
Role of NFE2L1 in the Regulation of Proteostasis: Implications for Aging and Neurodegenerative Diseases

{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction: The Ubiquitin Proteasome System
2. Role of the UPS in Aging and Neurodegenerative Diseases
3. Degradation of Amyloidogenic Proteins: A Role for the UPS vs. ALP
4. Inhibition of the UPS by Amyloidogenic Proteins
5. Upregulation of the UPS as a Therapeutic Strategy for NDs (I): Stimulation of Proteasome Activity
6. Upregulation of the UPS as a Therapeutic Strategy for NDs (II): Induction of Proteasome Subunit Expression
7. mTORC1-Mediated Control of Proteasome Subunit Expression and Assembly: NFE2L1-Dependent and -Independent Mechanisms
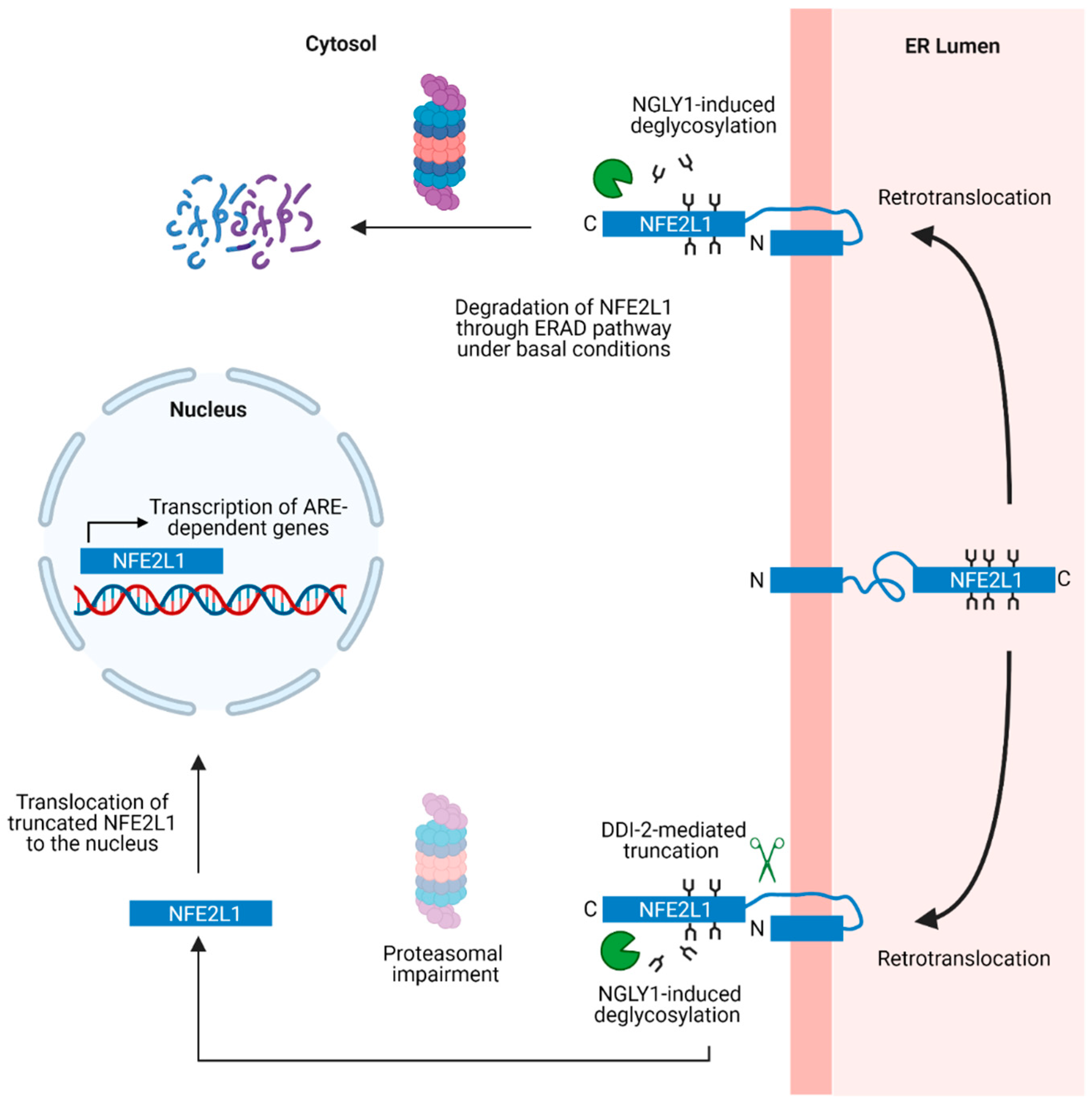
8. ER Stress-Induced NFE2L1 Activation

9. Structural Domains and Post-Translational Modifications of NFE2L1
10. Functional Interplay between NFE2L1 and Other Nrf Family Members
11. NFE2L1- and Nrf2-Dependent Regulation of Other Proteostasis Mechanisms
12. Role of NFE2L1 in Age-Related Phenotypes
13. Role of NFE2L1 in NDs
14. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Rothman, S. How Is the Balance between Protein Synthesis and Degradation Achieved? Theor. Biol. Med. Model. 2010, 7, 25. [Google Scholar] [CrossRef] [PubMed]
- Suraweera, A.; Münch, C.; Hanssum, A.; Bertolotti, A. Failure of Amino Acid Homeostasis Causes Cell Death Following Proteasome Inhibition. Mol. Cell 2012, 48, 242–253. [Google Scholar] [CrossRef] [PubMed]
- Young, V.R.; Steffee, W.P.; Pencharz, P.B.; Winterer, J.C.; Scrimshaw, N.S. Total Human Body Protein Synthesis in Relation to Protein Requirements at Various Ages. Nature 1975, 253, 192–194. [Google Scholar] [CrossRef] [PubMed]
- Naujokat, C.; Hoffmann, S. Role and Function of the 26S Proteasome in Proliferation and Apoptosis. Lab. Investig. 2002, 82, 965–980. [Google Scholar] [CrossRef]
- Bader, M.; Steller, H. Regulation of Cell Death by the Ubiquitin–Proteasome System. Curr. Opin. Cell Biol. 2009, 21, 878–884. [Google Scholar] [CrossRef]
- Rousseau, A.; Bertolotti, A. Regulation of Proteasome Assembly and Activity in Health and Disease. Nat. Rev. Mol. Cell Biol. 2018, 19, 697–712. [Google Scholar] [CrossRef]
- Pickart, C.M. Mechanisms Underlying Ubiquitination. Annu. Rev. Biochem. 2001, 70, 503–533. [Google Scholar] [CrossRef]
- Saeki, Y. Ubiquitin Recognition by the Proteasome. J. Biochem. 2017, 161, 113–124. [Google Scholar] [CrossRef]
- Ehlinger, A.; Walters, K.J. Structural Insights into Proteasome Activation by the 19S Regulatory Particle. Biochemistry 2013, 52, 3618–3628. [Google Scholar] [CrossRef]
- Braun, B.C.; Glickman, M.; Kraft, R.; Dahlmann, B.; Kloetzel, P.M.; Finley, D.; Schmidt, M. The Base of the Proteasome Regulatory Particle Exhibits Chaperone-like Activity. Nat. Cell Biol. 1999, 1, 221–226. [Google Scholar] [CrossRef]
- Inobe, T.; Matouschek, A. Paradigms of Protein Degradation by the Proteasome. Curr. Opin. Struct. Biol. 2014, 24, 156–164. [Google Scholar] [CrossRef] [PubMed]
- Aufderheide, A.; Unverdorben, P.; Baumeister, W.; Förster, F. Structural Disorder and Its Role in Proteasomal Degradation. FEBS Lett. 2015, 589, 2552–2560. [Google Scholar] [CrossRef]
- Prakash, S.; Tian, L.; Ratliff, K.S.; Lehotzky, R.E.; Matouschek, A. An Unstructured Initiation Site Is Required for Efficient Proteasome-Mediated Degradation. Nat. Struct. Mol. Biol. 2004, 11, 830–837. [Google Scholar] [CrossRef] [PubMed]
- Fabre, B.; Lambour, T.; Garrigues, L.; Ducoux-Petit, M.; Amalric, F.; Monsarrat, B.; Burlet-Schiltz, O.; Bousquet-Dubouch, M.P. Label-Free Quantitative Proteomics Reveals the Dynamics of Proteasome Complexes Composition and Stoichiometry in a Wide Range of Human Cell Lines. J. Proteome Res. 2014, 13, 3027–3037. [Google Scholar] [CrossRef] [PubMed]
- Baugh, J.M.; Viktorova, E.G.; Pilipenko, E.V. Proteasomes Can Degrade a Significant Proportion of Cellular Proteins Independent of Ubiquitination. J. Mol. Biol. 2009, 386, 814–827. [Google Scholar] [CrossRef]
- Aki, M.; Shimbara, N.; Takashina, M.; Akiyama, K.; Kagawa, S.; Tamura, T.; Tanahashi, N.; Yoshimura, T.; Tanaka, K.; Ichihara, A. Interferon-γ Induces Different Subunit Organizations and Functional Diversity of Proteasomes. J. Biochem. 1994, 115, 257–269. [Google Scholar] [CrossRef]
- Murata, S.; Takahama, Y.; Kasahara, M.; Tanaka, K. The Immunoproteasome and Thymoproteasome: Functions, Evolution and Human Disease. Nat. Immunol. 2018, 19, 923–931. [Google Scholar] [CrossRef]
- Stadtmueller, B.M.; Hill, C.P. Proteasome Activators. Mol. Cell 2011, 41, 8–19. [Google Scholar] [CrossRef]
- DeMartino, G.N.; Gillette, T.G. Proteasomes: Machines for All Reasons. Cell 2007, 129, 659–662. [Google Scholar] [CrossRef]
- Strehl, B.; Seifert, U.; Krüger, E.; Heink, S.; Kuckelkorn, U.; Kloetzel, P.M. Interferon-γ, the Functional Plasticity of the Ubiquitin-Proteasome System, and MHC Class I Antigen Processing. Immunol. Rev. 2005, 207, 19–30. [Google Scholar] [CrossRef]
- Mattson, M.P.; Arumugam, T.V. Hallmarks of Brain Aging: Adaptive and Pathological Modification by Metabolic States. Cell Metab. 2018, 27, 1176–1199. [Google Scholar] [CrossRef] [PubMed]
- Vilchez, D.; Saez, I.; Dillin, A. The Role of Protein Clearance Mechanisms in Organismal Ageing and Age-Related Diseases. Nat. Commun. 2014, 5, 5659. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.K.; Klopp, R.G.; Weindruch, R.; Prolla, T.A. Gene Expression Profile of Aging and Its Retardation by Caloric Restriction. Science 1999, 285, 1390–1393. [Google Scholar] [CrossRef] [PubMed]
- Jung, S.H.; Jae, S.H.; Chang, I.; Kim, S. Age-Associated Decrease in Proteasome Content and Activities in Human Dermal Fibroblasts: Restoration of Normal Level of Proteasome Subunits Reduces Aging Markers in Fibroblasts from Elderly Persons. J. Gerontol. Biol. Sci. 2007, 62, 490–499. [Google Scholar] [CrossRef]
