ABC Transporter C1 Prevents Dimethyl Fumarate from Targeting Alzheimer’s Disease

 , , , , , and
, , , , , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
1.1. Alzheimer’s Disease: Background and Therapy
1.2. Drug Repurposing
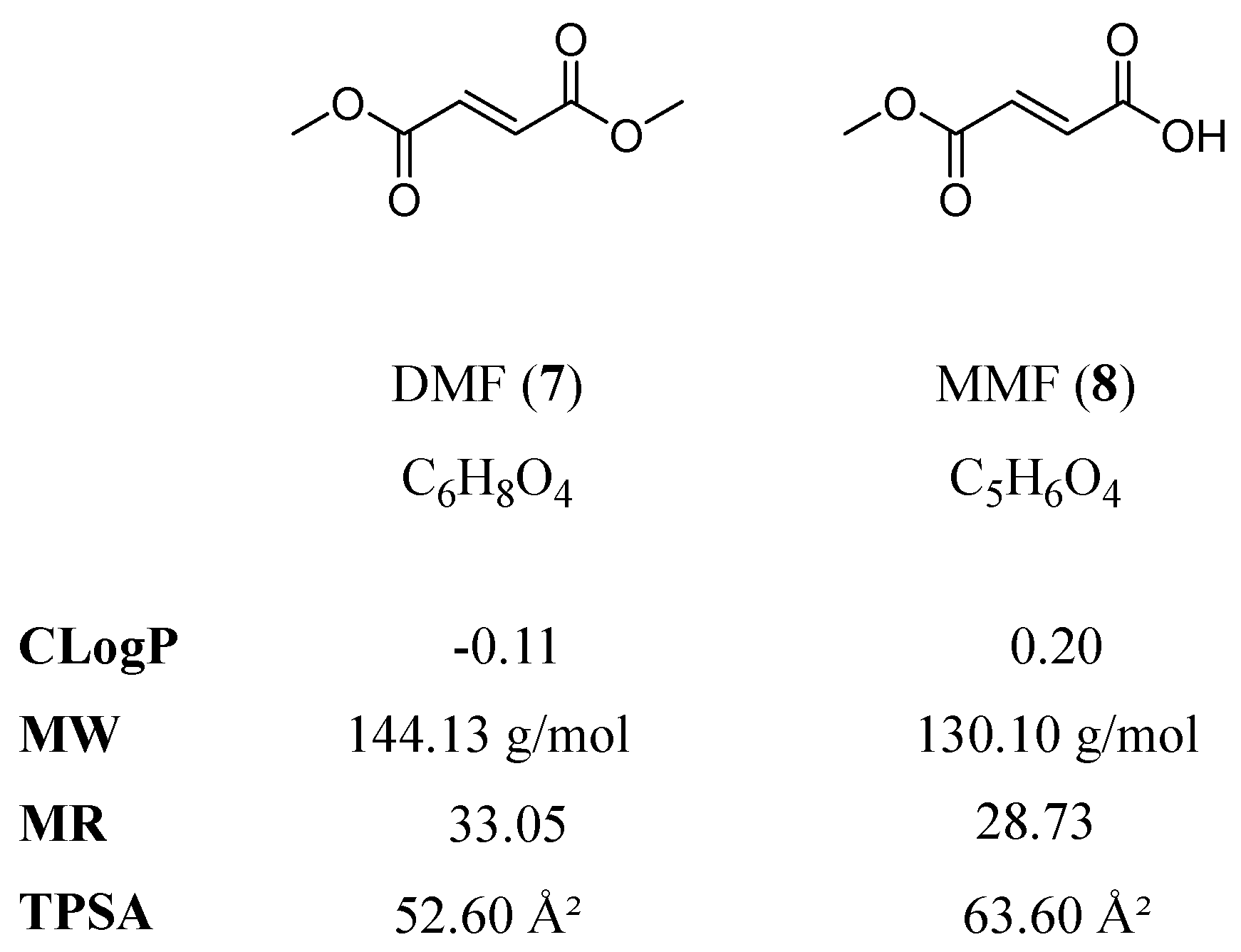
1.3. Dimethyl Fumarate
2. Material and Methods
2.1. Animal Models and Breeding Scheme
2.2. Treatment Scheme
2.3. Spectroscopic Measurement of DMF
2.4. Tissue Harvesting
2.5. Protein Extraction and Quantification
2.6. ABC Transporter Assays: Cell Culture
2.7. ABC Transporter Assays: Membrane Preparation
2.8. ABC Transporter Assays—Experimental Protocols
- Calcein AM: Fluorescence (excitation: 485 nm; emission: 520 nm) was measured for 30 min at 30 s intervals using a Paradigm® microplate reader (Beckman Coulter Biomaterials, Munich, Germany);
- Daunorubicin: Fluorescence (excitation: 488 nm; emission: 695/50 nm) was measured after 180 min incubation on an Attune NxT flow cytometer (Invitrogen, Waltham, MA, USA);
- Rhodamine 123 and pheophorbide A: Fluorescence (excitation: 488 nm; emission: 695/50 nm) was measured after 120 min incubation on an Attune NxT flow cytometer;
- Hoechst 33342: Fluorescence (excitation: 360 nm; emission: 460 nm) was measured after 120 min incubation using a Paradigm® microplate reader.
