Simple Summary
Chinese tongue sole (Cynoglossus semilaevis) is an important aquatic fish in northeast Asia that displays sexual growth morphism, with females being 2–4 times larger than males. Tongue sole has a ZZ/ZW sex determination system, and the genotypic female (ZW) can be sex-reversed into phenotypic males, namely, pseudomales. Pseudomales show similar growth to male fish and are disadvantageous in aquaculture. Moreover, pseudomale fish can produce sperm but only Z-type sperm, and its epigenetic information makes its ZW offspring prone to become pseudomale. Thus, screening the key genes for W absence would provide clues for producing high female ratio or all female fry, benefiting the tongue sole industry. In this study, we compared the transcriptomic profiles of pseudomale and male sperm and found that the FoxO signalling pathway, especially the foxo3a and foxo6-like genes, may play important roles in spermatogenesis. In addition, we identified an ~1 M bp area on the Z chromosome enriched with eight DEGs. The study has provided valuable data for screening candidate genes involved in pseudomale sperm abnormity, and their functional study in the future would shed light on sex control in the tongue sole industry.
Abstract
Chinese tongue sole (Cynoglossus semilaevis) has a ZZ/ZW sex determination system, but the genotypic female (ZW) can be sex-reversed into phenotypic males, namely, pseudomales. Pseudomale fish can produce only Z-type sperm but not W sperm. However, the molecular mechanism is unclear. To screen the key genes involved in pseudomale sperm abnormalities, we analysed the transcriptomic profiles of pseudomale and male sperm. In comparison to male sperm, 592 differentially expressed genes (DEGs) were identified in pseudomale sperm, including 499 upregulated and 93 downregulated genes. KEGG analysis indicated that the FoxO signalling pathway, especially the foxo3a and foxo6-like genes, may play an important role in spermatogenesis. The DEGs were mainly distributed on sex chromosomes, with 158 downregulated genes on the Z chromosome and 41 upregulated genes on the W chromosome. A specific area (14–15 M) on the Z chromosome was identified, which enriched eight DEGs inside the ~1 M region. In addition, there were five gene alleles on the sex chromosomes, which showed the opposite transcription pattern (upregulated for the W allele, downregulated for the Z allele). This study has provided valuable data for screening candidate genes involved in the pseudomale sperm abnormality.
1. Introduction
Chinese tongue sole (Cynoglossus semilaevis) belongs to order Pleuronectiformes, family Cynoglossidae. Due to its delicious taste, it is now an economically important marine flatfish in Northeast Asia, such as China, South Korea and Japan. Chinese tongue sole exhibits dramatic sex dimorphism, where female individuals (ZW type) can grow 2–4 times larger than males (ZZ type) [1], so a high-female-ratio or all-female fry would benefit tongue sole aquaculture. However, under special environmental conditions (e.g., high temperature), genotypic females of Chinese tongue sole can be sex-reversed into phenotypic males, namely, pseudomales [2,3]. Pseudomales display slow growth similar to that of male fish, which is disadvantageous for aquaculture. In theory, pseudomale fish should produce both Z and W sperm, and crossing them with females would increase the female ratio from 50% to 75% (ZZ:ZW:WW = 1:2:1). However, pseudomale sperm contains only Z-type, and W sperm are lacking. In addition, the pseudomale sperm inherit parental epigenetic information, which results in the ZW offspring of pseudomales being prone to becoming pseudomales [4].
In humans, over 2000 genes are involved in spermatogenesis, and defects in these genes may lead to sperm abnormalities or male infertility [5]. However, the lack of a specific type of sperm (W sperm absence) is a unique phenomenon in vertebrates. Thus, screening key genes and dissecting their regulatory network during spermatogenesis would provide clues for the W sperm absence, which has great application potential for producing high-female-ratio or all-female fry.