- Keller, J.N.; Hanni, K.B.; Markesbery, W.R. Possible Involvement of Proteasome Inhibition in Aging: Implications for Oxidative Stress. Mech. Ageing Dev. 2000, 113, 61–70. [Google Scholar] [CrossRef]
- Keller, J.N.; Huang, F.F.; Markesbery, W.R. Decreased Levels of Proteasome Activity and Proteasome Expression in Aging Spinal Cord. Neuroscience 2000, 98, 149–156. [Google Scholar] [CrossRef]
- Kelmer Sacramento, E.; Kirkpatrick, J.M.; Mazzetto, M.; Baumgart, M.; Bartolome, A.; Di Sanzo, S.; Caterino, C.; Sanguanini, M.; Papaevgeniou, N.; Lefaki, M.; et al. Reduced Proteasome Activity in the Aging Brain Results in Ribosome Stoichiometry Loss and Aggregation. Mol. Syst. Biol. 2020, 16, e9596. [Google Scholar] [CrossRef]
- Shimura, H.; Hattori, N.; Kubo, S.I.; Mizuno, Y.; Asakawa, S.; Minoshima, S.; Shimizu, N.; Iwai, K.; Chiba, T.; Tanaka, K.; et al. Familial Parkinson Disease Gene Product, Parkin, Is a Ubiquitin-Protein Ligase. Nat. Genet. 2000, 25, 302–305. [Google Scholar] [CrossRef]
- Graham, S.H.; Liu, H. Life and Death in the Trash Heap: The Ubiquitin Proteasome Pathway and UCHL1 in Brain Aging, Neurodegenerative Disease and Cerebral Ischemia. Ageing Res. Rev. 2017, 34, 30–38. [Google Scholar] [CrossRef]
- Leroy, E.; Boyer, R.; Auburger, G.; Leube, B.; Ulm, G.; Mezey, E.; Harta, G.; Brownstein, M.J.; Jonnalagada, S.; Chernova, T.; et al. The Ubiquitin Pathway in Parkinson’s Disease. Nature 1998, 395, 451–452. [Google Scholar] [CrossRef]
- Bilguvar, K.; Tyagi, N.K.; Ozkara, C.; Tuysuz, B.; Bakircioglu, M.; Choi, M.; Delil, S.; Caglayan, A.O.; Baranoski, J.F.; Erturk, O.; et al. Recessive Loss of Function of the Neuronal Ubiquitin Hydrolase UCHL1 Leads to Early-Onset Progressive Neurodegeneration. Proc. Natl. Acad. Sci. USA 2013, 110, 3489–3494. [Google Scholar] [CrossRef] [PubMed]
- Deng, H.X.; Chen, W.; Hong, S.T.; Boycott, K.M.; Gorrie, G.H.; Siddique, N.; Yang, Y.; Fecto, F.; Shi, Y.; Zhai, H.; et al. Mutations in UBQLN2 Cause Dominant X-Linked Juvenile and Adult-Onset ALS and ALS/Dementia. Nature 2011, 477, 211–215. [Google Scholar] [CrossRef]
- Renaud, L.; Picher-Martel, V.; Codron, P.; Julien, J.P. Key Role of UBQLN2 in Pathogenesis of Amyotrophic Lateral Sclerosis and Frontotemporal Dementia. Acta Neuropathol. Commun. 2019, 7, 103. [Google Scholar] [CrossRef]
- van den Boom, J.; Meyer, H. VCP/P97-Mediated Unfolding as a Principle in Protein Homeostasis and Signaling. Mol. Cell 2018, 69, 182–194. [Google Scholar] [CrossRef]
- Johnson, J.O.; Mandrioli, J.; Benatar, M.; Abramzon, Y.; Van Deerlin, V.M.; Trojanowski, J.Q.; Gibbs, J.R.; Brunetti, M.; Gronka, S.; Wuu, J.; et al. Exome Sequencing Reveals VCP Mutations as a Cause of Familial ALS. Neuron 2010, 68, 857–864. [Google Scholar] [CrossRef] [PubMed]
- Lefaki, M.; Papaevgeniou, N.; Chondrogianni, N. Redox Regulation of Proteasome Function. Redox Biol. 2017, 13, 452–458. [Google Scholar] [CrossRef] [PubMed]
- Jung, T.; Höhn, A.; Grune, T. The Proteasome and the Degradation of Oxidized Proteins: Part III-Redox Regulation of the Proteasomal System. Redox Biol. 2014, 2, 388–394. [Google Scholar] [CrossRef]
- Grune, T.; Merker, K.; Sandig, G.; Davies, K.J.A. Selective Degradation of Oxidatively Modified Protein Substrates by the Proteasome. Biochem. Biophys. Res. Commun. 2003, 305, 709–718. [Google Scholar] [CrossRef]
- Jung, T.; Höhn, A.; Grune, T. The Proteasome and the Degradation of Oxidized Proteins: Part II—Protein Oxidation and Proteasomal Degradation. Redox Biol. 2014, 2, 99–104. [Google Scholar] [CrossRef]
- Wang, X.; Yen, J.; Kaiser, P.; Huang, L. Regulation of the 26S Proteasome Complex during Oxidative Stress. Sci. Signal. 2010, 3, ra88. [Google Scholar] [CrossRef]
- Reinheckel, T.; Sitte, N.; Ullrich, O.; Kuckelkorn, U.; Davies, K.J.A.; Grune, T. Comparative Resistance of the 20 S and 26 S Proteasome to Oxidative Stress. Biochem. J. 1998, 335, 637–642. [Google Scholar] [CrossRef] [PubMed]
- Alam, P.; Bousset, L.; Melki, R.; Otzen, D.E. α-Synuclein Oligomers and Fibrils: A Spectrum of Species, a Spectrum of Toxicities. J. Neurochem. 2019, 150, 522–534. [Google Scholar] [CrossRef]
- Tycko, R. Molecular Structure of Aggregated Amyloid-β: Insights from Solid-State Nuclear Magnetic Resonance. Cold Spring Harb. Perspect. Med. 2016, 6, a024083. [Google Scholar] [CrossRef] [PubMed]
- Knowles, T.P.J.; Vendruscolo, M.; Dobson, C.M. The Amyloid State and Its Association with Protein Misfolding Diseases. Nat. Rev. Mol. Cell Biol. 2014, 15, 384–396. [Google Scholar] [CrossRef] [PubMed]
- Dear, A.J.; Meisl, G.; Šarić, A.; Michaels, T.C.T.; Kjaergaard, M.; Linse, S.; Knowles, T.P.J. Identification of On- and off-Pathway Oligomers in Amyloid Fibril Formation. Chem. Sci. 2020, 11, 6236–6247. [Google Scholar] [CrossRef]
- Muschol, M.; Hoyer, W. Amyloid Oligomers as On-Pathway Precursors or off-Pathway Competitors of Fibrils. Front. Mol. Biosci. 2023, 10, 1120416. [Google Scholar] [CrossRef]
- Ingelsson, M. Alpha-Synuclein Oligomers—Neurotoxic Molecules in Parkinson’s Disease and Other Lewy Body Disorders. Front. Neurosci. 2016, 10, 408. [Google Scholar] [CrossRef]
- Kayed, R.; Head, E.; Thompson, J.L.; McIntire, T.M.; Milton, S.C.; Cotman, C.W.; Glabel, C.G. Common Structure of Soluble Amyloid Oligomers Implies Common Mechanism of Pathogenesis. Science 2003, 300, 486–489. [Google Scholar] [CrossRef]
- Guerrero-Muñoz, M.J.; Gerson, J.; Castillo-Carranza, D.L. Tau Oligomers: The Toxic Player at Synapses in Alzheimer’s Disease. Front. Cell. Neurosci. 2015, 9, 464. [Google Scholar] [CrossRef]