2.9. ABC Transporter C1 ATPase Assay
2.10. Multi-Drug Resistance Reversal Assay
2.11. Cell Viability Assay
2.12. Statistical Analysis
3. Results
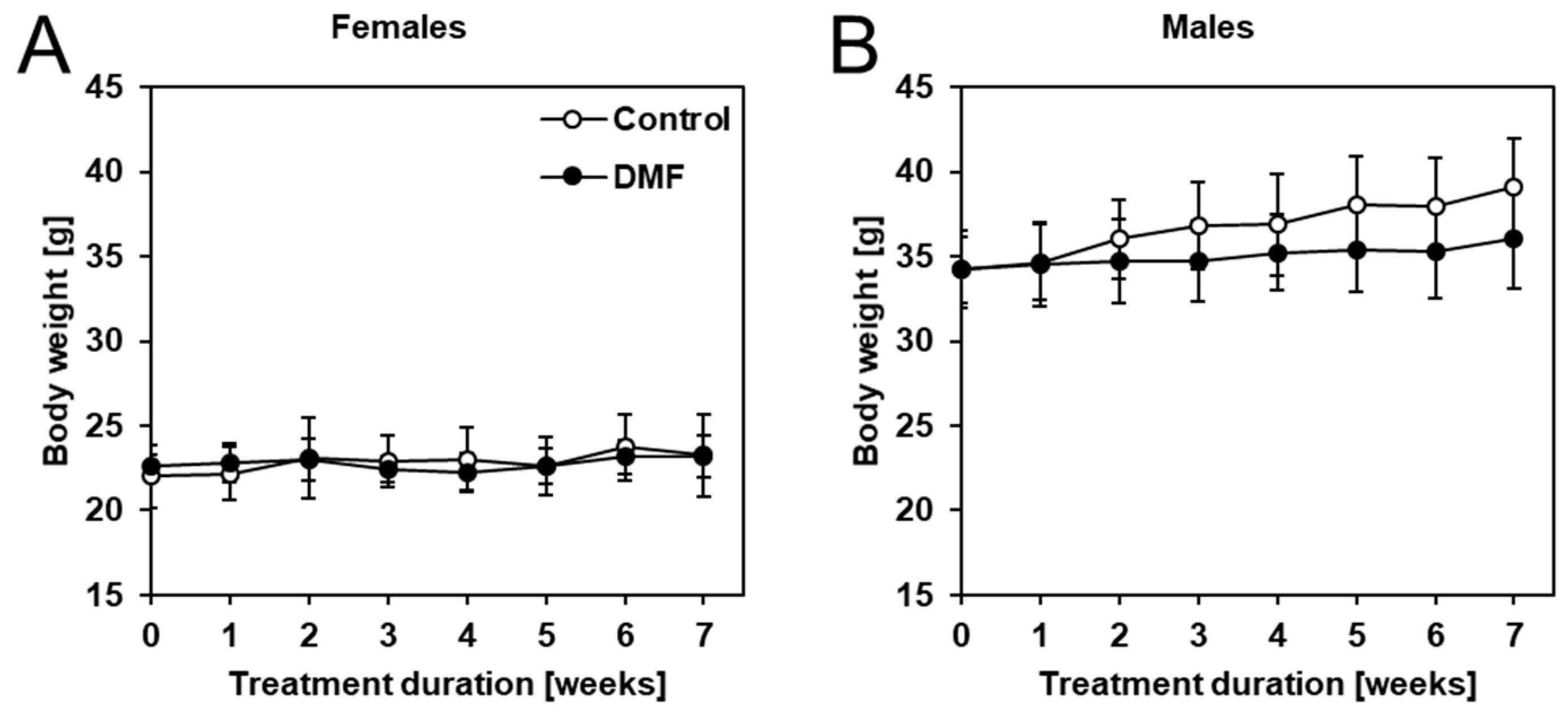
3.1. The Impact of DMF on Body Weight and Drinking Water Consumption
3.2. The Impact of DMF on Aβ Deposition
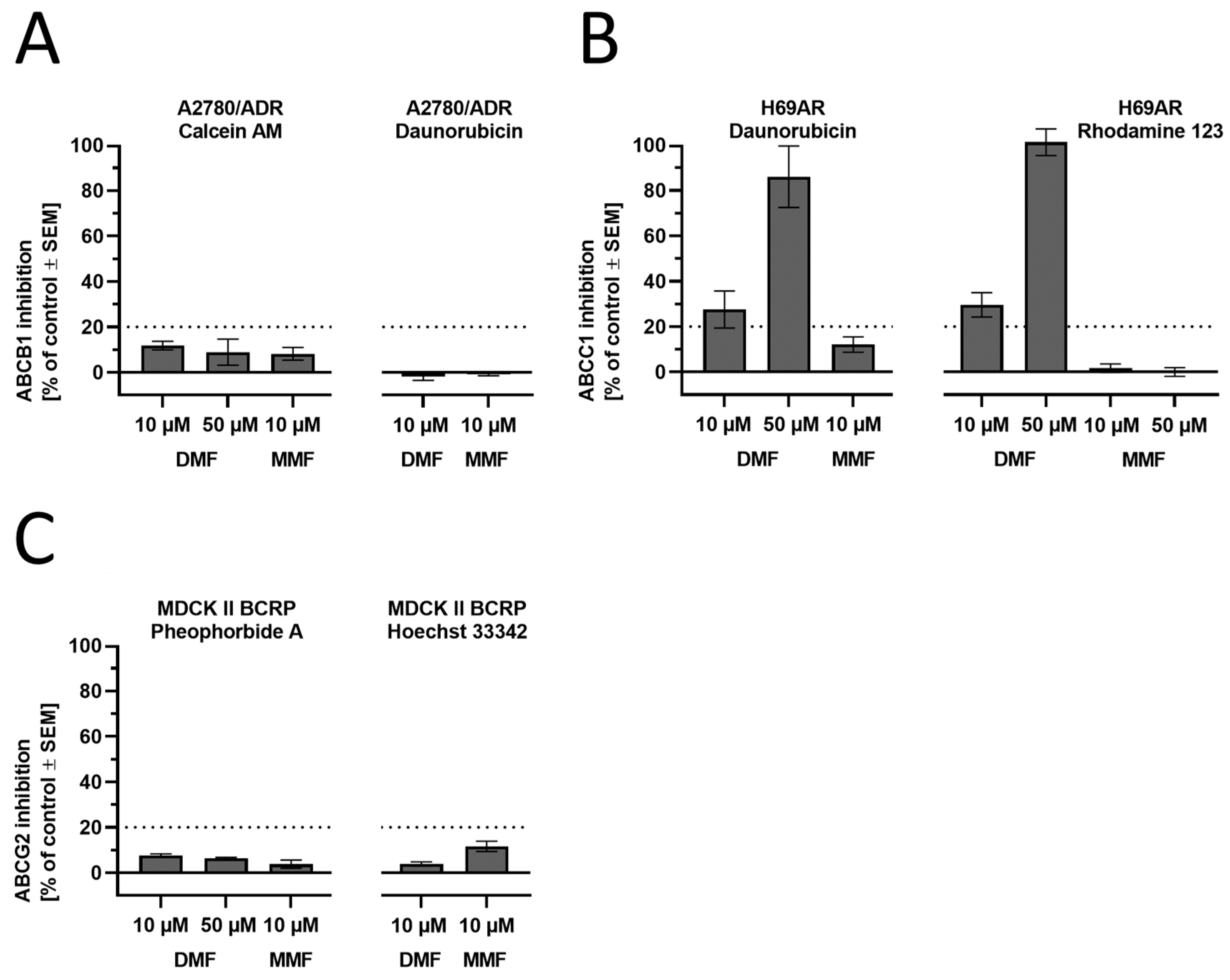
3.3. The Effect of DMF on ABC Transporter Function
3.4. The Effect of DMF and Its Glutathione Conjugate on ABCC1 ATPase Activity
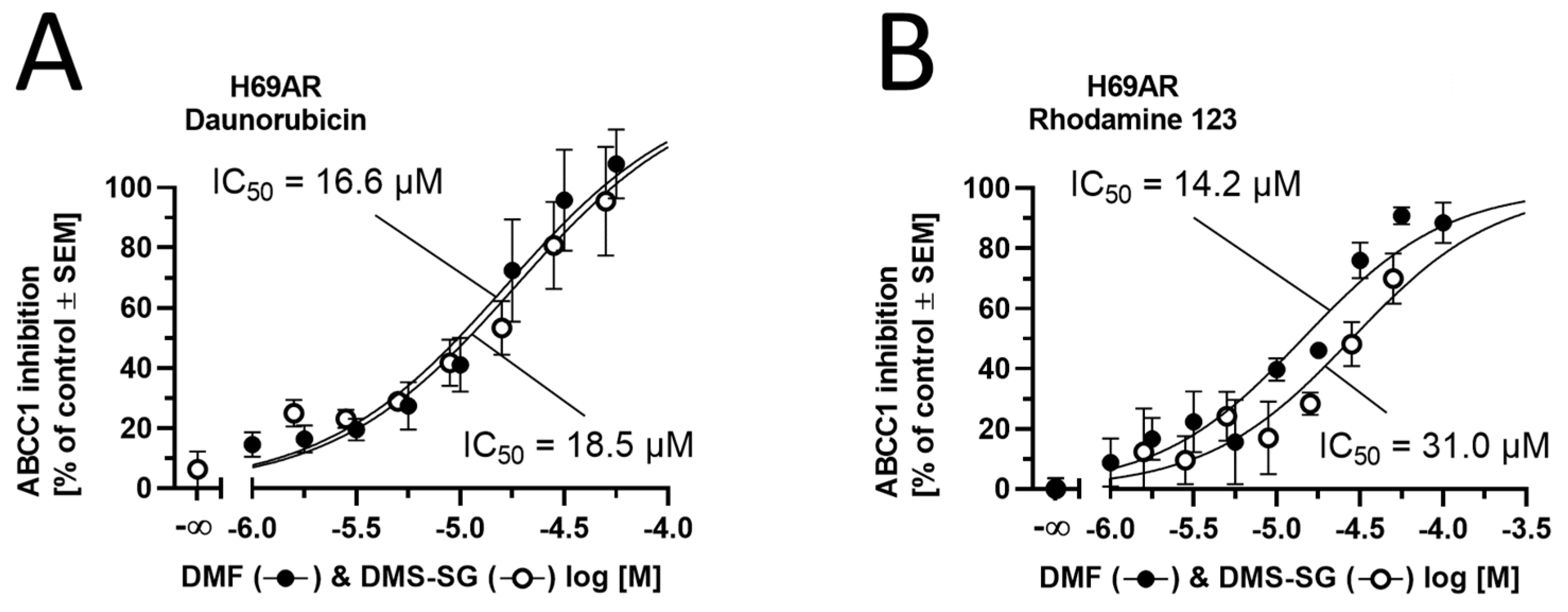
3.5. The Effect of DMF and DMS-SG on ABCC1 Transport Activity
3.6. Efficacy of DMF in Cancer Cells Expressing ABCC1
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
Appendix A. DMF Stability Tests

References
- Alzheimer’s Association. 2022 Alzheimer’s disease facts and figures. Alzheimers Dement 2022, 18, 700–789. [Google Scholar] [CrossRef] [PubMed]
- Wortmann, M. Dementia: A global health priority—Highlights from an ADI and World Health Organization report. Alzheimer’s Res. Ther. 2012, 4, 40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohle, L.; Brackhan, M.; Bascunana, P.; Pahnke, J. Dimethyl fumarate does not mitigate cognitive decline and beta-amyloidosis in female APPPS1 mice. Brain Res. 2021, 1768, 147579. [Google Scholar] [CrossRef] [PubMed]
- Vassar, R.; Bennett, B.D.; Babu-Khan, S.; Kahn, S.; Mendiaz, E.A.; Denis, P.; Teplow, D.B.; Ross, S.; Amarante, P.; Loeloff, R.; et al. Beta-secretase cleavage of Alzheimer’s amyloid precursor protein by the transmembrane aspartic protease BACE. Science 1999, 286, 735–741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lichtenthaler, S.F.; Wang, R.; Grimm, H.; Uljon, S.N.; Masters, C.L.; Beyreuther, K. Mechanism of the cleavage specificity of Alzheimer’s disease gamma-secretase identified by phenylalanine-scanning mutagenesis of the transmembrane domain of the amyloid precursor protein. Proc. Natl. Acad Sci. USA 1999, 96, 3053–3058. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, D.M., 3rd; Cookson, M.R.; Van Den Bosch, L.; Zetterberg, H.; Holtzman, D.M.; Dewachter, I. Hallmarks of neurodegenerative diseases. Cell 2023, 186, 693–714. [Google Scholar] [CrossRef]
- Masters, C.L.; Bateman, R.; Blennow, K.; Rowe, C.C.; Sperling, R.A.; Cummings, J.L. Alzheimer’s disease. Nat. Rev. Dis. Prim. 2015, 1, 15056. [Google Scholar] [CrossRef]
- Pahnke, J.; Frohlich, C.; Paarmann, K.; Krohn, M.; Bogdanovic, N.; Arsland, D.; Winblad, B. Cerebral ABC transporter-common mechanisms may modulate neurodegenerative diseases and depression in elderly subjects. Arch. Med. Res. 2014, 45, 738–743. [Google Scholar] [CrossRef]
- Pagonabarraga, J.; Alamo, C.; Castellanos, M.; Diaz, S.; Manzano, S. Depression in Major Neurodegenerative Diseases and Strokes: A Critical Review of Similarities and Differences among Neurological Disorders. Brain Sci. 2023, 13, 318. [Google Scholar] [CrossRef]
