


The Role of Mitochondrial Dysfunction in Idiopathic Pulmonary Fibrosis: New Perspectives for a Challenging Disease
 , ,
, ,
Abstract
:Simple Summary
Abstract
1. Introduction
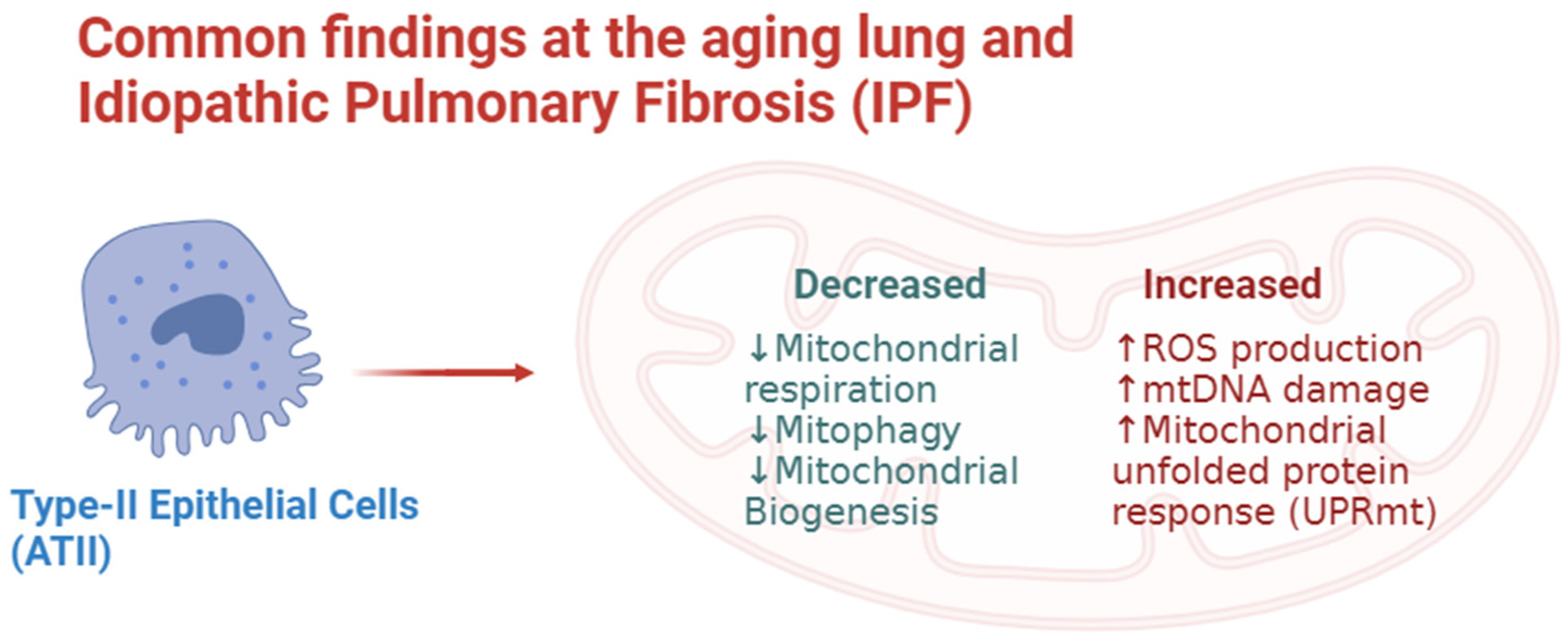
2. The Aging Lung
2.1. Mitochondrial Function in the Normal Lung
2.2. Adjustments in Respiratory Mechanics, Flow, and Lung Volumes
2.3. Cellular Changes in the Aging Lung
3. Mitochondria and Idiopathic Pulmonary Fibrosis
4. Newest Findings on Research for Mitochondrial Dysfunction
5. New Treatment Perspectives
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Martinez, F.J.; Collard, H.R.; Pardo, A.; Raghu, G.; Richeldi, L.; Selman, M.; Swigris, J.J.; Taniguchi, H.; Wells, A.U. Idiopathic pulmonary fibrosis. Nat. Rev. Dis. Prim. 2017, 3, 17074. [Google Scholar] [CrossRef] [PubMed]
- Shaghaghi, H.; Cuevas-Mora, K.; Para, R.; Tran, C.; Roque, W.; Robertson, M.J.; Rosas, I.O.; Summer, R.; Romero, F. A model of the aged lung epithelium in idiopathic pulmonary fibrosis. Aging 2021, 13, 16922–16937. [Google Scholar] [CrossRef]
- Roque, W.; Romero, F. Cellular metabolomics of pulmonary fibrosis, from amino acids to lipids. Am. J. Physiol. Cell Physiol. 2021, 320, C689–C695. [Google Scholar] [CrossRef]
- Para, R.; Romero, F.; George, G.; Summer, R. Metabolic Reprogramming as a Driver of Fibroblast Activation in PulmonaryFibrosis. Am. J. Med. Sci. 2019, 357, 394–398. [Google Scholar] [CrossRef] [PubMed]
- Schneider, J.L.; Rowe, J.H.; Garcia-de-Alba, C.; Kim, C.F.; Sharpe, A.H.; Haigis, M.C. The aging lung: Physiology, disease, and immunity. Cell 2021, 184, 1990–2019. [Google Scholar] [CrossRef] [PubMed]
- Navarro, S.; Driscoll, B. Regeneration of the Aging Lung: A Mini-Review. Gerontology 2017, 63, 270–280. [Google Scholar] [CrossRef]
- Zank, D.C.; Bueno, M.; Mora, A.L.; Rojas, M. Idiopathic pulmonary fibrosis: Aging, mitochondrial dysfunction, and cellular bioenergetics. Front. Med. 2018, 5, 10. [Google Scholar] [CrossRef]