- Shafiei, S.S.; Guerrero-Muñoz, M.J.; Castillo-Carranza, D.L. Tau Oligomers: Cytotoxicity, Propagation, and Mitochondrial Damage. Front. Aging Neurosci. 2017, 9, 83. [Google Scholar] [CrossRef]
- Pieri, L.; Madiona, K.; Bousset, L.; Melki, R. Fibrillar α-Synuclein and Huntingtin Exon 1 Assemblies Are Toxic to the Cells. Biophys. J. 2012, 102, 2894–2905. [Google Scholar] [CrossRef]
- Bousset, L.; Pieri, L.; Ruiz-Arlandis, G.; Gath, J.; Jensen, P.H.; Habenstein, B.; Madiona, K.; Olieric, V.; Böckmann, A.; Meier, B.H.; et al. Structural and Functional Characterization of Two Alpha-Synuclein Strains. Nat. Commun. 2013, 4, 2575. [Google Scholar] [CrossRef] [PubMed]
- Peelaerts, W.; Bousset, L.; Van Der Perren, A.; Moskalyuk, A.; Pulizzi, R.; Giugliano, M.; Van Den Haute, C.; Melki, R.; Baekelandt, V. α-Synuclein Strains Cause Distinct Synucleinopathies after Local and Systemic Administration. Nature 2015, 522, 340–344. [Google Scholar] [CrossRef] [PubMed]
- Flavin, W.P.; Bousset, L.; Green, Z.C.; Chu, Y.; Skarpathiotis, S.; Chaney, M.J.; Kordower, J.H.; Melki, R.; Campbell, E.M. Endocytic Vesicle Rupture Is a Conserved Mechanism of Cellular Invasion by Amyloid Proteins. Acta Neuropathol. 2017, 134, 629–653. [Google Scholar] [CrossRef] [PubMed]
- Mahul-Mellier, A.-L.; Vercruysse, F.; Maco, B.; Ait-Bouziad, N.; De Roo, M.; Muller, D.; Lashuel, H.A. Fibril Growth and Seeding Capacity Play Key Roles in α-Synuclein-Mediated Apoptotic Cell Death. Cell Death Differ. 2015, 22, 2107–2122. [Google Scholar] [CrossRef]
- Ciechanover, A.; Brundin, P. The Ubiquitin Proteasome System in Neurodegenerative Diseases: Sometimes the Chicken, Sometimes the Egg. Neuron 2003, 40, 427–446. [Google Scholar] [CrossRef]
- Cook, C.; Stetler, C.; Petrucelli, L. Disruption of Protein Quality Control in Parkinson’s Disease. Cold Spring Harb. Perspect. Med. 2012, 2, a009423. [Google Scholar] [CrossRef]
- Medinas, D.B.; Valenzuela, V.; Hetz, C. Proteostasis Disturbance in Amyotrophic Lateral Sclerosis. Hum. Mol. Genet. 2017, 26, R91–R104. [Google Scholar] [CrossRef]
- Aman, Y.; Schmauck-Medina, T.; Hansen, M.; Morimoto, R.I.; Simon, A.K.; Bjedov, I.; Palikaras, K.; Simonsen, A.; Johansen, T.; Tavernarakis, N.; et al. Autophagy in Healthy Aging and Disease. Nat. Aging 2021, 1, 634–650. [Google Scholar] [CrossRef]
- Stefanis, L.; Emmanouilidou, E.; Pantazopoulou, M.; Kirik, D.; Vekrellis, K.; Tofaris, G.K. How Is Alpha-Synuclein Cleared from the Cell? J. Neurochem. 2019, 150, 577–590. [Google Scholar] [CrossRef]
- Lee, M.J.; Lee, J.H.; Rubinsztein, D.C. Tau Degradation: The Ubiquitin-Proteasome System versus the Autophagy-Lysosome System. Prog. Neurobiol. 2013, 105, 49–59. [Google Scholar] [CrossRef]
- Tofaris, G.K.; Layfield, R.; Spillantini, M.G. α-Synuclein Metabolism and Aggregation Is Linked to Ubiquitin-Independent Degradation by the Proteasome. FEBS Lett. 2001, 509, 22–26. [Google Scholar] [CrossRef]
- Shabek, N.; Herman-Bachinsky, Y.; Buchsbaum, S.; Lewinson, O.; Haj-Yahya, M.; Hejjaoui, M.; Lashuel, H.A.; Sommer, T.; Brik, A.; Ciechanover, A. The Size of the Proteasomal Substrate Determines Whether Its Degradation Will Be Mediated by Mono- or Polyubiquitylation. Mol. Cell 2012, 48, 87–97. [Google Scholar] [CrossRef]
- Abeywardana, T.; Lin, Y.H.; Rott, R.; Engelender, S.; Pratt, M.R. Site-Specific Differences in Proteasome-Dependent Degradation of Monoubiquitinated α-Synuclein. Chem. Biol. 2013, 20, 1207–1213. [Google Scholar] [CrossRef] [PubMed]
- Rott, R.; Szargel, R.; Haskin, J.; Bandopadhyay, R.; Lees, A.J.; Shani, V.; Engelender, S. α-Synuclein Fate Is Determined by USP9X-Regulated Monoubiquitination. Proc. Natl. Acad. Sci. USA 2011, 108, 18666–18671. [Google Scholar] [CrossRef] [PubMed]
- Webb, J.L.; Ravikumar, B.; Atkins, J.; Skepper, J.N.; Rubinsztein, D.C. α-Synuclein Is Degraded by Both Autophagy and the Proteasome. J. Biol. Chem. 2003, 278, 25009–25013. [Google Scholar] [CrossRef] [PubMed]
- Emmanouilidou, E.; Stefanis, L.; Vekrellis, K. Cell-Produced α-Synuclein Oligomers Are Targeted to, and Impair, the 26S Proteasome. Neurobiol. Aging 2010, 31, 953–968. [Google Scholar] [CrossRef]
- Cliffe, R.; Sang, J.C.; Kundel, F.; Finley, D.; Klenerman, D.; Ye, Y. Filamentous Aggregates Are Fragmented by the Proteasome Holoenzyme. Cell Rep. 2019, 26, 2140–2149.e3. [Google Scholar] [CrossRef]
- Pantazopoulou, M.; Brembati, V.; Kanellidi, A.; Bousset, L.; Melki, R.; Stefanis, L. Distinct Alpha-Synuclein Species Induced by Seeding Are Selectively Cleared by the Lysosome or the Proteasome in Neuronally Differentiated SH-SY5Y Cells. J. Neurochem. 2021, 156, 880–896. [Google Scholar] [CrossRef]
- David, D.C.; Layfield, R.; Serpell, L.; Narain, Y.; Goedert, M.; Spillantini, M.G. Proteasomal Degradation of Tau Protein. J. Neurochem. 2002, 83, 176–185. [Google Scholar] [CrossRef]
- Han, D.H.; Na, H.K.; Choi, W.H.; Lee, J.H.; Kim, Y.K.; Won, C.; Lee, S.H.; Kim, K.P.; Kuret, J.; Min, D.H.; et al. Direct Cellular Delivery of Human Proteasomes to Delay Tau Aggregation. Nat. Commun. 2014, 5, 5633. [Google Scholar] [CrossRef] [PubMed]
- Scotter, E.L.; Vance, C.; Nishimura, A.L.; Lee, Y.B.; Chen, H.J.; Urwin, H.; Sardone, V.; Mitchell, J.C.; Rogelj, B.; Rubinsztein, D.C.; et al. Differential Roles of the Ubiquitin Proteasome System and Autophagy in the Clearance of Soluble and Aggregated TDP-43 Species. J. Cell Sci. 2014, 127, 1263–1278. [Google Scholar] [CrossRef] [PubMed]
- Kabuta, T.; Suzuki, Y.; Wada, K. Degradation of Amyotrophic Lateral Sclerosis-Linked Mutant Cu,Zn-Superoxide Dismutase Proteins by Macroautophagy and the Proteasome. J. Biol. Chem. 2006, 281, 30524–30533. [Google Scholar] [CrossRef]
