnidarian-Dinoflagellate Symbioses. Biol. Bull. 2011, 221, 261–269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yao, Q. Nucleotide-binding oligomerization domain containing 2: Structure, function, and diseases. Semin. Arthritis Rheum. 2013, 43, 125–130. [Google Scholar] [CrossRef]
- Brennan, J.J.; Gilmore, T.D. Evolutionary Origins of Toll-like Receptor Signaling. Mol. Biol. Evol. 2018, 35, 1576–1587. [Google Scholar] [CrossRef] [Green Version]
- Hemmrich, G.; Miller, D.J.; Bosch, T.C.G. The evolution of immunity: A low-life perspective. Trends Immunol. 2007, 28, 449–454. [Google Scholar] [CrossRef] [PubMed]
- Brennan, J.J.; Messerschmidt, J.L.; Williams, L.M.; Matthews, B.J.; Reynoso, M.; Gilmore, T.D. Sea anemone model has a single Toll-like receptor that can function in pathogen detection, NF-κB signal transduction, and development. Proc. Natl. Acad. Sci. USA 2017, 114, E10122–E10131. [Google Scholar] [CrossRef] [Green Version]
- Perez, S.; Weis, V. Nitric oxide and cnidarian bleaching: An eviction notice mediates breakdown of a symbiosis. J. Exp. Biol. 2006, 209, 2804–2810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mansfield, K.M.; Carter, N.; Nguyen, L.; Cleves, P.A.; Alshanbayeva, A.; Williams, L.M.; Crowder, C.; Penvose, A.R.; Finnerty, J.R.; Weis, V.M.; et al. Transcription factor NF-κB is modulated by symbiotic status in a sea anemone model of cnidarian bleaching. Sci. Rep. 2017, 7, 16025. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeSalvo, M.K.; Sunagawa, S.; Voolstra, C.R.; Medina, M. Transcriptomic responses to heat stress and bleaching in the elkhorn coral Acropora palmata. Mar. Ecol. Prog. Ser. 2010, 402, 97–113. [Google Scholar] [CrossRef] [Green Version]
- Chen, M.-C.; Hong, M.-C.; Huang, Y.-S.; Liu, M.-C.; Cheng, Y.-M.; Fang, L.-S. ApRab11, a cnidarian homologue of the recycling regulatory protein Rab11, is involved in the establishment and maintenance of the Aiptasia–Symbiodinium endosymbiosis. Biochem. Biophys. Res. Commun. 2005, 338, 1607–1616. [Google Scholar] [CrossRef]







| Metrics | Value |
|---|---|
| Polymerase read bases (G) | 43.11 |
| Polymerase reads | 699,225 |
| Polymerase read length (mean) | 61,652 |
| Polymerase read N50 | 106,108 |
| Enrichment Class | Description | p-Value |
|---|---|---|
| KEGG | Mucin type O-glycan biosynthesis | 0 |
| KEGG | Pyrimidine metabolism | 0.029366305 |
| KEGG | Other types of O-glycan biosynthesis | 0.03529412 |
| KEGG | Glycosphingolipid biosynthesis | 0.046674445 |
| GO-MF | Acetylglucosaminyltransferase activity | 0 |
| GO-MF | Acetylgalactosaminyltransferase activity | 0.001059322 |
| GO-MF | Galactosyltransferase activity | 0.001064963 |
| GO-MF | Transferase activity, transferring glycosyl groups | 0.002166847 |
| GO-MF | Transferase activity, transferring hexosyl groups | 0.045801528 |
| Components | Number of Transcriptions | Average Expression (TPM) | Average Number of Isoforms |
|---|---|---|---|
| Complement and its receptor | 4 | 20.5 | 6 |
| Lectin | 70 | 24.32 | 1.54 |
| Rab | 84 | 30.7 | 1.27 |
| Scavenger receptors | 14 | 17.75 | 1.14 |
| Sphingosine rheostat | 4 | 15.67 | 1.5 |
| Thrombospondin type I repeats | 23 | 11.88 | 1 |
| Toll-like receptors | 8 | 12.45 | 1 |
| Transforming growth factor beta | 17 | 25.92 | 1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, Y.; Liao, X.; Han, T.; Su, A.; Guo, Z.; Lu, N.; He, C.; Lu, Z. Full-Length Transcriptome Sequencing of the Scleractinian Coral Montipora foliosa Reveals the Gene Expression Profile of Coral–Zooxanthellae Holobiont. Biology 2021, 10, 1274. https://doi.org/10.3390/biology10121274
Liu Y, Liao X, Han T, Su A, Guo Z, Lu N, He C, Lu Z. Full-Length Transcriptome Sequencing of the Scleractinian Coral Montipora foliosa Reveals the Gene Expression Profile of Coral–Zooxanthellae Holobiont. Biology. 2021; 10(12):1274. https://doi.org/10.3390/biology10121274
Chicago/Turabian StyleLiu, Yunqing, Xin Liao, Tingyu Han, Ao Su, Zhuojun Guo, Na Lu, Chunpeng He, and Zuhong Lu. 2021. "Full-Length Transcriptome Sequencing of the Scleractinian Coral Montipora foliosa Reveals the Gene Expression Profile of Coral–Zooxanthellae Holobiont" Biology 10, no. 12: 1274. https://doi.org/10.3390/biology10121274
APA StyleLiu, Y., Liao, X., Han, T., Su, A., Guo, Z., Lu, N., He, C., & Lu, Z. (2021). Full-Length Transcriptome Sequencing of the Scleractinian Coral Montipora foliosa Reveals the Gene Expression Profile of Coral–Zooxanthellae Holobiont. Biology, 10(12), 1274. https://doi.org/10.3390/biology10121274