
Heterologous Expression and Rational Design of l-asparaginase from Rhizomucor miehei to Improve Thermostability

 ,
, 
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Plasmids, Strains, and Media
2.2. Expression of l-asparaginase Genes from Various Sources in E. coli
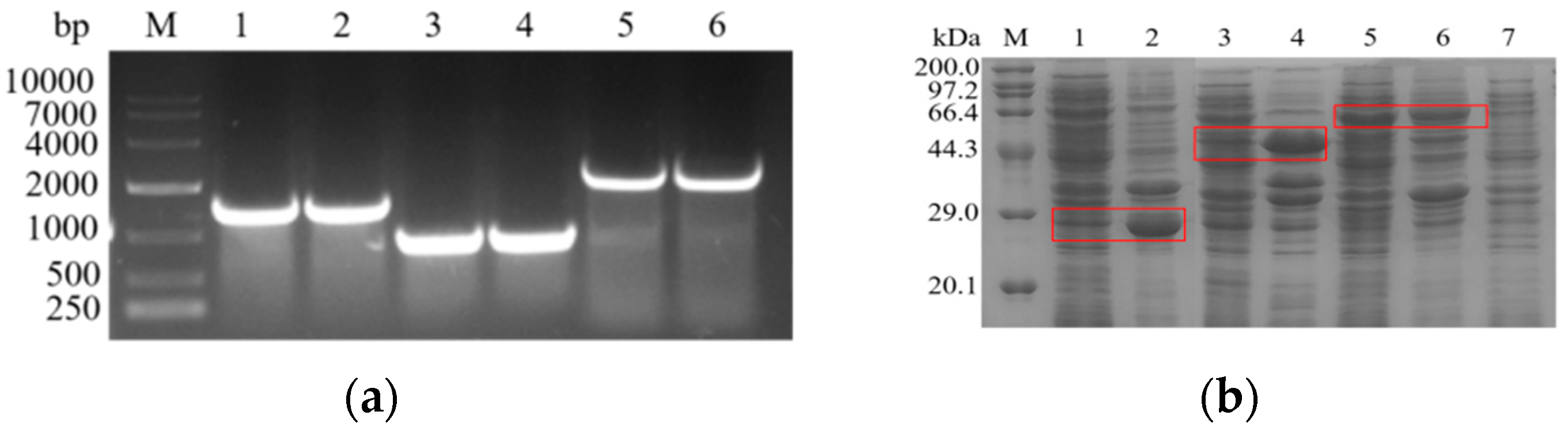
2.3. SDS-PAGE Analysis of Recombinant l-asparaginase
2.4. Purification and Quantification of Recombinant l-asparaginase
2.5. Determination of l-asparaginase Activity
2.6. Simulation and Analysis of the Three-Dimensional Structure of l-asparaginase
2.7. Site-Directed Mutagenesis
2.8. Characterization of the Enzymatic Properties of Recombinant l-asparaginase and Mutants
2.9. Fermentation in a 5-L Fermenter
3. Results
3.1. Cloning and Expression of l-asparaginase Genes from Different Sources
3.2. Selection of Mutagenesis Sites for Improving Thermostability of l-asparaginase
3.3. Enzyme Activity Assays of Recombinant Mutants
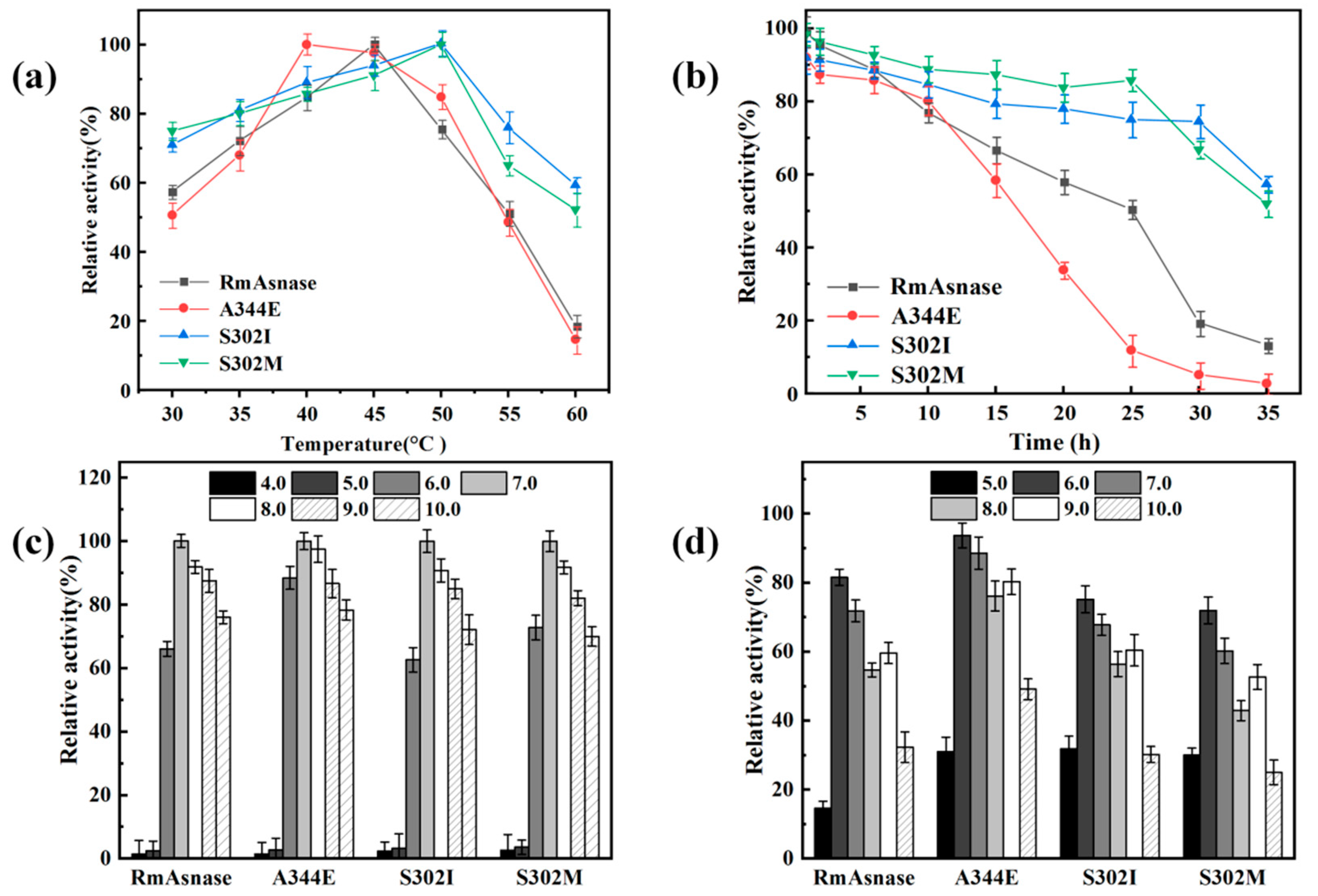
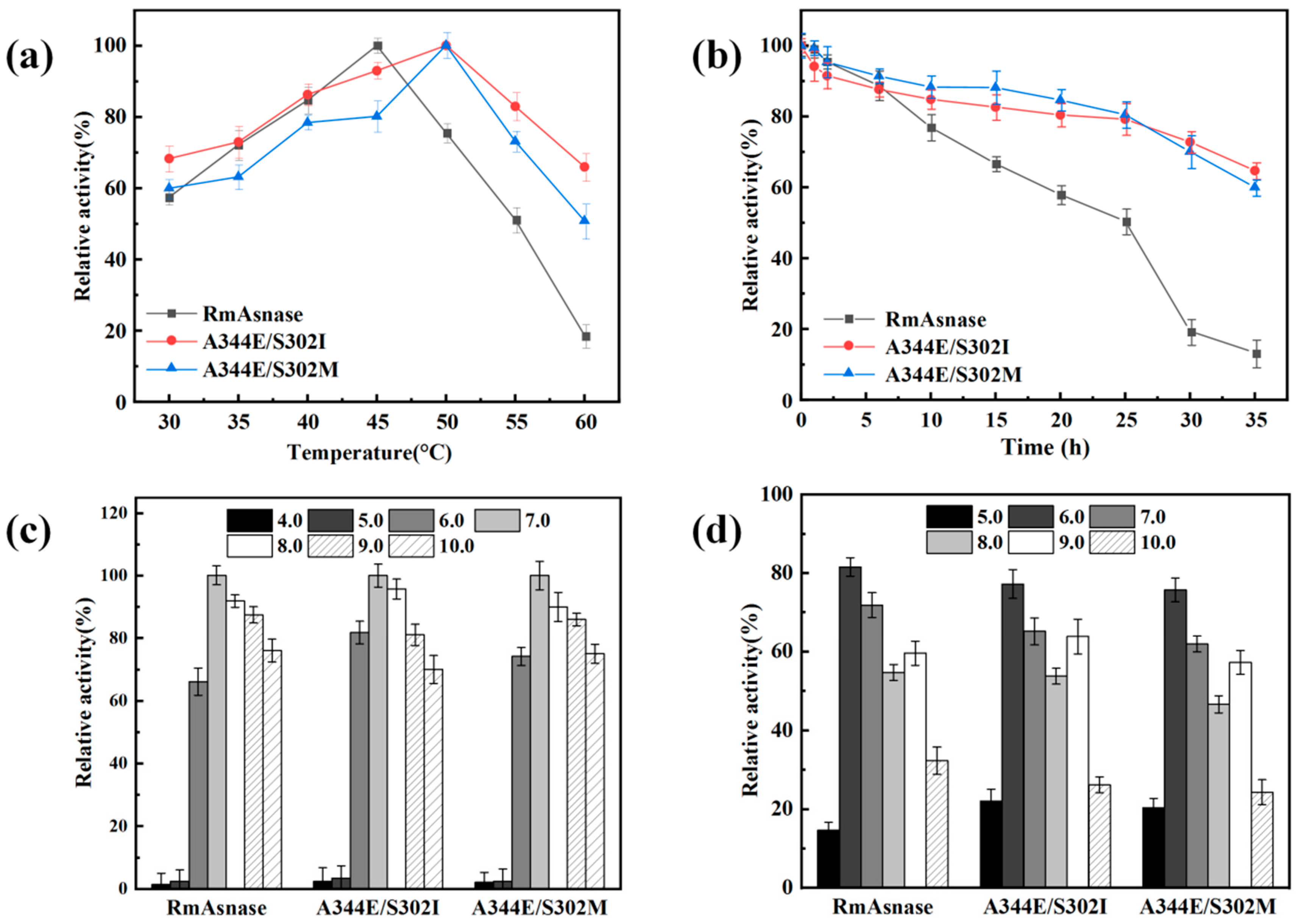
3.4. Optimal Temperature and Thermostability of Recombinant Mutants
3.5. Optimal pH and pH Stability of Recombinant Mutants
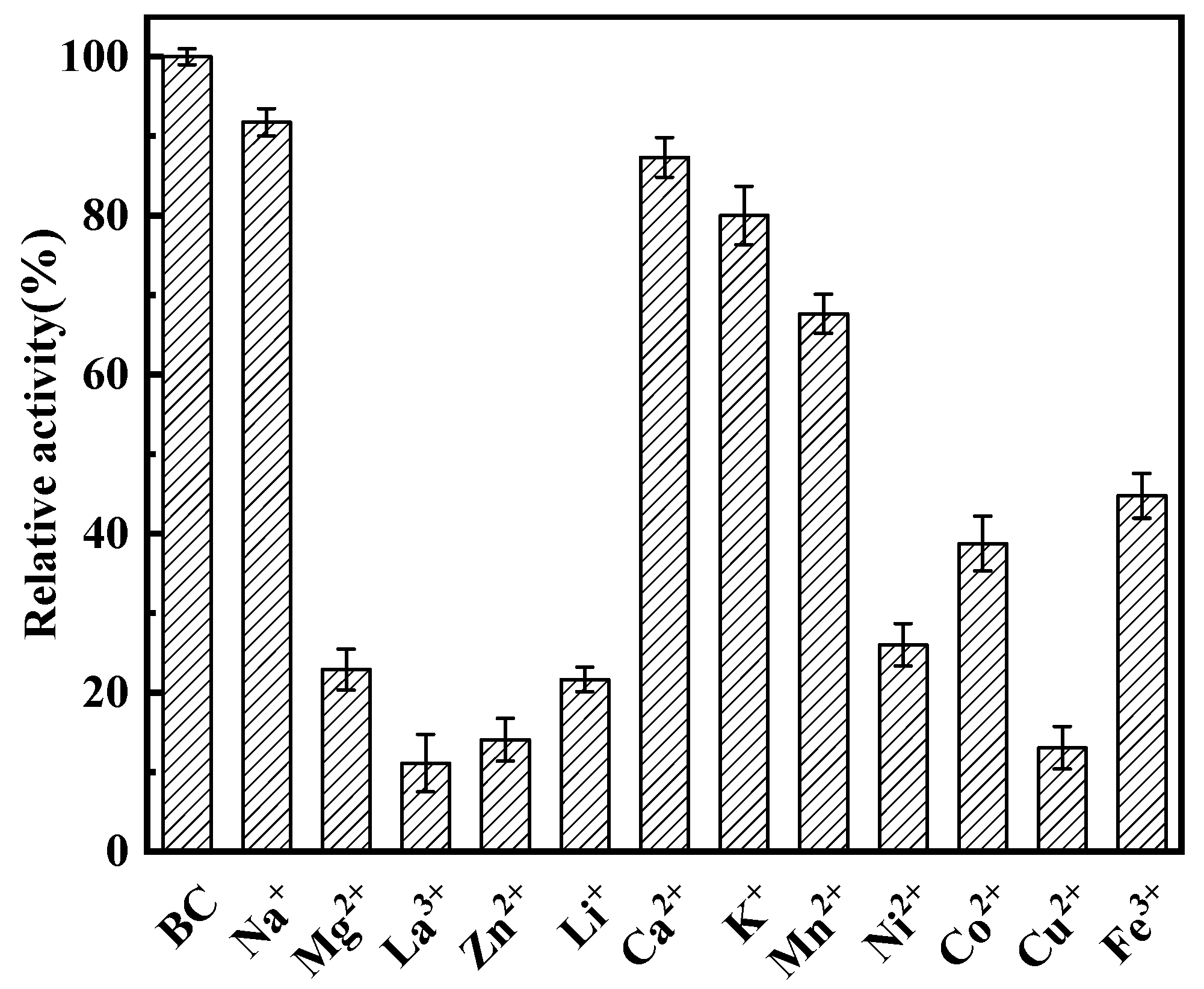
3.6. Influence of Metal Ions on l-asparaginase Activity
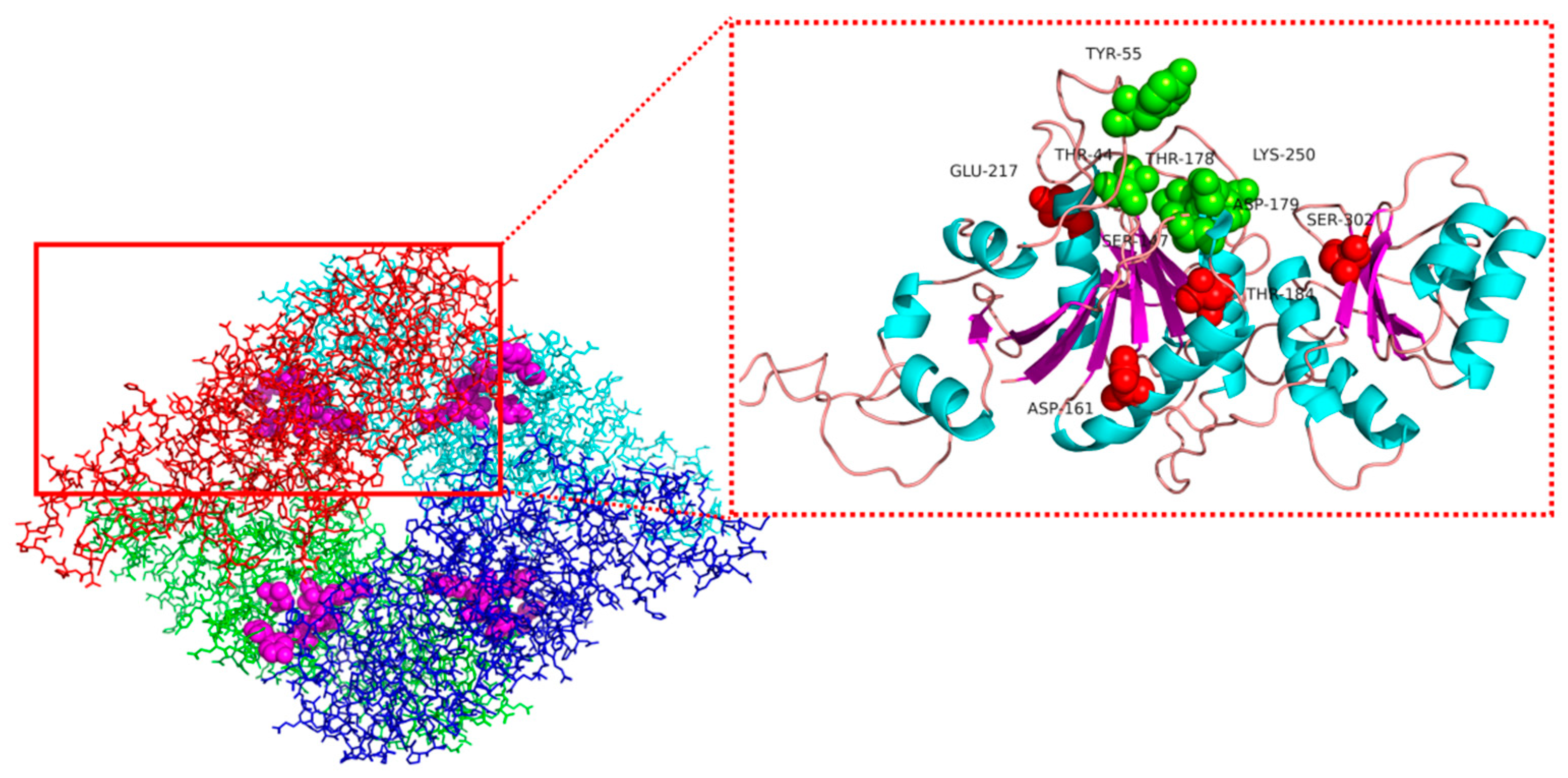
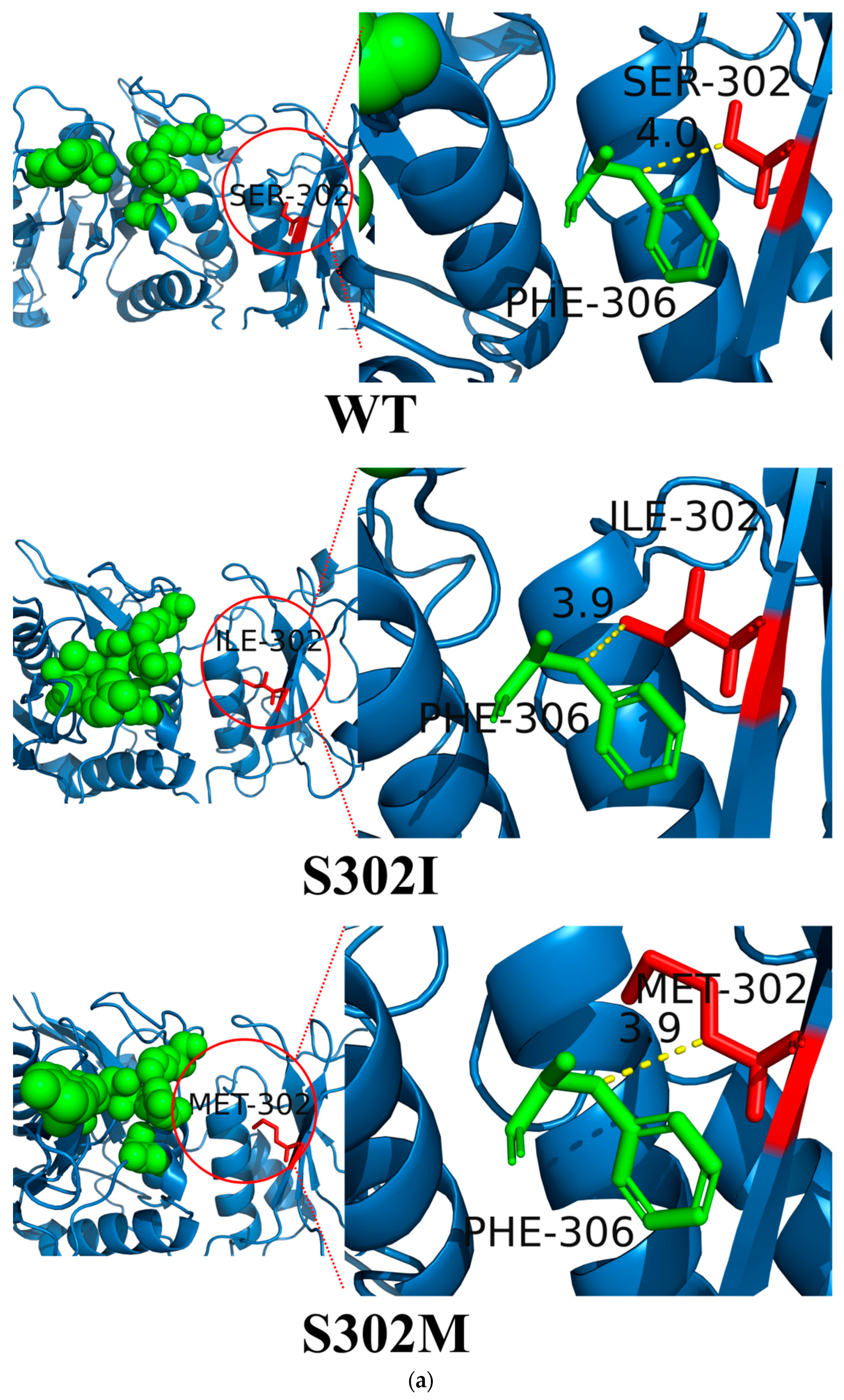
3.7. Structural Simulation and Analysis of l-asparaginase Mutants
3.8. Construction of Combination Mutants and Determination of General Properties
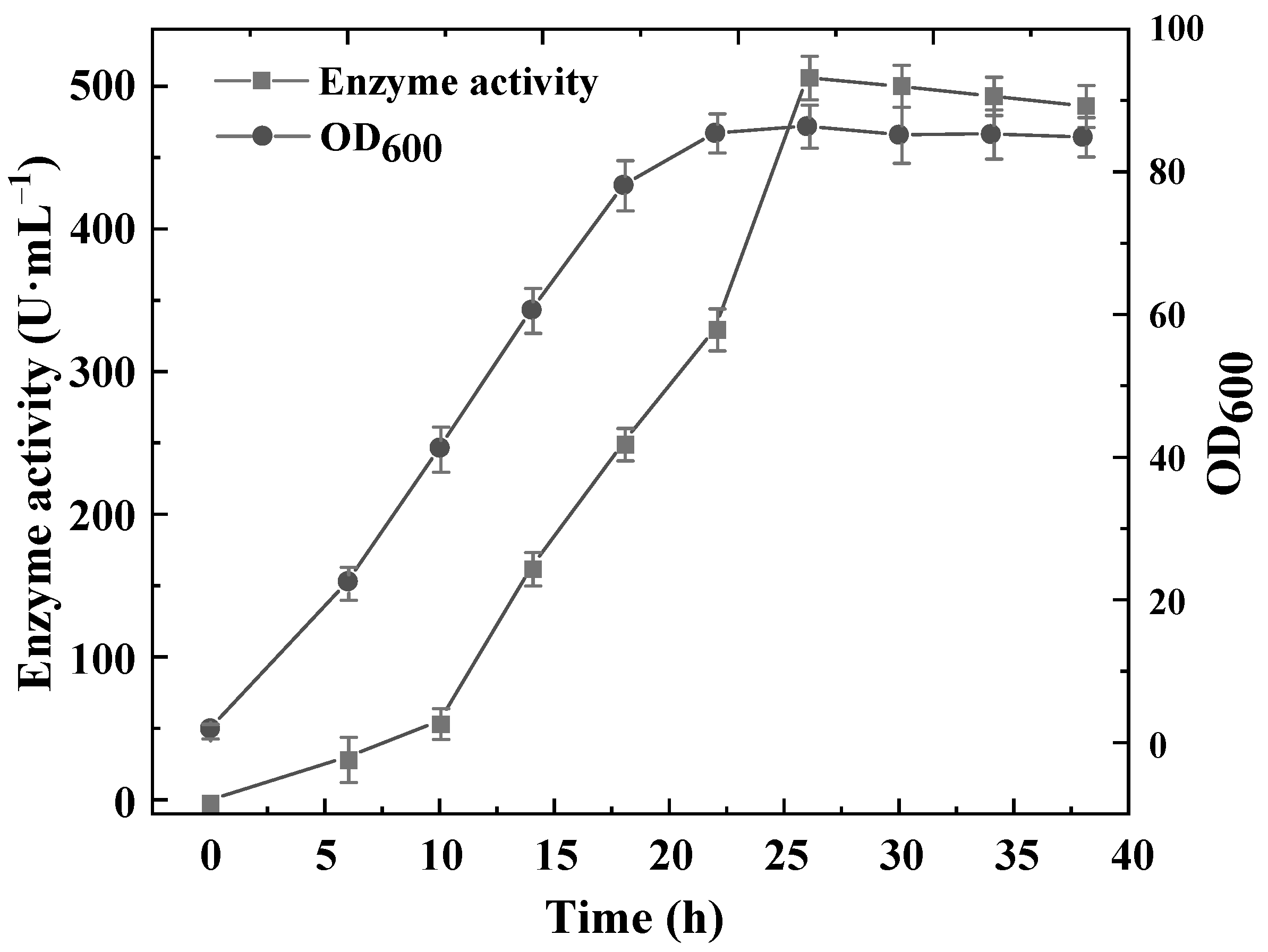
3.9. Construction of a High-Yield l-asparaginase B. subtilis Recombinant and Production in a 5-L Fermenter