- Deriziotis, P.; André, R.; Smith, D.M.; Goold, R.; Kinghorn, K.J.; Kristiansen, M.; Nathan, J.A.; Rosenzweig, R.; Krutauz, D.; Glickman, M.H.; et al. Misfolded PrP Impairs the UPS by Interaction with the 20S Proteasome and Inhibition of Substrate Entry. EMBO J. 2011, 30, 3065–3077. [Google Scholar] [CrossRef] [PubMed]
- Kristiansen, M.; Deriziotis, P.; Dimcheff, D.E.; Jackson, G.S.; Ovaa, H.; Naumann, H.; Clarke, A.R.; van Leeuwen, F.W.B.; Menéndez-Benito, V.; Dantuma, N.P.; et al. Disease-Associated Prion Protein Oligomers Inhibit the 26S Proteasome. Mol. Cell 2007, 26, 175–188. [Google Scholar] [CrossRef]
- Thibaudeau, T.A.; Anderson, R.T.; Smith, D.M. A Common Mechanism of Proteasome Impairment by Neurodegenerative Disease-Associated Oligomers. Nat. Commun. 2018, 9, 1097. [Google Scholar] [CrossRef] [PubMed]
- Myeku, N.; Clelland, C.L.; Emrani, S.; Kukushkin, N.V.; Yu, W.H.; Goldberg, A.L.; Duff, K.E. Tau-Driven 26S Proteasome Impairment and Cognitive Dysfunction Can Be Prevented Early in Disease by Activating CAMP-PKA Signaling. Nat. Med. 2016, 22, 46–53. [Google Scholar] [CrossRef]
- Tanaka, Y.; Engelender, S.; Igarashi, S.; Rao, R.K.; Wanner, T.; Tanzi, R.E.; Sawa, A.; Dawson, V.L.; Dawson, T.M.; Ross, C.A. Inducible Expression of Mutant α-Synuclein Decreases Proteasome Activity and Increases Sensitivity to Mitochondria-Dependent Apoptosis. Hum. Mol. Genet. 2001, 10, 919–926. [Google Scholar] [CrossRef]
- McKinnon, C.; De Snoo, M.L.; Gondard, E.; Neudorfer, C.; Chau, H.; Ngana, S.G.; O’Hara, D.M.; Brotchie, J.M.; Koprich, J.B.; Lozano, A.M.; et al. Early-Onset Impairment of the Ubiquitin-Proteasome System in Dopaminergic Neurons Caused by α-Synuclein. Acta Neuropathol. Commun. 2020, 8, 17. [Google Scholar] [CrossRef]
- Cheroni, C.; Peviani, M.; Cascio, P.; DeBiasi, S.; Monti, C.; Bendotti, C. Accumulation of Human SOD1 and Ubiquitinated Deposits in the Spinal Cord of SOD1G93A Mice during Motor Neuron Disease Progression Correlates with a Decrease of Proteasome. Neurobiol. Dis. 2005, 18, 509–522. [Google Scholar] [CrossRef] [PubMed]
- Cheroni, C.; Marino, M.; Tortarolo, M.; Veglianese, P.; De Biasi, S.; Fontana, E.; Zuccarello, L.V.; Maynard, C.J.; Dantuma, N.P.; Bendotti, C. Functional Alterations of the Ubiquitin-Proteasome System in Motor Neurons of a Mouse Model of Familial Amyotrophic Lateral Sclerosis. Hum. Mol. Genet. 2009, 18, 82–96. [Google Scholar] [CrossRef] [PubMed]
- Boland, B.; Yu, W.H.; Corti, O.; Mollereau, B.; Henriques, A.; Bezard, E.; Pastores, G.M.; Rubinsztein, D.C.; Nixon, R.A.; Duchen, M.R.; et al. Promoting the Clearance of Neurotoxic Proteins in Neurodegenerative Disorders of Ageing. Nat. Rev. Drug Discov. 2018, 17, 660–688. [Google Scholar] [CrossRef] [PubMed]
- VerPlank, J.J.S.; Goldberg, A.L. Regulating Protein Breakdown through Proteasome Phosphorylation. Biochem. J. 2017, 474, 3355–3371. [Google Scholar] [CrossRef] [PubMed]
- Lokireddy, S.; Kukushkin, N.V.; Goldberg, A.L. CAMP-Induced Phosphorylation of 26S Proteasomes on Rpn6/PSMD11 Enhances Their Activity and the Degradation of Misfolded Proteins. Proc. Natl. Acad. Sci. USA 2015, 112, E7176–E7185. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H.; Park, Y.; Yoon, S.K.; Yoon, J.B. Osmotic Stress Inhibits Proteasome by P38 MAPK-Dependent Phosphorylation. J. Biol. Chem. 2010, 285, 41280–41289. [Google Scholar] [CrossRef]
- Leestemaker, Y.; de Jong, A.; Witting, K.F.; Penning, R.; Schuurman, K.; Rodenko, B.; Zaal, E.A.; van de Kooij, B.; Laufer, S.; Heck, A.J.R.; et al. Proteasome Activation by Small Molecules. Cell Chem. Biol. 2017, 24, 725–736. [Google Scholar] [CrossRef]
- Lee, B.H.; Lee, M.J.; Park, S.; Oh, D.C.; Elsasser, S.; Chen, P.C.; Gartner, C.; Dimova, N.; Hanna, J.; Gygi, S.P.; et al. Enhancement of Proteasome Activity by a Small-Molecule Inhibitor of USP14. Nature 2010, 467, 179–184. [Google Scholar] [CrossRef]
- Chu, T.T.; Gao, N.; Li, Q.Q.; Chen, P.G.; Yang, X.F.; Chen, Y.X.; Zhao, Y.F.; Li, Y.M. Specific Knockdown of Endogenous Tau Protein by Peptide-Directed Ubiquitin-Proteasome Degradation. Cell Chem. Biol. 2016, 23, 453–461. [Google Scholar] [CrossRef]
- Kargbo, R.B. PROTAC Compounds Targeting α-Synuclein Protein for Treating Neurogenerative Disorders: Alzheimer’s and Parkinson’s Diseases. ACS Med. Chem. Lett. 2020, 11, 1086–1087. [Google Scholar] [CrossRef]
- Qu, J.; Ren, X.; Xue, F.; He, Y.; Zhang, R.; Zheng, Y.; Huang, H.; Wang, W.; Zhang, J. Specific Knockdown of α-Synuclein by Peptide-Directed Proteasome Degradation Rescued Its Associated Neurotoxicity. Cell Chem. Biol. 2020, 27, 751–762. [Google Scholar] [CrossRef] [PubMed]
- Meiners, S.; Heyken, D.; Weller, A.; Ludwig, A.; Stangl, K.; Kloetzel, P.M.; Krüger, E. Inhibition of Proteasome Activity Induces Concerted Expression of Proteasome Genes and de Novo Formation of Mammalian Proteasomes. J. Biol. Chem. 2003, 278, 21517–21525. [Google Scholar] [CrossRef] [PubMed]
- Bugno, M.; Daniel, M.; Chepelev, N.L.; Willmore, W.G. Changing Gears in Nrf1 Research, from Mechanisms of Regulation to Its Role in Disease and Prevention. Biochim. Biophys. Acta Gene Regul. Mech. 2015, 1849, 1260–1276. [Google Scholar] [CrossRef] [PubMed]
- Radhakrishnan, S.K.; den Besten, W.; Deshaies, R.J. P97-Dependent Retrotranslocation and Proteolytic Processing Govern Formation of Active Nrf1 upon Proteasome Inhibition. eLife 2014, 3, e01856. [Google Scholar] [CrossRef] [PubMed]
- Sha, Z.; Goldberg, A.L. Proteasome-Mediated Processing of Nrf1 Is Essential for Coordinate Induction of All Proteasome Subunits and P97. Curr. Biol. 2014, 24, 1573–1583. [Google Scholar] [CrossRef] [PubMed]
- Koizumi, S.; Irie, T.; Hirayama, S.; Sakurai, Y.; Yashiroda, H.; Naguro, I.; Ichijo, H.; Hamazaki, J.; Murata, S. The Aspartyl Protease DDI2 Activates Nrf1 to Compensate for Proteasome Dysfunction. eLife 2016, 5, e18357. [Google Scholar] [CrossRef]