In Chinese tongue sole, there are many genes reported to be involved in spermatogenesis, especially those localized on the Z chromosome, such as dmrt1, tesk1, and neurl3 [3,6,7]. Large-scale analysis was performed to study the molecular mechanism of sex reversal [8,9,10]. However, most research attention is focused on gonadal differentiation, and a comparison of transcriptomic profiles between pseudomale and male sperm is still lacking. In this study, we conducted transcriptomic analysis of pseudomale and male sperm, and the differentially expressed genes displayed a special distribution pattern.
2. Materials and Methods
2.1. Sample Preparation
Chinese tongue sole was purchased from Haiyang High-Tech Experimental Base (Haiyang, Shandong Province, China). Sperm was obtained from male and pseudomale fish (1.5 years old, each sex included four individuals; average body weight and length for male 176.2 g, 30.8 cm, and for pseudomale, 177.4 g, 30.9 cm); it was transferred to liquid nitrogen immediately and then stored at −80 °C until RNA isolation. Regarding the sample size, we actually obtained 13 pseudomale individuals. However, pseudomale produced less sperm, and at last, only four samples were qualified for both quality and quantity. Despite this, the transcriptomic data showed high consistency, and qPCR results showed their reliability, so we conducted the analysis by using four males and four pseudomales. Fins were also collected from the tail for genomic DNA extraction. All experimental procedures were approved by the Yellow Sea Fisheries Research Institute’s animal care and use committee.
2.2. Genetic Sex Identification
Genomic DNA was extracted from the fins and stored in absolute ethanol using a TIANamp Marine Animals DNA kit (Tiangen, Beijing, China) following the manufacturer’s instructions. Genetic sex identification was performed using specific primers as previously described [11]. In brief, specific primers were designed for PCR (Sex F: CCTAAATGATGGATGTAGATTCTGTC; Sex R: GATCCAGAGAAAATAAACCCAGG), and the resultant product was examined by 4% agarose gel electrophoresis. For female samples, two bands (169 and 134 bp) could be visualized, while only one band (169 bp) could be observed for male samples.
2.3. Total RNA Extraction and Library Preparation
Total RNA was extracted using TRIzol (Invitrogen, Carlsbad, CA, USA) reagent based on the manufacturer’s protocol. RNA purity and quantification were evaluated using a NanoDrop 2000 spectrophotometer (Thermo Scientific, Waltham, MA, USA). Within each group, four males and four pseudomales were used for RNA isolation. Thus, eight cDNA libraries of M1-4 and PM1-4 were constructed.
2.4. RNA Sequencing and Differentially Expressed Gene (DEG) Analysis
Transcriptome sequencing and analysis were conducted using the Illumina platform by OE Biotech Co., Ltd. (OE Biotech, Shanghai, China), and the raw data (raw reads) were submitted to NCBI (Accession: PRJNA894095). Raw data were processed using Trimmomatic (0.36) [12]. The reads containing poly-N and the low-quality reads were removed to obtain clean reads. Then, the clean reads were mapped to the reference genome using hisat2 [2,13] under the default settings except ‘--rna-strandness rf --fr’. The fragments per kilobase million (FPKM) value of each gene was calculated using Cufflinks [14] and the read counts of each gene were obtained by htseq-count (0.9.1) [15] with the parameter ‘-s reverse’. Differential expression was analyzed using the DESeq (1.18.0) R package functions estimateSizeFactors and nbinomTest (http://www.bioconductor.org/packages/release/bioc/html/DESeq.html (accessed on 16 November 2022)). Genes with fold change >2 or fold change <0.5 and p < 0.05 were considered DEGs. The DEGs were then enriched by Gene Ontology (GO) terms and Kyoto Encyclopedia of Genes and Genomes (KEGG) categories using the clusterProfiler (4.6.0) R package based on the hypergeometric distribution (https://www.bioconductor.org/packages/release/bioc/html/clusterProfiler.html (accessed on 16 November 2022). Transcriptomic data of testis pseudomales and males were obtained from a previous study by our group [16].