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Batool, T.; Makky, E.A.; Jalal, M.; Yusoff, M.M. A Comprehensive Review on L-Asparaginase and Its Applications. Appl. Biochem. Biotechnol. 2016, 178, 900–923. [Google Scholar] [CrossRef] [Green Version]
- Dumina, M.V.; Eldarov, M.A.; Zdanov, D.D.; Sokolov, N.N. L-asparaginases of extremophilic microorganisms in biomedicine. Biomeditsinskaia Khimiia 2020, 66, 105–123. [Google Scholar] [CrossRef]
- Kidd, J.G. Regression of transplanted lymphomas induced in vivo by means of normal guinea pig serum. I. Course of transplanted cancers of various kinds in mice and rats given guinea pig serum, horse serum, or rabbit serum. J. Exp. Med. 1953, 98, 565–582. [Google Scholar] [CrossRef] [PubMed]
- Broome, J.D. Evidence that the L-asparaginase of guinea pig serum is responsible for its antilymphoma effects. I. Properties of the L-asparaginase of guinea pig serum in relation to those of the antilymphoma substance. J. Exp. Med. 1963, 118, 99–120. [Google Scholar] [CrossRef] [Green Version]
- Stadler, R.H.; Scholz, G. Acrylamide: An update on current knowledge in analysis, levels in food, mechanisms of formation, and potential strategies of control. Nutr. Rev. 2004, 62, 449–467. [Google Scholar] [CrossRef]
- Kukurova, K.; Morales, F.J.; Bednarikova, A.; Ciesarova, Z. Effect of L-asparaginase on acrylamide mitigation in a fried-dough pastry model. Mol. Nutr. Food Res. 2009, 53, 1532–1539. [Google Scholar] [CrossRef] [PubMed]
- Hendriksen, H.V.; Kornbrust, B.A.; Ostergaard, P.R.; Stringer, M.A. Evaluating the Potential for Enzymatic Acrylamide Mitigation in a Range of Food Products Using an Asparaginase from Aspergillus oryzae. J. Agric. Food Chem. 2009, 57, 4168–4176. [Google Scholar] [CrossRef] [PubMed]