- Lehrbach, N.J.; Ruvkun, G. Proteasome Dysfunction Triggers Activation of SKN-1A/Nrf1 by the Aspartic Protease DDI-1. eLife 2016, 5, e17721. [Google Scholar] [CrossRef]
- Radhakrishnan, S.K.; Lee, C.S.; Young, P.; Beskow, A.; Chan, J.Y.; Deshaies, R.J. Transcription Factor Nrf1 Mediates the Proteasome Recovery Pathway after Proteasome Inhibition in Mammalian Cells. Mol. Cell 2010, 38, 17–28. [Google Scholar] [CrossRef]
- Kwak, M.-K.; Wakabayashi, N.; Greenlaw, J.L.; Yamamoto, M.; Kensler, T.W. Antioxidants Enhance Mammalian Proteasome Expression through the Keap1-Nrf2 Signaling Pathway. Mol. Cell. Biol. 2003, 23, 8786–8794. [Google Scholar] [CrossRef]
- Kraft, D.C.; Deocaris, C.C.; Wadhwa, R.; Rattan, S.I.S. Preincubation with the Proteasome Inhibitor MG-132 Enhances Proteasome Activity via the Nrf2 Transcription Factor in Aging Human Skin Fibroblasts. Ann. N. Y. Acad. Sci. 2006, 1067, 420–424. [Google Scholar] [CrossRef]
- Liu, Y.; Hettinger, C.L.; Zhang, D.; Rezvani, K.; Wang, X.; Wang, H. Sulforaphane Enhances Proteasomal and Autophagic Activities in Mice and Is a Potential Therapeutic Reagent for Huntington’s Disease. J. Neurochem. 2014, 129, 539–547. [Google Scholar] [CrossRef]
- Steffen, J.; Seeger, M.; Koch, A.; Krüger, E. Proteasomal Degradation Is Transcriptionally Controlled by TCF11 via an ERAD-Dependent Feedback Loop. Mol. Cell 2010, 40, 147–158. [Google Scholar] [CrossRef] [PubMed]
- Baird, L.; Tsujita, T.; Kobayashi, E.H.; Funayama, R.; Nagashima, T.; Nakayama, K.; Yamamoto, M. A Homeostatic Shift Facilitates Endoplasmic Reticulum Proteostasis through Transcriptional Integration of Proteostatic Stress Response Pathways. Mol. Cell. Biol. 2017, 37, e00439-16. [Google Scholar] [CrossRef] [PubMed]
- Lehrbach, N.J.; Breen, P.C.; Ruvkun, G. Protein Sequence Editing of SKN-1A/Nrf1 by Peptide:N-Glycanase Controls Proteasome Gene Expression. Cell 2019, 177, 737–750.e15. [Google Scholar] [CrossRef]
- Mannhaupt, G.; Schnall, R.; Karpov, V.; Vetter, I.; Feldmann, H. Rpn4p Acts as a Transcription Factor by Binding to PACE, a Nonamer Box Found Upstream of 26S Proteasomal and Other Genes in Yeast. FEBS Lett. 1999, 450, 27–34. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Fu, J.; Ha, S.W.; Ju, D.; Zheng, J.; Li, L.; Xie, Y. The CCAAT Box-Binding Transcription Factor NF-Y Regulates Basal Expression of Human Proteasome Genes. Biochim. Biophys. Acta 2012, 1823, 818–825. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Qiao, F.; Leiferman, P.C.; Ross, A.; Schlenker, E.H.; Wang, H. FOXOs Modulate Proteasome Activity in Human-Induced Pluripotent Stem Cells of Huntington’s Disease and Their Derived Neural Cells. Hum. Mol. Genet. 2017, 26, 4416–4428. [Google Scholar] [CrossRef]
- Vilchez, D.; Boyer, L.; Morantte, I.; Lutz, M.; Merkwirth, C.; Joyce, D.; Spencer, B.; Page, L.; Masliah, E.; Travis Berggren, W.; et al. Increased Proteasome Activity in Human Embryonic Stem Cells Is Regulated by PSMD11. Nature 2012, 489, 304–308. [Google Scholar] [CrossRef]
- Vangala, J.R.; Dudem, S.; Jain, N.; Kalivendi, S.V. Regulation of Psmb5 Protein and β Subunits of Mammalian Proteasome by Constitutively Activated Signal Transducer and Activator of Transcription 3 (Stat3): Potential Role in Bortezomib-Mediated Anticancer Therapy. J. Biol. Chem. 2014, 289, 12612–12622. [Google Scholar] [CrossRef]
- Zhang, Y.; Nicholatos, J.; Dreier, J.R.; Ricoult, S.J.H.; Widenmaier, S.B.; Hotamisligil, G.S.; Kwiatkowski, D.J.; Manning, B.D. Coordinated Regulation of Protein Synthesis and Degradation by MTORC1. Nature 2014, 513, 440–443. [Google Scholar] [CrossRef]
- Zhang, Y.; Manning, B.D. MTORC1 Signaling Activates NRF1 to Increase Cellular Proteasome Levels. Cell Cycle 2015, 14, 2011–2017. [Google Scholar] [CrossRef]
- Meul, T.; Berschneider, K.; Schmitt, S.; Mayr, C.H.; Mattner, L.F.; Schiller, H.B.; Yazgili, A.S.; Wang, X.; Lukas, C.; Schlesser, C.; et al. Mitochondrial Regulation of the 26S Proteasome. Cell Rep. 2020, 32, 108059. [Google Scholar] [CrossRef] [PubMed]
- Rousseau, A.; Bertolotti, A. An Evolutionarily Conserved Pathway Controls Proteasome Homeostasis. Nature 2016, 536, 184–189. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Zhai, B.; Gygi, S.P.; Goldberg, A.L. MTOR Inhibition Activates Overall Protein Degradation by the Ubiquitin Proteasome System as Well as by Autophagy. Proc. Natl. Acad. Sci. USA 2015, 112, 15790–15797. [Google Scholar] [CrossRef] [PubMed]
- Beese, C.J.; Brynjólfsdóttir, S.H.; Frankel, L.B. Selective Autophagy of the Protein Homeostasis Machinery: Ribophagy, Proteaphagy and ER-Phagy. Front. Cell Dev. Biol. 2020, 7, 373. [Google Scholar] [CrossRef] [PubMed]
- Robida-Stubbs, S.; Glover-Cutter, K.; Lamming, D.W.; Mizunuma, M.; Narasimhan, S.D.; Neumann-Haefelin, E.; Sabatini, D.M.; Blackwell, T.K. TOR Signaling and Rapamycin Influence Longevity by Regulating SKN-1/Nrf and DAF-16/FoxO. Cell Metab. 2012, 15, 713–724. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.H.; Walter, P.; Yen, T.S.B. Endoplasmic Reticulum Stress in Disease Pathogenesis. Annu. Rev. Pathol. 2008, 3, 399–425. [Google Scholar] [CrossRef]
- Hetz, C.; Saxena, S. ER Stress and the Unfolded Protein Response in Neurodegeneration. Nat. Rev. Neurol. 2017, 13, 477–491. [Google Scholar] [CrossRef]
- Görlach, A.; Bertram, K.; Hudecova, S.; Krizanova, O. Calcium and ROS: A Mutual Interplay. Redox Biol. 2015, 6, 260–271. [Google Scholar] [CrossRef]
- Glover-Cutter, K.M.; Lin, S.; Blackwell, T.K. Integration of the Unfolded Protein and Oxidative Stress Responses through SKN-1/Nrf. PLoS Genet. 2013, 9, e1003701. [Google Scholar] [CrossRef]
- Li, F.; Gao, B.; Dong, H.; Shi, J.; Fang, D. Icariin Induces Synoviolin Expression through NFE2L1 to Protect Neurons from ER Stress-Induced Apoptosis. PLoS ONE 2015, 10, e0119955. [Google Scholar] [CrossRef]