- Crismon, M.L. Tacrine: First drug approved for Alzheimer’s disease. Ann. Pharm. 1994, 28, 744–751. [Google Scholar] [CrossRef]
- de Los Rios, C.; Marco-Contelles, J. Tacrines for Alzheimer’s disease therapy. III. The PyridoTacrines. Eur. J. Med. Chem. 2019, 166, 381–389. [Google Scholar] [CrossRef]
- Doody, R.S.; Raman, R.; Farlow, M.; Iwatsubo, T.; Vellas, B.; Joffe, S.; Kieburtz, K.; He, F.; Sun, X.; Thomas, R.G.; et al. A phase 3 trial of semagacestat for treatment of Alzheimer’s disease. N. Engl. J. Med. 2013, 369, 341–350. [Google Scholar] [CrossRef]
- MacManus, D.G.; Miller, D.H.; Kappos, L.; Gold, R.; Havrdova, E.; Limmroth, V.; Polman, C.H.; Schmierer, K.; Yousry, T.A.; Eraksoy, M.; et al. BG-12 reduces evolution of new enhancing lesions to T1-hypointense lesions in patients with multiple sclerosis. J. Neurol. 2011, 258, 449–456. [Google Scholar] [CrossRef] [Green Version]
- Blair, H.A. Dimethyl Fumarate: A Review in Moderate to Severe Plaque Psoriasis. Drugs 2018, 78, 123–130. [Google Scholar] [CrossRef]
- McGuire, V.A.; Ruiz-Zorrilla Diez, T.; Emmerich, C.H.; Strickson, S.; Ritorto, M.S.; Sutavani, R.V.; Weibeta, A.; Houslay, K.F.; Knebel, A.; Meakin, P.J.; et al. Dimethyl fumarate blocks pro-inflammatory cytokine production via inhibition of TLR induced M1 and K63 ubiquitin chain formation. Sci. Rep. 2016, 6, 31159. [Google Scholar] [CrossRef] [Green Version]
- Montes Diaz, G.; Fraussen, J.; Van Wijmeersch, B.; Hupperts, R.; Somers, V. Dimethyl fumarate induces a persistent change in the composition of the innate and adaptive immune system in multiple sclerosis patients. Sci. Rep. 2018, 8, 8194. [Google Scholar] [CrossRef] [Green Version]
- Longbrake, E.E.; Mao-Draayer, Y.; Cascione, M.; Zielinski, T.; Bame, E.; Brassat, D.; Chen, C.; Kapadia, S.; Mendoza, J.P.; Miller, C.; et al. Dimethyl fumarate treatment shifts the immune environment toward an anti-inflammatory cell profile while maintaining protective humoral immunity. Mult. Scler. 2021, 27, 883–894. [Google Scholar] [CrossRef]
- Fox, R.J.; Miller, D.H.; Phillips, J.T.; Hutchinson, M.; Havrdova, E.; Kita, M.; Yang, M.; Raghupathi, K.; Novas, M.; Sweetser, M.T.; et al. Placebo-controlled phase 3 study of oral BG-12 or glatiramer in multiple sclerosis. N. Engl. J. Med. 2012, 367, 1087–1097. [Google Scholar] [CrossRef] [Green Version]
- Gold, R.; Kappos, L.; Arnold, D.L.; Bar-Or, A.; Giovannoni, G.; Selmaj, K.; Tornatore, C.; Sweetser, M.T.; Yang, M.; Sheikh, S.I.; et al. Placebo-controlled phase 3 study of oral BG-12 for relapsing multiple sclerosis. N. Engl. J. Med. 2012, 367, 1098–1107. [Google Scholar] [CrossRef] [Green Version]