- Gurunathan, B.; Sahadevan, R. Optimization of Culture Conditions and Bench-Scale Production of L-Asparaginase by Submerged Fermentation of Aspergillus terreus MTCC 1782. J. Microbiol. Biotechnol. 2012, 22, 923–929. [Google Scholar] [CrossRef] [Green Version]
- Balakrishnan, K.; Nair, A.; Kumar, R. Screening of microbial isolates for the fermentative production of L-asparaginase in submerged fermentation. Res. Rev. J. Pharm. Pharm. Sci. 2013, 2, 95–100. [Google Scholar]
- Mashburn, L.T.; Wriston, J.C., Jr. Tumor inhibitory effect of L-asparaginase from escherichia coli. Arch. Biochem. Biophys. 1964, 105, 450–452. [Google Scholar] [CrossRef]
- Kobrinsky, N.L.; Sposto, R.; Shah, N.R.; Anderson, J.R.; DeLaat, C.; Morse, M.; Warkentin, P.; Gilchrist, G.S.; Cohen, M.D.; Shina, D.; et al. Outcomes of treatment of children and adolescents with recurrent non-Hodgkin’s lymphoma and Hodgkin’s disease with dexamethasone, etoposide, cisplatin, cytarabine, and l-asparaginase, maintenance chemotherapy, and transplantation: Children’s Cancer Group study CCG-5912. J. Clin. Oncol. 2001, 19, 2390–2396. [Google Scholar] [CrossRef]
- Pieters, R.; Hunger, S.P.; Boos, J.; Rizzari, C.; Silverman, L.; Baruchel, A.; Goekbuget, N.; Schrappe, M.; Pui, C.-H. L-Asparaginase Treatment in Acute Lymphoblastic Leukemia. Cancer 2011, 117, 238–249. [Google Scholar] [CrossRef] [Green Version]
- Duval, M.; Suciu, S.; Ferster, A.; Rialland, X.; Nelken, B.; Lutz, P.; Benoit, Y.; Robert, A.; Manel, A.M.; Vilmer, E.; et al. Comparison of Escherichia coli-asparaginase with Erwinia-asparaginase in the treatment of childhood lymphoid malignancies: Results of a randomized European Organisation for Research and Treatment of Cancer-Children’s Leukemia Group phase 3 trial. Blood 2002, 99, 2734–2739. [Google Scholar] [CrossRef] [Green Version]