2.5. qPCR Validation
To validate the transcriptomic data, seven genes were randomly selected (the primers are shown in Supplementary Table S1), and qPCR was performed with a 7500 Fast machine (Applied Biosystems, Foster City, CA, USA) using TB Green Premix Ex Taq (Tli RNaseH Plus) (Takara, Japan) according to the manufacturer’s’ protocols. Β-actin was used as an internal reference for normalization. Each individual had four repetitions, and the relative transcription level was calculated by the 2−ΔΔCt method. All data are presented as the mean ± SD, and differences between the means were tested by analysis of variance (ANOVA) followed by Duncan’s post hoc test using SPSS software. Significant differences were accepted when the p value ≤ 0.05. In addition, qPCR was also performed to analyze the transcriptomic profile of five foxo genes (the primers are shown in Supplementary Table S1).
3. Results
3.1. Transcriptomic Overview and Identification of DEGs
As shown in Supplementary Table S2, eight sperm samples (four male and four pseudomale individuals) were used, producing raw data ranging from 6.43 to 6.90 Gb. The average rate of valid bases was 96.70%, and the clean reads of Q30 were all above 92%. As shown in Supplementary Table S3, we obtained 592 DEGs, including 499 upregulated and 93 downregulated genes (pseudomale versus male). From the volcano plot, we found that most DEGs showed fold change ranging from 2–8 (Figure 1A). In Figure 1B, heatmap indicated the repeatability among samples and those genes showing high expression in male but low expression in pseudomale should be focused upon. The qPCR data exhibited a profile consistent with the FPKM value, indicating the reliability of the transcriptomic data (Figure 1C).
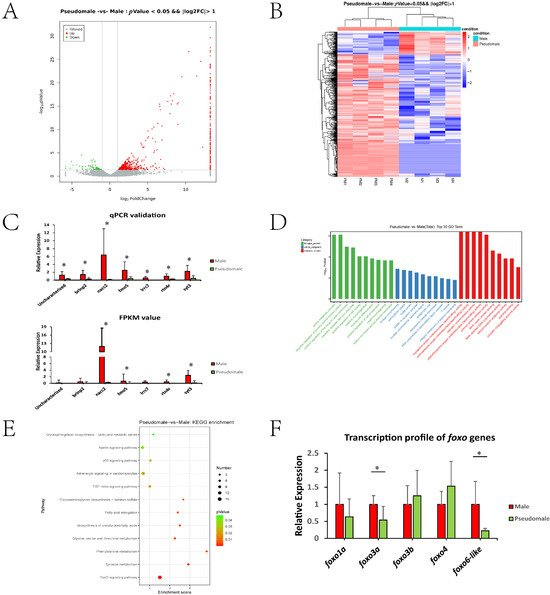
Figure 1.
(A) Volcano plot of DEGs. (B) Heat map of DEGs. (C) qPCR and transcriptomic profile of seven randomly selected genes between male and pseudomale sperm. GO (D) and KEGG analysis (E) of DEGs between the pseudomale and male sperm transcriptomes. (F) The transcription profile of five foxo genes between the male and pseudomale sperm. * indicated significant differences (p < 0.05).
3.2. GO and KEGG Enrichment
As shown in Figure 1D, the top 30 GO terms were listed based on three categories: biological process, cellular component, and molecular function. The GO terms were mainly involved in basic metabolic processes, such as amine metabolism and different amino acid deaminating processes. However, several terms (predominantly in the cellular component category, such as nuclear chromosome, extracellular space, Golgi lumen and so on) were also enriched, which might be related to the formation or maintenance of the sperm structure. The KEGG enrichment data are shown in Figure 1E. The pathways could be mainly categorized into three groups: fatty acid metabolism (Biosynthesis of unsaturated fatty acids, Fatty acid elongation, Apelin signaling pathway, Glycosphingolipid biosynthesis), amino acid metabolism (Tyrosine metabolism, Phenylalanine metabolism, Glycine, serine and threonine metabolism), and other pathways (Glycosaminoglycan biosynthesis, TGF-beta signaling pathway, Adrenergic signaling, p53 signaling pathway, FoxO signaling pathway). It is worth noting that the FoxO signalling pathway exhibited significant enrichment. Based on these observations, the tongue sole genome was screened, and five foxo genes were identified: foxo1a, foxo3a, foxo3b, foxo4, and foxo6-like. Their transcription profiles were examined by qPCR, and foxo3a and foxo6-like showed a significant decline in pseudomale sperm, suggesting their role in pseudomale sperm abnormalities (Figure 1F).