- Gold, R.; Arnold, D.L.; Bar-Or, A.; Hutchinson, M.; Kappos, L.; Havrdova, E.; MacManus, D.G.; Yousry, T.A.; Pozzilli, C.; Selmaj, K.; et al. Long-term effects of delayed-release dimethyl fumarate in multiple sclerosis: Interim analysis of ENDORSE, a randomized extension study. Mult. Scler. 2017, 23, 253–265. [Google Scholar] [CrossRef] [Green Version]
- Lanzillo, R.; Moccia, M.; Palladino, R.; Signoriello, E.; Carotenuto, A.; Maniscalco, G.T.; Sacca, F.; Bonavita, S.; Russo, C.V.; Iodice, R.; et al. Clinical predictors of Dimethyl Fumarate response in multiple sclerosis: A real life multicentre study. Mult. Scler. Relat. Disord. 2020, 38, 101871. [Google Scholar] [CrossRef] [PubMed]
- Suneetha, A.; Raja Rajeswari, K. Role of dimethyl fumarate in oxidative stress of multiple sclerosis: A review. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2016, 1019, 15–20. [Google Scholar] [CrossRef] [PubMed]
- Campolo, M.; Casili, G.; Biundo, F.; Crupi, R.; Cordaro, M.; Cuzzocrea, S.; Esposito, E. The Neuroprotective Effect of Dimethyl Fumarate in an MPTP-Mouse Model of Parkinson’s Disease: Involvement of Reactive Oxygen Species/Nuclear Factor-kappaB/Nuclear Transcription Factor Related to NF-E2. Antioxid. Redox Signal. 2017, 27, 453–471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campolo, M.; Casili, G.; Lanza, M.; Filippone, A.; Paterniti, I.; Cuzzocrea, S.; Esposito, E. Multiple mechanisms of dimethyl fumarate in amyloid beta-induced neurotoxicity in human neuronal cells. J. Cell Mol. Med. 2018, 22, 1081–1094. [Google Scholar] [CrossRef]
- Casili, G.; Campolo, M.; Paterniti, I.; Lanza, M.; Filippone, A.; Cuzzocrea, S.; Esposito, E. Dimethyl Fumarate Attenuates Neuroinflammation and Neurobehavioral Deficits Induced by Experimental Traumatic Brain Injury. J. Neurotrauma 2018, 35, 1437–1451. [Google Scholar] [CrossRef] [Green Version]
- Safari, A.; Fazeli, M.; Namavar, M.R.; Tanideh, N.; Jafari, P.; Borhani-Haghighi, A. Therapeutic effects of oral dimethyl fumarate on stroke induced by middle cerebral artery occlusion: An animal experimental study. Restor. Neurol. Neurosci. 2017, 35, 265–274. [Google Scholar] [CrossRef]
- Zhao, X.; Sun, G.; Zhang, J.; Ting, S.M.; Gonzales, N.; Aronowski, J. Dimethyl Fumarate Protects Brain From Damage Produced by Intracerebral Hemorrhage by Mechanism Involving Nrf2. Stroke 2015, 46, 1923–1928. [Google Scholar] [CrossRef] [Green Version]
- Majkutewicz, I.; Kurowska, E.; Podlacha, M.; Myslinska, D.; Grembecka, B.; Rucinski, J.; Plucinska, K.; Jerzemowska, G.; Wrona, D. Dimethyl fumarate attenuates intracerebroventricular streptozotocin-induced spatial memory impairment and hippocampal neurodegeneration in rats. Behav. Brain Res. 2016, 308, 24–37. [Google Scholar] [CrossRef]