- Li, L.-Z.; Xie, T.-H.; Li, H.-J.; Qing, C.; Zhang, G.-M.; Sun, M.-S. Enhancing the thermostability of Escherichia coli L-asparaginase II by substitution with pro in predicted hydrogen-bonded turn structures. Enzym. Microb. Technol. 2007, 41, 523–527. [Google Scholar] [CrossRef]
- Kotzia, G.A.; Labrou, N.E. Engineering thermal stability of L-asparaginase by in vitro directed evolution. FEBS J. 2009, 276, 1750–1761. [Google Scholar] [CrossRef] [PubMed]
- Vidya, J.; Ushasree, M.V.; Pandey, A. Effect of surface charge alteration on stability of l-asparaginase II from Escherichia sp. Enzym. Microb. Technol. 2014, 56, 15–19. [Google Scholar] [CrossRef] [PubMed]
- Verma, S.; Mehta, R.K.; Maiti, P.; Röhm, K.-H.; Sonawane, A. Improvement of stability and enzymatic activity by site-directed mutagenesis of E. coli asparaginase II. Biochim. Et Biophys. Acta (BBA)-Proteins Proteom. 2014, 1844, 1219–1230. [Google Scholar] [CrossRef]
- Long, S.; Zhang, X.; Rao, Z.; Chen, K.; Xu, M.; Yang, T.; Yang, S. Amino acid residues adjacent to the catalytic cavity of tetramer L-asparaginase II contribute significantly to its catalytic efficiency and thermostability. Enzym. Microb. Technol. 2016, 82, 15–22. [Google Scholar] [CrossRef]
- Sudhir, A.P.; Agarwaal, V.V.; Dave, B.R.; Patel, D.H.; Subramanian, R.B. Enhanced catalysis of L-asparaginase from Bacillus licheniformis by a rational redesign. Enzym. Microb. Technol. 2016, 86, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Schumann, W. Production of recombinant proteins in Bacillus subtilis. Adv. Appl. Microbiol. 2007, 62, 137–189. [Google Scholar] [PubMed]