- Ho, D.V.; Chan, J.Y. Induction of Herpud1 Expression by ER Stress Is Regulated by Nrf1. FEBS Lett. 2015, 589, 615–620. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Ren, Y.; Li, S.; Hayes, J.D. Transcription Factor Nrf1 Is Topologically Repartitioned across Membranes to Enable Target Gene Transactivation through Its Acidic Glucose-Responsive Domains. PLoS ONE 2014, 9, e93458. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, Y.; Asahina, M.; Murakami, A.; Kawawaki, J.; Yoshida, M.; Fujinawa, R.; Iwai, K.; Tozawa, R.; Matsuda, N.; Tanaka, K.; et al. Loss of Peptide: N-Glycanase Causes Proteasome Dysfunction Mediated by a Sugar-Recognizing Ubiquitin Ligase. Proc. Natl. Acad. Sci. USA 2021, 118, e2102902118. [Google Scholar] [CrossRef] [PubMed]
- Biswas, M.; Kwong, E.K.; Park, E.; Nagra, P.; Chan, J.Y. Glycogen Synthase Kinase 3 Regulates Expression of Nuclear Factor-Erythroid-2 Related Transcription Factor-1 (Nrf1) and Inhibits pro-Survival Function of Nrf1. Exp. Cell Res. 2013, 319, 1922–1931. [Google Scholar] [CrossRef] [PubMed]
- Tsuchiya, Y.; Taniguchi, H.; Ito, Y.; Morita, T.; Karim, M.R.; Ohtake, N.; Fukagai, K.; Ito, T.; Okamuro, S.; Iemura, S.; et al. The Casein Kinase 2-Nrf1 Axis Controls the Clearance of Ubiquitinated Proteins by Regulating Proteasome Gene Expression. Mol. Cell. Biol. 2013, 33, 3461–3472. [Google Scholar] [CrossRef]
- Tsuchiya, Y.; Morita, T.; Kim, M.; Iemura, S.-I.; Natsume, T.; Yamamoto, M.; Kobayashi, A. Dual Regulation of the Transcriptional Activity of Nrf1 by β-TrCP- and Hrd1-Dependent Degradation Mechanisms. Mol. Cell. Biol. 2011, 31, 4500–4512. [Google Scholar] [CrossRef]
- Chen, J.; Liu, X.; Lü, F.; Liu, X.; Ru, Y.; Ren, Y.; Yao, L.; Zhang, Y. Transcription Factor Nrf1 Is Negatively Regulated by Its O-GlcNAcylation Status. FEBS Lett. 2015, 589, 2347–2358. [Google Scholar] [CrossRef]
- Han, J.W.; Valdez, J.L.; Ho, D.V.; Lee, C.S.; Kim, H.M.; Wang, X.; Huang, L.; Chan, J.Y. Nuclear Factor-Erythroid-2 Related Transcription Factor-1 (Nrf1) Is Regulated by O-GlcNAc Transferase. Free. Radic. Biol. Med. 2017, 110, 196–205. [Google Scholar] [CrossRef]
- Sekine, H.; Okazaki, K.; Kato, K.; Alam, M.M.; Shima, H.; Katsuoka, F.; Tsujita, T.; Suzuki, N.; Kobayashi, A.; Igarashi, K.; et al. O-GlcNAcylation Signal Mediates Proteasome Inhibitor Resistance in Cancer Cells by Stabilizing NRF1. Mol. Cell. Biol. 2018, 38, e00252-18. [Google Scholar] [CrossRef]
- Chan, J.Y.; Han, X.L.; Kan, Y.W. Cloning of Nrf1, an NF-E2-Related Transcription Factor, by Genetic Selection in Yeast. Proc. Natl. Acad. Sci. USA 1993, 90, 11371–11375. [Google Scholar] [CrossRef]
- McKie, J.; Johnstone, K.; Mattei, M.G.; Scambler, P. Cloning and Mapping of Murine Nfe2l1. Genomics 1995, 25, 716–719. [Google Scholar] [CrossRef] [PubMed]
- Caterina, J.J.; Donze, D.; Sun, C.W.; Ciavatta, D.J.; Townes, T.M. Cloning and Functional Characterization of LCR-F1: A BZIP Transcription Factor That Activates Erythroid-Specific, Human Globin Gene Expression. Nucleic Acids Res. 1994, 22, 2383–2391. [Google Scholar] [CrossRef]
- Blackwell, T.K.; Steinbaugh, M.J.; Hourihan, J.M.; Ewald, C.Y.; Isik, M. SKN-1/Nrf, Stress Responses, and Aging in Caenorhabditis Elegans. Free. Radic. Biol. Med. 2015, 88, 290–301. [Google Scholar] [CrossRef]
- Zhang, Y.; Xiang, Y. Molecular and Cellular Basis for the Unique Functioning of Nrf1, an Indispensable Transcription Factor for Maintaining Cell Homoeostasis and Organ Integrity. Biochem. J. 2016, 473, 961–1000. [Google Scholar] [CrossRef] [PubMed]
- Biswas, M.; Phan, D.; Watanabe, M.; Chan, J.Y. The Fbw7 Tumor Suppressor Regulates Nuclear Factor E2-Related Dactor 1 Transcription Factor Turnover through Proteasome-Mediated Proteolysis. J. Biol. Chem. 2011, 286, 39282–39289. [Google Scholar] [CrossRef]
- Han, J.J.W.; Ho, D.V.; Kim, H.M.; Lee, J.Y.; Jeon, Y.S.; Chan, J.Y. The Deubiquitinating Enzyme USP7 Regulates the Transcription Factor Nrf1 by Modulating Its Stability in Response to Toxic Metal Exposure. J. Biol. Chem. 2021, 296, 100732. [Google Scholar] [CrossRef] [PubMed]
- Hu, S.; Xiang, Y.; Qiu, L.; Wang, M.; Zhang, Y. Activation of the Membrane-Bound Nrf1 Transcription Factor by USP19, a Ubiquitin-Specific Protease C-Terminally Anchored in the Endoplasmic Reticulum. Biochim. Biophys. Acta Mol. Cell Res. 2022, 1869, 119299. [Google Scholar] [CrossRef]
- Fukagai, K.; Waku, T.; Chowdhury, A.M.M.A.; Kubo, K.; Matsumoto, M.; Kato, H.; Natsume, T.; Tsuruta, F.; Chiba, T.; Taniguchi, H.; et al. USP15 Stabilizes the Transcription Factor Nrf1 in the Nucleus, Promoting the Proteasome Gene Expression. Biochem. Biophys. Res. Commun. 2016, 478, 363–370. [Google Scholar] [CrossRef]
- Tomlin, F.M.; Gerling-Driessen, U.I.M.; Liu, Y.C.; Flynn, R.A.; Vangala, J.R.; Lentz, C.S.; Clauder-Muenster, S.; Jakob, P.; Mueller, W.F.; Ordoñez-Rueda, D.; et al. Inhibition of NGLY1 Inactivates the Transcription Factor Nrf1 and Potentiates Proteasome Inhibitor Cytotoxicity. ACS Cent. Sci. 2017, 3, 1143–1155. [Google Scholar] [CrossRef]
- Mergenthaler, P.; Lindauer, U.; Dienel, G.A.; Meisel, A. Sugar for the Brain: The Role of Glucose in Physiological and Pathological Brain Function. Trends Neurosci. 2013, 36, 587–597. [Google Scholar] [CrossRef]
- Hammond, T.C.; Xing, X.; Wang, C.; Ma, D.; Nho, K.; Crane, P.K.; Elahi, F.; Ziegler, D.A.; Liang, G.; Cheng, Q.; et al. β-Amyloid and Tau Drive Early Alzheimer’s Disease Decline While Glucose Hypometabolism Drives Late Decline. Commun. Biol. 2020, 3, 352. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Lucocq, J.M.; Hayes, J.D. The Nrf1 CNC/BZIP Protein Is a Nuclear Envelope-Bound Transcription Factor That Is Activated by t-Butyl Hydroquinone but Not by Endoplasmic Reticulum Stressors. Biochem. J. 2009, 418, 293–310. [Google Scholar] [CrossRef] [PubMed]