- Majkutewicz, I.; Kurowska, E.; Podlacha, M.; Myslinska, D.; Grembecka, B.; Rucinski, J.; Pierzynowska, K.; Wrona, D. Age-dependent effects of dimethyl fumarate on cognitive and neuropathological features in the streptozotocin-induced rat model of Alzheimer’s disease. Brain Res. 2018, 1686, 19–33. [Google Scholar] [CrossRef]
- Wrona, D.; Majkutewicz, I.; Swiatek, G.; Dunacka, J.; Grembecka, B.; Glac, W. Dimethyl Fumarate as the Peripheral Blood Inflammatory Mediators Inhibitor in Prevention of Streptozotocin-Induced Neuroinflammation in Aged Rats. J. Inflamm. Res. 2022, 15, 33–52. [Google Scholar] [CrossRef]
- Wang, X.; Campos, C.R.; Peart, J.C.; Smith, L.K.; Boni, J.L.; Cannon, R.E.; Miller, D.S. Nrf2 upregulates ATP binding cassette transporter expression and activity at the blood-brain and blood-spinal cord barriers. J. Neurosci. 2014, 34, 8585–8593. [Google Scholar] [CrossRef] [Green Version]
- Ji, L.; Li, H.; Gao, P.; Shang, G.; Zhang, D.D.; Zhang, N.; Jiang, T. Nrf2 pathway regulates multidrug-resistance-associated protein 1 in small cell lung cancer. PLoS ONE 2013, 8, e63404. [Google Scholar] [CrossRef] [Green Version]
- Pahnke, J.; Bascunana, P.; Brackhan, M.; Stefan, K.; Namasivayam, V.; Koldamova, R.; Wu, J.; Mohle, L.; Stefan, S.M. Strategies to gain novel Alzheimer’s disease diagnostics and therapeutics using modulators of ABCA transporters. Free Neuropathol. 2021, 2, 33. [Google Scholar] [CrossRef]
- Krohn, M.; Lange, C.; Hofrichter, J.; Scheffler, K.; Stenzel, J.; Steffen, J.; Schumacher, T.; Bruning, T.; Plath, A.S.; Alfen, F.; et al. Cerebral amyloid-beta proteostasis is regulated by the membrane transport protein ABCC1 in mice. J. Clin. Investig. 2011, 121, 3924–3931. [Google Scholar] [CrossRef]
- Namasivayam, V.; Stefan, K.; Gorecki, L.; Korabecny, J.; Soukup, O.; Jansson, P.J.; Pahnke, J.; Stefan, S.M. Physicochemistry shapes bioactivity landscape of pan-ABC transporter modulators: Anchor point for innovative Alzheimer’s disease therapeutics. Int. J. Biol. Macromol. 2022, 217, 775–791. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [Green Version]
- Radde, R.; Bolmont, T.; Kaeser, S.A.; Coomaraswamy, J.; Lindau, D.; Stoltze, L.; Calhoun, M.E.; Jaggi, F.; Wolburg, H.; Gengler, S.; et al. Abeta42-driven cerebral amyloidosis in transgenic mice reveals early and robust pathology. EMBO Rep. 2006, 7, 940–946. [Google Scholar] [CrossRef] [Green Version]
- Bascunana, P.; Brackhan, M.; Mohle, L.; Wu, J.; Bruning, T.; Eiriz, I.; Jansone, B.; Pahnke, J. Time- and Sex-Dependent Effects of Fingolimod Treatment in a Mouse Model of Alzheimer’s Disease. Biomolecules 2023, 13, 331. [Google Scholar] [CrossRef]
- Scheffler, K.; Stenzel, J.; Krohn, M.; Lange, C.; Hofrichter, J.; Schumacher, T.; Bruning, T.; Plath, A.S.; Walker, L.; Pahnke, J. Determination of spatial and temporal distribution of microglia by 230nm-high-resolution, high-throughput automated analysis reveals different amyloid plaque populations in an APP/PS1 mouse model of Alzheimer’s disease. Curr. Alzheimer Res. 2011, 8, 781–788. [Google Scholar] [CrossRef]