- Meena, B.; Anburajan, L.; Sathish, T.; Raghavan, R.V.; Dharani, G.; Vinithkumar, N.V.; Kirubagaran, R. l-Asparaginase from Streptomyces griseus NIOT-VKMA29: Optimization of process variables using factorial designs and molecular characterization of L-asparaginase gene. Sci. Rep. 2015, 5, 12404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chityala, S.; Dasu, V.V.; Ahmad, J.; Prakasham, R.S. High yield expression of novel glutaminase free l-asparaginase II of Pectobacterium carotovorum MTCC 1428 in Bacillus subtilis WB800N. Bioprocess Biosyst. Eng. 2015, 38, 2271–2284. [Google Scholar] [CrossRef]
- Feng, Y. Bacillus Subtilis Asparaginase Molecular Modification and High-Efficiency Expression. Ph.D. Thesis, JiangNan University, Wuxi, China, 2018. [Google Scholar]
- Nguyen, T.C.; Do, T.T.; Nguyen, T.H.T.; Quyen, D.T. Expression, purification and evaluation of recombinant L-asparaginase in mehthylotrophic yeast Pichia pastoris. J. Vietnam. Environ. 2014, 6, 288–292. [Google Scholar] [CrossRef]
- Ferrara, M.A.; Severino, N.M.B.; Siani, A.C.; Pereira, N.; Torres, F.A.G.; Bon, E.P.S. Asparaginase production by a recombinant Pichia pastoris strain harbouring Saccharomyces cerevisiae ASP3 gene. J. Biotechnol. 2005, 118, S69. [Google Scholar] [CrossRef]
- Li, X. Molecular Modification and Expression Optimization of Pyrococcus Yayanosii CH1 l-Asparaginase. Ph.D. Thesis, Jiangnan University, Wuxi, China, 2019. [Google Scholar]