3.3. Chromosomal Distribution Pattern of DEGs
The chromosomal distribution of DEGs was further analyzed. As shown in Figure 2, the number of DEGs showed no big fluctuation among autosomes, while a biased DEG distribution on the sex chromosomes was observed. Among the 499 upregulated genes, 158 were localized on the W chromosome. Of the 93 downregulated genes, 41 were localized on the Z chromosome. Moreover, we found that the DEGs were not evenly distributed on the sex chromosomes and instead were specifically enriched in the 14–15 M region of the Z chromosome (Figure 3A). As shown in Figure 3B, eight genes were mapped in this region, including one upregulated and seven downregulated genes. In fact, there were also eight genes upregulated in the corresponding region of the W chromosome. The accumulation of DEGs in this “hot area” might be the result of epigenetic regulation, as pseudomale testis showed hypermethylation in this area compared to male (Supplementary Figure S3 and unpublished data). Interestingly, in pseudomales, we also identified five gene alleles on the sex chromosomes displaying opposite expression patterns, that is, upregulation for the W allele but downregulation for the Z allele (Table 1).
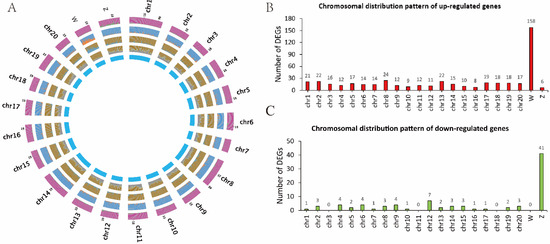
Figure 2.
The chromosomal distribution of DEGs from sperm transcriptomes. (A) The circus plot of the genome features. From outer to inner: A distribution of downregulated genes; B, distribution of upregulated genes; C, distribution of DEGs; D, gene density; E, representing the 24 chromosomes. (B) The number of upregulated DEGs on each chromosome. (C) The number of downregulated DEGs on each chromosome.
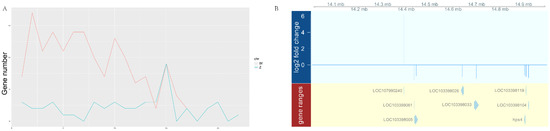
Figure 3.
The distribution pattern of sperm DEGs on the Z chromosome. (A) The number of DEGs every 1 M. (B) Genes localized in the 14–15 M region of the Z chromosome.

Table 1.
Information on five gene alleles on the sex chromosomes.
4. Discussion
Transcriptome analysis has been widely used as an exploratory experimental method to reveal key genes of biological traits. Corresponding software, such as Hisat2 (for reads mapping) and DESeq (for DEGs analysis), has been used to provide new insights into the mechanism of disease resistance, stress resistance, sex determination and differentiation or sex reversal of tongue sole [9,17,18,19].
As an important marine fish in northeast Asia, the tongue sole industry is limited by its female ratio in the aquatic population. Gynogenesis is the most frequently used method in sex control. Here we use tongue sole as example; gynogenesis should produce ZZ and WW fry. However, WW fry is unable to survive and develop [1]. Thus, pseudomale could be an alternative for breeding. In theory, pseudomale crossed with female should produce three genotypes (ZZ:ZW:WW = 1:2:1). Although WW fry cannot survive, the female ratio should reach 66.7% (ZZ:ZW = 1:2). Unfortunately, we have found there was no W sperm in pseudomales, so uncovering the mechanism of W sperm absence has both scientific significance and applicable potential. Azoospermia is frequently reported in mammals, while a lack of specific sperm, such as pseudomale tongue sole producing Z-type sperm but no W-type sperm, is an uncommon phenomenon. To elucidate the mechanism of W sperm absence, studies have focused on the gonadal level, not on sperm level. Since the pseudomale could produce sperm, we compared the transcriptomic profile between pseudomale and male sperm. The W chromosome only existed in the pseudomale, so the transcription level of W-linked genes was not comparable between males and pseudomales. For this reason, we predominantly focused on DEGs of the Z chromosome. In pseudomale sperm, most of the downregulated genes were enriched on the Z chromosome. A similar pattern was also observed in pseudomale testis, with 75 of 132 downregulated genes on the Z chromosome (Supplementary Figure S1). According to our previous postulation, many Z chromosome genes are involved in male determination/differentiation and spermatogenesis, so their downregulation might account for abnormal pseudomale spermatogenesis.