- Habib, A.A.; Hammad, S.F.; Amer, M.M.; Kamal, A.H. Stability indicating RP-HPLC method for determination of dimethyl fumarate in presence of its main degradation products: Application to degradation kinetics. J. Sep. Sci. 2021, 44, 726–734. [Google Scholar] [CrossRef]
- Pathak, D.; Patel, S.; Patel, D. UV Spectroscopy Assay Method Development and Validation of Dimethyl Fumarate and Cyclosporine Drugs in Nano Dosage Forms. Int. J. Pharm. Qual. Assur. 2020, 11, 196–204. [Google Scholar] [CrossRef]
- Puri, S.; Stefan, K.; Khan, S.L.; Pahnke, J.; Stefan, S.M.; Juvale, K. Indole Derivatives as New Structural Class of Potent and Antiproliferative Inhibitors of Monocarboxylate Transporter 1 (MCT1; SLC16A1). J. Med. Chem. 2023, 66, 657–676. [Google Scholar] [CrossRef] [PubMed]
- Silbermann, K.; Li, J.; Namasivayam, V.; Baltes, F.; Bendas, G.; Stefan, S.M.; Wiese, M. Superior Pyrimidine Derivatives as Selective ABCG2 Inhibitors and Broad-Spectrum ABCB1, ABCC1, and ABCG2 Antagonists. J. Med. Chem. 2020, 63, 10412–10432. [Google Scholar] [CrossRef] [PubMed]
- Sterz, K.; Mollmann, L.; Jacobs, A.; Baumert, D.; Wiese, M. Activators of P-glycoprotein: Structure-activity relationships and investigation of their mode of action. ChemMedChem 2009, 4, 1897–1911. [Google Scholar] [CrossRef]
- Pajeva, I.K.; Sterz, K.; Christlieb, M.; Steggemann, K.; Marighetti, F.; Wiese, M. Interactions of the multidrug resistance modulators tariquidar and elacridar and their analogues with P-glycoprotein. ChemMedChem 2013, 8, 1701–1713. [Google Scholar] [CrossRef]
- Stefan, K.; Schmitt, S.M.; Wiese, M. 9-Deazapurines as Broad-Spectrum Inhibitors of the ABC Transport Proteins P-Glycoprotein, Multidrug Resistance-Associated Protein 1, and Breast Cancer Resistance Protein. J. Med. Chem. 2017, 60, 8758–8780. [Google Scholar] [CrossRef]
- Bakos, E.; Evers, R.; Sinko, E.; Varadi, A.; Borst, P.; Sarkadi, B. Interactions of the human multidrug resistance proteins MRP1 and MRP2 with organic anions. Mol. Pharmacol. 2000, 57, 760–768. [Google Scholar] [CrossRef] [Green Version]
- Kraege, S.; Stefan, K.; Kohler, S.C.; Wiese, M. Optimization of Acryloylphenylcarboxamides as Inhibitors of ABCG2 and Comparison with Acryloylphenylcarboxylates. ChemMedChem 2016, 11, 2547–2558. [Google Scholar] [CrossRef]
- Spindler, A.; Stefan, K.; Wiese, M. Synthesis and Investigation of Tetrahydro-beta-carboline Derivatives as Inhibitors of the Breast Cancer Resistance Protein (ABCG2). J. Med. Chem. 2016, 59, 6121–6135. [Google Scholar] [CrossRef]
- Kraege, S.; Stefan, K.; Juvale, K.; Ross, T.; Willmes, T.; Wiese, M. The combination of quinazoline and chalcone moieties leads to novel potent heterodimeric modulators of breast cancer resistance protein (BCRP/ABCG2). Eur. J. Med. Chem. 2016, 117, 212–229. [Google Scholar] [CrossRef]