- Zhu, M.; Zhang, X.; Wang, Z.; Lin, W.; Xu, M.; Yang, T.; Shao, M.; Rao, Z. Molecular modification and high-efficiency expression of L-asparaginase from Rhizomucor Miehei. Chin. J. Biotechnol. 2021, 37, 3242–3252. [Google Scholar]
- Jiajia, Y.; Chen, Y.; Xuewei, P.; Mengkai, H.; Yuxuan, D.; Osire, T.; Taowei, Y.; Zhiming, R. Metabolic engineering of Bacillus subtilis for enhancing riboflavin production by alleviating dissolved oxygen limitation. Bioresour. Technol. 2021, 333, 125228. [Google Scholar] [CrossRef]
- Hu, L.L.; Feng, K.Y.; Gu, L.; Liu, X.J. Prediction of Protein Quaternary Structure with Feature Selection and Analysis Based on Protein Biological Features. Protein Pept. Lett. 2012, 19, 23–28. [Google Scholar] [CrossRef] [PubMed]
- Schalk, A.M.; Antansijevic, A.; Caffrey, M.; Lavie, A. Experimental data in support of a direct displacement mechanism for type I/II L-asparaginases. J. Biol. Chem. 2016, 291, 5088–5100. [Google Scholar] [CrossRef] [Green Version]
- Bi, J.; Chen, S.; Zhao, X.; Nie, Y.; Xu, Y. Computation-aided engineering of starch-debranching pullulanase fromBacillus thermoleovoransfor enhanced thermostability. Appl. Microbiol. Biotechnol. 2020, 104, 7551–7562. [Google Scholar] [CrossRef]
- Khan, S.; Vihinen, M. Performance of Protein Stability Predictors. Hum. Mutat. 2010, 31, 675–684. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.; Liu, Y.; Shin, H.-D.; Chen, R.R.; Wang, N.S.; Li, J.; Du, G.; Chen, J. Developing Bacillus spp. as a cell factory for production of microbial enzymes and industrially important biochemicals in the context of systems and synthetic biology. Appl. Microbiol. Biotechnol. 2013, 97, 6113–6127. [Google Scholar] [CrossRef] [PubMed]