Furthermore, we identified a specific region on the Z chromosome, and eight DEGs were enriched within an approximately 1 M area (Figure 3 and Supplementary Table S4). However, these genes were mainly involved in energy metabolism and transcription based on their annotation, and their functionality in spermatogenesis requires further investigation. Interestingly, another eight DEGs were also identified to be enriched in this same area of the Z chromosome in pseudomale testis, but they were totally different from these DEGs from pseudomale sperm compared to male testis (Supplementary Figure S2 and Supplementary Table S5), so this region might accumulate genes accounting for gonadal differentiation and spermatogenesis. Furthermore, we identified five gene alleles showing the opposite expression pattern: upregulation on the W allele but downregulation on the Z allele (Table 1). To our knowledge, only the transcriptional activator protein Pur-beta-like (LOC103396891) has been suggested to be associated with sexual dimorphism (data not shown), while its role in spermatogenesis is unclear. Therefore, functional study of these genes during spermatogenesis is still needed.
It is interesting that the FoxO signalling pathway was identified via KEGG analysis. There have been a number of reports about the participation of the FoxO signalling pathway and foxo genes in spermatogenesis in mammals. Among them, foxo1 is conserved in mammalian species and has been suggested to play an important role in spermatogonial stem differentiation and spermatogenesis [20,21,22,23,24,25]. We also analyzed the expression patterns of the five foxo genes in early gonadal stages, all of which showed male-biased expression (data not shown). Thus, we postulate that the FoxO signalling pathway might not only function in spermatogenesis but also participate in early testicular differentiation (Figure 1C,D, and Supplementary Figure S4).
5. Conclusions
Comparative transcriptomic analysis of tongue sole sperm identified 592 DEGs (pseudomale versus male, 499 up and 93 down). The DEGs were predominantly enriched on sex chromosomes, with 158 downregulated genes on the Z chromosome and 41 upregulated genes on the W chromosome. A specific ~1 M region (14–15 M) on the Z chromosome harbored eight DEGs. Moreover, the FoxO signalling pathway plays an important role in spermatogenesis, and the functions of foxo3a and foxo6-like should be considered.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/biology11121716/s1, Figure S1: Bioinformatic analysis of DEGs; Figure S2: The chromosomal distribution of DEGS from testis transcriptomes; Figure S3: The distribution pattern of testis DEGs on Z chromosome; Figure S4: The DEG information of FoxO signaling pathway. Table S1: Primers used in this study; Table S2: Summary of sperm transcriptomes; Table S3: The chromosomal distribution of DEGs from sperm transcriptome; Table S4: The information of eight genes enriched in 14–15 M region of Z chromosomes; Table S5: The chromosomal distribution of DEGs from testis transcriptome.
Author Contributions
Y.S., M.L., Z.C., M.Z., T.Z., L.L. and X.X. conducted the experiments; W.X. and N.W. designed the experiments; W.X. and M.W. wrote and reviewed the article; W.X. secured the funding. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by [National Natural Science Foundation], grant number [32072955] and [Taishan Scholar Climbing Project Fund of Shandong]. And APC was funded by [National Natural Science Foundation].
Institutional Review Board Statement
All experimental procedures were approved by the Yellow Sea Fisheries Research Institute’s animal care and use committee (Approval number, YSFRI-2022035).
Informed Consent Statement
Not applicable.
Data Availability Statement
The transcriptomic data were submitted to NCBI and available (Accession: PRJNA894095).