- Enns, G.M.; Shashi, V.; Bainbridge, M.; Gambello, M.J.; Zahir, F.R.; Bast, T.; Crimian, R.; Schoch, K.; Platt, J.; Cox, R.; et al. Mutations in NGLY1 Cause an Inherited Disorder of the Endoplasmic Reticulum-Associated Degradation Pathway. Genet. Med. 2014, 16, 751–758. [Google Scholar] [CrossRef]
- Owings, K.G.; Lowry, J.B.; Bi, Y.; Might, M.; Chow, C.Y. Transcriptome and Functional Analysis in a Drosophila Model of NGLY1 Deficiency Provides Insight into Therapeutic Approaches. Hum. Mol. Genet. 2018, 27, 1055–1066. [Google Scholar] [CrossRef]
- Ma, Q. Role of Nrf2 in Oxidative Stress and Toxicity. Annu. Rev. Pharmacol. Toxicol. 2013, 53, 401–426. [Google Scholar] [CrossRef] [PubMed]
- Boas, S.M.; Joyce, K.L.; Cowell, R.M. The NRF2-Dependent Transcriptional Regulation of Antioxidant Defense Pathways: Relevance for Cell Type-Specific Vulnerability to Neurodegeneration and Therapeutic Intervention. Antioxidants 2022, 11, 8. [Google Scholar] [CrossRef]
- Liu, P.; Kerins, M.J.; Tian, W.; Neupane, D.; Zhang, D.D.; Ooi, A. Differential and Overlapping Targets of the Transcriptional Regulators NRF1, NRF2, and NRF3 in Human Cells. J. Biol. Chem. 2019, 294, 18131–18149. [Google Scholar] [CrossRef]
- Itoh, K.; Chiba, T.; Takahashi, S.; Ishii, T.; Igarashi, K.; Katoh, Y.; Oyake, T.; Hayashi, N.; Satoh, K.; Hatayama, I.; et al. An Nrf2/Small Maf Heterodimer Mediates the Induction of Phase II Detoxifying Enzyme Genes through Antioxidant Response Elements. Biochem. Biophys. Res. Commun. 1997, 236, 313–322. [Google Scholar] [CrossRef]
- Johnsen, Ø.; Murphy, P.; Prydz, H.; Kolstø, A.B. Interaction of the CNC-BZIP Factor TCF11/LCR-F1/Nrf1 with MafG: Binding-Site Selection and Regulation of Transcription. Nucleic Acids Res. 1998, 26, 512–520. [Google Scholar] [CrossRef]
- Sankaranarayanan, K.; Jaiswal, A.K. Nrf3 Negatively Regulates Antioxidant-Response Element-Mediated Expression and Antioxidant Induction of NAD(P)H:Quinone Oxidoreductase1 Gene. J. Biol. Chem. 2004, 279, 50810–50817. [Google Scholar] [CrossRef]
- Farmer, S.C.; Sun, C.W.; Winnier, G.E.; Hogan, B.L.M.; Townes, T.M. The BZIP Transcription Factor LCR-F1 Is Essential for Mesoderm Formation in Mouse Development. Genes Dev. 1997, 11, 786–798. [Google Scholar] [CrossRef]
- Chan, J.Y.; Kwong, M.; Lu, R.; Chang, J.; Wang, B.; Yen, T.S.B.; Kan, Y.W. Targeted Disruption of the Ubiquitous CNC-BZIP Transcription Factor, Nrf-1, Results in Anemia and Embryonic Lethality in Mice. EMBO J. 1998, 17, 1779–1787. [Google Scholar] [CrossRef]
- Chen, L.; Kwong, M.; Lu, R.; Ginzinger, D.; Lee, C.; Leung, L.; Chan, J.Y. Nrf1 Is Critical for Redox Balance and Survival of Liver Cells during Development. Mol. Cell. Biol. 2003, 23, 4673–4686. [Google Scholar] [CrossRef]
- Chan, K.; Lu, R.; Chang, J.C.; Kan, Y.W. NRF2, a Member of the NFE2 Family of Transcription Factors, Is Not Essential for Murine Erythropoiesis, Growth, and Development. Proc. Natl. Acad. Sci. USA 1996, 93, 13943–13948. [Google Scholar] [CrossRef]
- Derjuga, A.; Gourley, T.S.; Holm, T.M.; Heng, H.H.Q.; Shivdasani, R.A.; Ahmed, R.; Andrews, N.C.; Blank, V. Complexity of CNC Transcription Factors as Revealed by Gene Targeting of the Nrf3 Locus. Mol. Cell. Biol. 2004, 24, 3286–3294. [Google Scholar] [CrossRef]
- Leung, L.; Kwong, M.; Hou, S.; Lee, C.; Chan, J.Y. Deficiency of the Nrf1 and Nrf2 Transcription Factors Results in Early Embryonic Lethality and Severe Oxidative Stress. J. Biol. Chem. 2003, 278, 48021–48029. [Google Scholar] [CrossRef]
- Ohtsuji, M.; Katsuoka, F.; Kobayashi, A.; Aburatani, H.; Hayes, J.D.; Yamamoto, M. Nrf1 and Nrf2 Play Distinct Roles in Activation of Antioxidant Response Element-Dependent Genes. J. Biol. Chem. 2008, 283, 33554–33562. [Google Scholar] [CrossRef]
- Bell, K.F.S.; Al-Mubarak, B.; Martel, M.A.; McKay, S.; Wheelan, N.; Hasel, P.; Márkus, N.M.; Baxter, P.; Deighton, R.F.; Serio, A.; et al. Neuronal Development Is Promoted by Weakened Intrinsic Antioxidant Defences Due to Epigenetic Repression of Nrf2. Nat. Commun. 2015, 6, 7066. [Google Scholar] [CrossRef]
- Lee, C.S.; Lee, C.; Hu, T.; Nguyen, J.M.; Zhang, J.; Martin, M.V.; Vawter, M.P.; Huang, E.J.; Chan, J.Y. Loss of Nuclear Factor E2-Related Factor 1 in the Brain Leads to Dysregulation of Proteasome Gene Expression and Neurodegeneration. Proc. Natl. Acad. Sci. USA 2011, 108, 8408–8413. [Google Scholar] [CrossRef]
- Forcina, G.C.; Pope, L.; Murray, M.; Dong, W.; Abu-Remaileh, M.; Bertozzi, C.R.; Dixon, S.J. Ferroptosis Regulation by the NGLY1/NFE2L1 Pathway. Proc. Natl. Acad. Sci. USA 2022, 119, e2118646119. [Google Scholar] [CrossRef]
- Kocaturk, N.M.; Gozuacik, D. Crosstalk between Mammalian Autophagy and the Ubiquitin-Proteasome System. Front. Cell Dev. Biol. 2018, 6, 128. [Google Scholar] [CrossRef] [PubMed]
- Sha, Z.; Schnell, H.M.; Ruoff, K.; Goldberg, A. Rapid Induction of P62 and GAB ARA PL1 upon Proteasome Inhibition Promotes Survival before Autophagy Activation. J. Cell Biol. 2018, 217, 1757–1776. [Google Scholar] [CrossRef] [PubMed]
- Cui, M.; Atmanli, A.; Morales, M.G.; Tan, W.; Chen, K.; Xiao, X.; Xu, L.; Liu, N.; Bassel-Duby, R.; Olson, E.N. Nrf1 Promotes Heart Regeneration and Repair by Regulating Proteostasis and Redox Balance. Nat. Commun. 2021, 12, 5270. [Google Scholar] [CrossRef] [PubMed]
- Jain, A.; Lamark, T.; Sjøttem, E.; Larsen, K.B.; Awuh, J.A.; Øvervatn, A.; McMahon, M.; Hayes, J.D.; Johansen, T. P62/SQSTM1 Is a Target Gene for Transcription Factor NRF2 and Creates a Positive Feedback Loop by Inducing Antioxidant Response Element-Driven Gene Transcription. J. Biol. Chem. 2010, 285, 22576–22591. [Google Scholar] [CrossRef] [PubMed]