- Aubets, J.; Jansat, J.M.; Salva, M.; Birks, V.M.; Cole, R.J.; Lewis, J.; Pitcher, A.; Hall, M. No evidence for interactions of dimethylfumarate (DMF) and its main metabolite monomethylfumarate (MMF) with human cytochrome P450 (CYP) enzymes and the P-glycoprotein (P-gp) drug transporter. Pharmacol. Res. Perspect. 2019, 7, e00540. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, T.J.; Ak, M.; Mrowietz, U. Reactivity of dimethyl fumarate and methylhydrogen fumarate towards glutathione and N-acetyl-L-cysteine--preparation of S-substituted thiosuccinic acid esters. Bioorg. Med. Chem. 2007, 15, 333–342. [Google Scholar] [CrossRef]
- Wiese, M.; Stefan, S.M. The A-B-C of small-molecule ABC transport protein modulators: From inhibition to activation-a case study of multidrug resistance-associated protein 1 (ABCC1). Med. Res. Rev. 2019, 39, 2031–2081. [Google Scholar] [CrossRef]
- Stefan, S.M.; Wiese, M. Small-molecule inhibitors of multidrug resistance-associated protein 1 and related processes: A historic approach and recent advances. Med. Res. Rev. 2019, 39, 176–264. [Google Scholar] [CrossRef] [Green Version]
- Heijn, M.; Hooijberg, J.H.; Scheffer, G.L.; Szabo, G.; Westerhoff, H.V.; Lankelma, J. Anthracyclines modulate multidrug resistance protein (MRP) mediated organic anion transport. Biochim. Biophys. Acta 1997, 1326, 12–22. [Google Scholar] [CrossRef] [Green Version]
- Silbermann, K.; Li, J.; Namasivayam, V.; Stefan, S.M.; Wiese, M. Rational drug design of 6-substituted 4-anilino-2-phenylpyrimidines for exploration of novel ABCG2 binding site. Eur. J. Med. Chem. 2021, 212, 113045. [Google Scholar] [CrossRef]
- Mela, V.; Sayd Gaban, A.; O’Neill, E.; Bechet, S.; Walsh, A.; Lynch, M.A. The Modulatory Effects of DMF on Microglia in Aged Mice Are Sex-Specific. Cells 2022, 11, 729. [Google Scholar] [CrossRef]
- Rojo, A.I.; Pajares, M.; Garcia-Yague, A.J.; Buendia, I.; Van Leuven, F.; Yamamoto, M.; Lopez, M.G.; Cuadrado, A. Deficiency in the transcription factor NRF2 worsens inflammatory parameters in a mouse model with combined tauopathy and amyloidopathy. Redox Biol. 2018, 18, 173–180. [Google Scholar] [CrossRef]
- Abd El-Fatah, I.M.; Abdelrazek, H.M.A.; Ibrahim, S.M.; Abdallah, D.M.; El-Abhar, H.S. Dimethyl fumarate abridged tauo-/amyloidopathy in a D-Galactose/ovariectomy-induced Alzheimer’s-like disease: Modulation of AMPK/SIRT-1, AKT/CREB/BDNF, AKT/GSK-3beta, adiponectin/Adipo1R, and NF-kappaB/IL-1beta/ROS trajectories. Neurochem. Int. 2021, 148, 105082. [Google Scholar] [CrossRef]
- Sun, X.; Suo, X.; Xia, X.; Yu, C.; Dou, Y. Dimethyl Fumarate is a Potential Therapeutic Option for Alzheimer’s Disease. J. Alzheimers Dis. 2022, 85, 443–456. [Google Scholar] [CrossRef]