- Xiao, J.U.; Peng, B.; Su, Z.; Liu, A.; Hu, Y.; Nomura, C.T.; Chen, S.; Wang, Q. Facilitating Protein Expression with Portable 5′-UTR Secondary Structures in Bacillus licheniformis. Acs Synth. Biol. 2020, 9, 1051–1058. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.-B.; Wu, Z.-L. Identification of amino acid residues responsible for increased thermostability of feruloyl esterase A from Aspergillus niger using the PoPMuSiC algorithm. Bioresour. Technol. 2011, 102, 2093–2096. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Yang, S.-X.; Liu, Z.-M.; Li, N.-N.; Li, L.; Mou, H.-J. Rational Design of Alginate Lyase from Microbulbifer sp. Q7 to Improve Thermal Stability. Mar. Drugs 2019, 17, 378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, L.; Deng, Z.M.; Yang, H.Q.; Li, J.H.; Shin, H.D.; Chen, R.R.; Du, G.C.; Chen, J. In Silico Rational Design and Systems Engineering of Disulfide Bridges in the Catalytic Domain of an Alkaline alpha-Amylase from Alkalimonas amylolytica To Improve Thermostability. Appl. Environ. Microbiol. 2014, 80, 798–807. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, L.; Liu, Y.; Sun, Y.; Yan, Q.; Jiang, Z. Biochemical Characterization of a Novel L-Asparaginase with Low Glutaminase Activity from Rhizomucor miehei and Its Application in Food Safety and Leukemia Treatment. Appl. Environ. Microbiol. 2014, 80, 1561–1569. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Position | Original Amino Acid | Mutant Amino Acid | ΔΔG Value (kcal·mol−1) |
|---|---|---|---|
| 302 | S | M | −2.42261 |
| 302 | S | L | −2.39485 |
| 161 | D | M | −1.94591 |
| 184 | D | M | −1.69645 |
| 302 | S | I | −1.61742 |
| 217 | E | M | −1.57523 |