- Pajares, M.; Jiménez-Moreno, N.; García-Yagüe, Á.J.; Escoll, M.; de Ceballos, M.L.; Van Leuven, F.; Rábano, A.; Yamamoto, M.; Rojo, A.I.; Cuadrado, A. Transcription Factor NFE2L2/NRF2 Is a Regulator of Macroautophagy Genes. Autophagy 2016, 12, 1902–1916. [Google Scholar] [CrossRef]
- Saibil, H. Chaperone Machines for Protein Folding, Unfolding and Disaggregation. Nat. Rev. Mol. Cell Biol. 2013, 14, 630–642. [Google Scholar] [CrossRef]
- Neef, D.W.; Jaeger, A.M.; Thiele, D.J. Heat Shock Transcription Factor 1 as a Therapeutic Target in Neurodegenerative Diseases. Nat. Rev. Drug Discov. 2011, 10, 930–944. [Google Scholar] [CrossRef]
- Oroń, M.; Grochowski, M.; Jaiswar, A.; Legierska, J.; Jastrzębski, K.; Nowak-Niezgoda, M.; Kołos, M.; Kaźmierczak, W.; Olesiński, T.; Lenarcik, M.; et al. The Molecular Network of the Proteasome Machinery Inhibition Response Is Orchestrated by HSP70, Revealing Vulnerabilities in Cancer Cells. Cell Rep. 2022, 40, 111428. [Google Scholar] [CrossRef]
- Kalmar, B.; Greensmith, L. Induction of Heat Shock Proteins for Protection against Oxidative Stress. Adv. Drug Deliv. Rev. 2009, 61, 310–318. [Google Scholar] [CrossRef]
- Paul, S.; Ghosh, S.; Mandal, S.; Sau, S.; Pal, M. NRF2 Transcriptionally Activates the Heat Shock Factor 1 Promoter under Oxidative Stress and Affects Survival and Migration Potential of MCF7 Cells. J. Biol. Chem. 2018, 293, 19303–19316. [Google Scholar] [CrossRef]
- Ibrahim, L.; Mesgarzadeh, J.; Xu, I.; Powers, E.T.; Luke Wiseman, R.; Bollong, M.J. Defining the Functional Targets of Capʼn‘collar Transcription Factors NRF1, NRF2, and NRF3. Antioxidants 2020, 9, 1025. [Google Scholar] [CrossRef] [PubMed]
- Lapierre, L.R.; Hansen, M. Lessons from C. Elegans: Signaling Pathways for Longevity. Trends Endocrinol. Metab. 2012, 23, 637–644. [Google Scholar] [CrossRef] [PubMed]
- An, J.H.; Blackwell, T.K. SKN-1 Links C. Elegans Mesendodermal Specification to a Conserved Oxidative Stress Response. Genes Dev. 2003, 17, 1882–1893. [Google Scholar] [CrossRef]
- Vilchez, D.; Morantte, I.; Liu, Z.; Douglas, P.M.; Merkwirth, C.; Rodrigues, A.P.C.; Manning, G.; Dillin, A. RPN-6 Determines C. Elegans Longevity under Proteotoxic Stress Conditions. Nature 2012, 489, 263–268. [Google Scholar] [CrossRef] [PubMed]
- Lehrbach, N.J.; Ruvkun, G. Endoplasmic Reticulum-Associated SKN-1A/Nrf1 Mediates a Cytoplasmic Unfolded Protein Response and Promotes Longevity. eLife 2019, 8, e44425. [Google Scholar] [CrossRef] [PubMed]
- Chondrogianni, N.; Georgila, K.; Kourtis, N.; Tavernarakis, N.; Gonos, E.S. 20S Proteasome Activation Promotes Life Span Extension and Resistance to Proteotoxicity in Caenorhabditis Elegans. FASEB J. 2015, 29, 611–622. [Google Scholar] [CrossRef]
- Bishop, N.A.; Guarente, L. Two Neurons Mediate Diet-Restriction-Induced Longevity in C. Elegans. Nature 2007, 447, 545–549. [Google Scholar] [CrossRef]
- Tullet, J.M.A.; Hertweck, M.; An, J.H.; Baker, J.; Hwang, J.Y.; Liu, S.; Oliveira, R.P.; Baumeister, R.; Blackwell, T.K. Direct Inhibition of the Longevity-Promoting Factor SKN-1 by Insulin-like Signaling in C. Elegans. Cell 2008, 132, 1025–1038. [Google Scholar] [CrossRef]
- Kobayashi, A.; Tsukide, T.; Miyasaka, T.; Morita, T.; Mizoroki, T.; Saito, Y.; Ihara, Y.; Takashima, A.; Noguchi, N.; Fukamizu, A.; et al. Central Nervous System-Specific Deletion of Transcription Factor Nrf1 Causes Progressive Motor Neuronal Dysfunction. Genes Cells 2011, 16, 692–703. [Google Scholar] [CrossRef]
- Villaescusa, J.C.; Li, B.; Toledo, E.M.; Rivetti di Val Cervo, P.; Yang, S.; Stott, S.R.; Kaiser, K.; Islam, S.; Gyllborg, D.; Laguna-Goya, R.; et al. A PBX1 Transcriptional Network Controls Dopaminergic Neuron Development and Is Impaired in Parkinson’s Disease. EMBO J. 2016, 35, 1963–1978. [Google Scholar] [CrossRef]
- Nath, S.R.; Yu, Z.; Gipson, T.A.; Marsh, G.B.; Yoshidome, E.; Robins, D.M.; Todi, S.V.; Housman, D.E.; Lieberman, A.P. Androgen Receptor Polyglutamine Expansion Drives Age-Dependent Quality Control Defects and Muscle Dysfunction. J. Clin. Investig. 2018, 128, 3630–3641. [Google Scholar] [CrossRef]
- Bott, L.C.; Badders, N.M.; Chen, K.L.; Harmison, G.G.; Bautista, E.; Shih, C.C.Y.; Katsuno, M.; Sobue, G.; Taylor, J.P.; Dantuma, N.P.; et al. A Small-Molecule Nrf1 and Nrf2 Activator Mitigates Polyglutamine Toxicity in Spinal and Bulbar Muscular Atrophy. Hum. Mol. Genet. 2016, 25, 1979–1989. [Google Scholar] [CrossRef] [PubMed]
- Hommen, F.; Bilican, S.; Vilchez, D. Protein Clearance Strategies for Disease Intervention. J. Neural Transm. 2022, 129, 141–172. [Google Scholar] [CrossRef] [PubMed]
- Koehler, A.N. A Complex Task? Direct Modulation of Transcription Factors with Small Molecules. Curr. Opin. Chem. Biol. 2010, 14, 331–340. [Google Scholar] [CrossRef] [PubMed]
- Iaconelli, J.; Ibrahim, L.; Chen, E.; Hull, M.; Schultz, P.G.; Bollong, M.J. Small-Molecule Stimulators of NRF1 Transcriptional Activity. ChemBioChem 2020, 21, 1816–1819. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chandran, A.; Oliver, H.J.; Rochet, J.-C. Role of NFE2L1 in the Regulation of Proteostasis: Implications for Aging and Neurodegenerative Diseases. Biology 2023, 12, 1169. https://doi.org/10.3390/biology12091169
Chandran A, Oliver HJ, Rochet J-C. Role of NFE2L1 in the Regulation of Proteostasis: Implications for Aging and Neurodegenerative Diseases. Biology. 2023; 12(9):1169. https://doi.org/10.3390/biology12091169
Chicago/Turabian StyleChandran, Aswathy, Haley Jane Oliver, and Jean-Christophe Rochet. 2023. "Role of NFE2L1 in the Regulation of Proteostasis: Implications for Aging and Neurodegenerative Diseases" Biology 12, no. 9: 1169. https://doi.org/10.3390/biology12091169