- Hollingworth, P.; Harold, D.; Sims, R.; Gerrish, A.; Lambert, J.C.; Carrasquillo, M.M.; Abraham, R.; Hamshere, M.L.; Pahwa, J.S.; Moskvina, V.; et al. Common variants at ABCA7, MS4A6A/MS4A4E, EPHA1, CD33 and CD2AP are associated with Alzheimer’s disease. Nat. Genet. 2011, 43, 429–435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holstege, H.; Hulsman, M.; Charbonnier, C.; Grenier-Boley, B.; Quenez, O.; Grozeva, D.; van Rooij, J.G.J.; Sims, R.; Ahmad, S.; Amin, N.; et al. Exome sequencing identifies rare damaging variants in ATP8B4 and ABCA1 as risk factors for Alzheimer’s disease. Nat. Genet. 2022, 54, 1786–1794. [Google Scholar] [CrossRef] [PubMed]
- Koldamova, R.; Staufenbiel, M.; Lefterov, I. Lack of ABCA1 considerably decreases brain ApoE level and increases amyloid deposition in APP23 mice. J. Biol. Chem. 2005, 280, 43224–43235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bossaerts, L.; Cacace, R.; Van Broeckhoven, C. The role of ATP-binding cassette subfamily A in the etiology of Alzheimer’s disease. Mol. Neurodegener. 2022, 17, 31. [Google Scholar] [CrossRef]
- Kumar, P.; Sharma, G.; Kumar, R.; Malik, R.; Singh, B.; Katare, O.P.; Raza, K. Enhanced Brain Delivery of Dimethyl Fumarate Employing Tocopherol-Acetate-Based Nanolipidic Carriers: Evidence from Pharmacokinetic, Biodistribution, and Cellular Uptake Studies. ACS Chem. Neurosci. 2017, 8, 860–865. [Google Scholar] [CrossRef]
- Chen, Z.S.; Tiwari, A.K. Multidrug resistance proteins (MRPs/ABCCs) in cancer chemotherapy and genetic diseases. FEBS J. 2011, 278, 3226–3245. [Google Scholar] [CrossRef] [Green Version]
- Miclea, A.; Leussink, V.I.; Hartung, H.P.; Gold, R.; Hoepner, R. Safety and efficacy of dimethyl fumarate in multiple sclerosis: A multi-center observational study. J. Neurol. 2016, 263, 1626–1632. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Möhle, L.; Stefan, K.; Bascuñana, P.; Brackhan, M.; Brüning, T.; Eiriz, I.; El Menuawy, A.; van Genderen, S.; Santos-García, I.; Górska, A.M.; et al. ABC Transporter C1 Prevents Dimethyl Fumarate from Targeting Alzheimer’s Disease. Biology 2023, 12, 932. https://doi.org/10.3390/biology12070932
Möhle L, Stefan K, Bascuñana P, Brackhan M, Brüning T, Eiriz I, El Menuawy A, van Genderen S, Santos-García I, Górska AM, et al. ABC Transporter C1 Prevents Dimethyl Fumarate from Targeting Alzheimer’s Disease. Biology. 2023; 12(7):932. https://doi.org/10.3390/biology12070932
Chicago/Turabian StyleMöhle, Luisa, Katja Stefan, Pablo Bascuñana, Mirjam Brackhan, Thomas Brüning, Ivan Eiriz, Ahmed El Menuawy, Sylvie van Genderen, Irene Santos-García, Anna Maria Górska, and et al. 2023. "ABC Transporter C1 Prevents Dimethyl Fumarate from Targeting Alzheimer’s Disease" Biology 12, no. 7: 932. https://doi.org/10.3390/biology12070932