| 217 | E | P | −1.56941 |
| 217 | E | R | −1.41972 |
| 302 | S | V | −1.17737 |
| Enzyme | Specific Enzyme Activity (U·mg−1) | Relative Enzyme Activity (100%) |
|---|---|---|
| WT | 509.1 ± 0.5 | 100.0 ± 1.0 |
| A344E | 786.8 ± 0.7 | 154.5 ± 1.4 |
| S302I | 672.2 ± 0.3 | 132.0 ± 0.6 |
| S302M | 650.1 ± 0.9 | 127.7 ± 1.8 |
| E217R | 581.5 ± 0.3 | 114.2 ± 0.7 |
| E217P | 571.4 ± 0.5 | 112.2 ± 1.1 |
| S302V | 412.6 ± 0.6 | 81.0 ± 1.3 |
| S302L | 410.9 ± 0.7 | 80.7 ± 1.5 |
| E217M | 152.1 ± 1.0 | 29.9 ± 2.1 |
| D161M | 100.3 ± 0.6 | 19.7 ± 1.3 |
| D184M | 82.3 ± 0.9 | 16.2 ± 1.8 |
| Enzyme | Specific Enzyme Activity (U·mg−1) | Relative Enzyme Activity (100%) |
|---|---|---|
| WT | 509.1 ± 0.6 | 100.0 ± 1.2 |
| A344E/S302I | 732.2 ± 0.3 | 143.8 ± 0.7 |
| A344E/S302M | 708.9 ± 1.2 | 139.2 ± 2.5 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, X.; Wang, Z.; Wang, Y.; Li, X.; Zhu, M.; Zhang, H.; Xu, M.; Yang, T.; Rao, Z. Heterologous Expression and Rational Design of l-asparaginase from Rhizomucor miehei to Improve Thermostability. Biology 2021, 10, 1346. https://doi.org/10.3390/biology10121346
Zhang X, Wang Z, Wang Y, Li X, Zhu M, Zhang H, Xu M, Yang T, Rao Z. Heterologous Expression and Rational Design of l-asparaginase from Rhizomucor miehei to Improve Thermostability. Biology. 2021; 10(12):1346. https://doi.org/10.3390/biology10121346
Chicago/Turabian StyleZhang, Xian, Zhi Wang, Yimai Wang, Xu Li, Manchi Zhu, Hengwei Zhang, Meijuan Xu, Taowei Yang, and Zhiming Rao. 2021. "Heterologous Expression and Rational Design of l-asparaginase from Rhizomucor miehei to Improve Thermostability" Biology 10, no. 12: 1346. https://doi.org/10.3390/biology10121346