Acknowledgments
This work was supported by the National Natural Science Foundation (32072955) and the Taishan Scholar Climbing Project Fund of Shandong, China.
Conflicts of Interest
The authors have declared that no competing financial interest exist. All data from this study are included in this article.
Correction Statement
This article has been republished with a minor correction to the Funding and Acknowledgements statement. This change does not affect the scientific content of the article.
References
- Chen, S.-L.; Tian, Y.-S.; Yang, J.-F.; Shao, C.-W.; Ji, X.-S.; Zhai, J.-M.; Liao, X.-L.; Zhuang, Z.-M.; Su, P.-Z.; Xu, J.-Y.; et al. Artificial Gynogenesis and Sex Determination in Half-Smooth Tongue Sole (Cynoglossus semilaevis). Mar. Biotechnol. 2009, 11, 243–251. [Google Scholar] [CrossRef]
- Chen, S.; Zhang, G.; Shao, C.; Huang, Q.; Liu, G.; Zhang, P.; Song, W.; An, N.; Chalopin, D.; Volff, J.-N.; et al. Whole-genome sequence of a flatfish provides insights into ZW sex chromosome evolution and adaptation to a benthic lifestyle. Nat. Genet. 2014, 46, 253–260. [Google Scholar] [CrossRef]
- Meng, L.; Zhu, Y.; Zhang, N.; Liu, W.; Liu, Y.; Shao, C.; Wang, N.; Chen, S. Cloning and characterization of tesk1, a novel spermatogenesis-related gene, in the tongue sole (Cynoglossus semilaevis). PloS ONE 2014, 9, e107922. [Google Scholar] [CrossRef] [PubMed]
- Shao, C.; Li, Q.; Chen, S.; Zhang, P.; Lian, J.; Hu, Q.; Sun, B.; Jin, L.; Liu, S.; Wang, Z.; et al. Epigenetic modification and inheritance in sexual reversal of fish. Genome Res. 2014, 24, 604–615. [Google Scholar] [CrossRef] [PubMed]
- Krausz, C.; Riera-Escamilla, A. Genetics of male infertility. Nat. Rev. Urol. 2018, 15, 369–384. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Li, H.; Dong, Z.; Cui, Z.; Zhang, N.; Meng, L.; Zhu, Y.; Liu, Y.; Li, Y.; Guo, H.; et al. Ubiquitin ligase gene neurl3 plays a role in spermatogenesis of half-smooth tongue sole (Cynoglossus semilaevis) by regulating testis protein ubiquitination. Gene 2016, 592, 215–220. [Google Scholar] [CrossRef] [PubMed]
- Cui, Z.; Liu, Y.; Wang, W.; Wang, Q.; Zhang, N.; Lin, F.; Wang, N.; Shao, C.; Dong, Z.; Li, Y.; et al. Genome editing reveals dmrt1 as an essential male sex-determining gene in Chinese tongue sole (Cynoglossus semilaevis). Sci. Rep. 2017, 7, 42213. [Google Scholar] [CrossRef] [PubMed]
- Lin, G.; Gao, D.; Lu, J.; Sun, X. Transcriptome Profiling Reveals the Sexual Dimorphism of Gene Expression Patterns during Gonad Differentiation in the Half-Smooth Tongue Sole (Cynoglossus semilaevis). Mar. Biotechnol. 2021, 23, 18–30. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Liu, X.; Jin, C.; Du, X.; He, Y.; Zhang, Q. Transcriptome Profiling Insights the Feature of Sex Reversal Induced by High Temperature in Tongue Sole Cynoglossus semilaevis. Front. Genet. 2019, 10, 522. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Liu, K.; Feng, B.; Zhang, Z.; Wang, R.; Tang, L.; Li, W.; Li, Q.; Piferrer, F.; Shao, C. Transcriptome of Gonads From High Temperature Induced Sex Reversal During Sex Determination and Differentiation in Chinese Tongue Sole, Cynoglossus semilaevis. Front. Genet. 2019, 10, 1128. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Chen, S.; Gao, F.; Meng, L.; Hu, Q.; Song, W.; Shao, C.; Lv, W. SCAR transformation of sex-specific SSR marker and its application in half-smooth tongue sole(Cynoglossus semiliaevis). J. Agric. Biotechnol. 2014, 22, 787–792. [Google Scholar]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; Van Baren, M.J.; Salzberg, S.L.; Wold, B.J.; Pachter, L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 28, 511–515. [Google Scholar] [CrossRef] [PubMed]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq—A Python framework to work with high-throughput sequencing data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef]
- Wang, N.; Yang, Q.; Wang, J.; Shi, R.; Li, M.; Gao, J.; Xu, W.; Yang, Y.; Chen, Y.; Chen, S. Integration of Transcriptome and Methylome Highlights the Roles of Cell Cycle and Hippo Signaling Pathway in Flatfish Sexual Size Dimorphism. Front. Cell Dev. Biol. 2021, 9, 743722. [Google Scholar] [CrossRef]
- Zhang, X.; Hao, X.; Ma, W.; Zhu, T.; Zhang, Z.; Wang, Q.; Liu, K.; Shao, C.; Wang, H.-Y. Transcriptome Analysis Indicates Immune Responses against Vibrio harveyi in Chinese Tongue Sole (Cynoglossus semilaevis). Animals 2022, 12, 1144. [Google Scholar] [CrossRef]
- Guo, L.; Wang, Y.; Liang, S.; Lin, G.; Chen, S.; Yang, G. Tissue-overlapping response of half-smooth tongue sole (Cynoglossus semilaevis) to thermostressing based on transcriptome profiles. Gene 2016, 586, 97–104. [Google Scholar] [CrossRef]
- Xu, W.; Cui, Z.; Wang, N.; Zhang, M.; Wang, J.; Xu, X.; Liu, Y.; Chen, S. Transcriptomic analysis revealed gene expression profiles during the sex differentiation of Chinese tongue sole (Cynoglossus semilaevis). Comp. Biochem. Physiol. Part D Genom. Proteom. 2021, 40, 100919. [Google Scholar] [CrossRef]
- Goertz, M.J.; Wu, Z.; Gallardo, T.D.; Hamra, F.K.; Castrillon, D.H. Foxo1 is required in mouse spermatogonial stem cells for their maintenance and the initiation of spermatogenesis. J. Clin. Investig. 2011, 121, 3456–3466. [Google Scholar] [CrossRef]
- Huang, P.; Zhou, Z.; Shi, F.; Shao, G.; Wang, R.; Wang, J.; Wang, K.; Ding, W. Effects of the IGF-1/PTEN/Akt/FoxO signaling pathway on male reproduction in rats subjected to water immersion and restraint stress. Mol. Med. Rep. 2016, 14, 5116–5124. [Google Scholar] [CrossRef] [PubMed]
- Liang, G.; Wang, Q.; Zhang, G.; Li, Z.; Wang, Q. Differentially expressed miRNAs and potential therapeutic targets for asthenospermia. Andrologia 2021, 54, e14265. [Google Scholar] [CrossRef] [PubMed]
- Hu, T.; Luo, S.; Xi, Y.; Tu, X.; Yang, X.; Zhang, H.; Feng, J.; Wang, C.; Zhang, Y. Integrative bioinformatics approaches for identifying potential biomarkers and pathways involved in non-obstructive azoospermia. Transl. Androl. Urol. 2021, 10, 243–257. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Guo, L.; Wang, J.; Li, M.; Appiah, M.O.; Liu, H.; Zhao, J.; Yang, L.; Lu, W. Photoperiodic effect on the testicular transcriptome in broiler roosters. J. Anim. Physiol. Anim. Nutr. 2020, 104, 918–927. [Google Scholar] [CrossRef]
- Ghaffari, R.; Di Bona, K.R.; Riley, C.L.; Richburg, J.H. Copper transporter 1 (CTR1) expression by mouse testicular germ cells, but not Sertoli cells, is essential for functional spermatogenesis. PLoS ONE 2019, 14, e0215522. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).