- Caldeira, D.D.A.F.; Weiss, D.J.; Rocco, P.R.M.; Silva, P.L.; Cruz, F.F. Mitochondria in Focus: From Function to Therapeutic Strategies in Chronic Lung Diseases. Front. Immunol. 2021, 12, 782074. [Google Scholar] [CrossRef]
- Giorgi, C.; Agnoletto, C.; Bononi, A.; Bonora, M.; De Marchi, E.; Marchi, S.; Missiroli, S.; Patergnani, S.; Poletti, F.; Rimessi, A.; et al. Mitochondrial calcium homeostasis as potential target for mitochondrial medicine. Mitochondrion 2012, 12, 77–85. [Google Scholar] [CrossRef]
- Martinou, J.C.; Youle, R.J. Mitochondria in Apoptosis: Bcl-2 family Members and Mitochondrial Dynamics. Dev. Cell 2011, 21, 92. [Google Scholar] [CrossRef]
- Schumacker, P.T.; Gillespie, M.N.; Nakahira, K.; Choi, A.M.K.; Crouser, E.D.; Piantadosi, C.A.; Bhattacharya, J. Mitochondria in lung biology and pathology: More than just a powerhouse. Am. J. Physiol. Lung Cell Mol. Physiol. 2014, 306, 962–974. [Google Scholar] [CrossRef] [PubMed]
- Archer, S.L.; Souil, E.; Dinh-Xuan, A.T.; Schremmer, B.; Mercier, J.C.; El Yaagoubi, A.; Nguyen-Huu, L.; Reeve, H.L.; Hampl, V. Molecular identification of the role of voltage-gated K+ channels, Kv1.5 and Kv2.1, in hypoxic pulmonary vasoconstriction and control of resting membrane potential in rat pulmonary artery myocytes. J. Clin Investig. 1998, 101, 2319–2330. [Google Scholar] [CrossRef] [PubMed]
- Waypa, G.B.; Schumacker, P.T. Hypoxia-induced changes in pulmonary and systemic vascular resistance: Where is the O2 sensor? Respir. Physiol. Neurobiol. 2010, 174, 201–211. [Google Scholar] [CrossRef] [PubMed]
- Dunham-Snary, K.J.; Wu, D.; Sykes, E.A.; Thakrar, A.; Parlow, L.R.; Mewburn, J.D.; Parlow, J.L.; Archer, S.L. Hypoxic Pulmonary Vasoconstriction: From Molecular Mechanisms to Medicine. Chest 2017, 151, 181–192. [Google Scholar] [CrossRef]
- Zhang, J.; Ney, P.A. Role of BNIP3 and NIX in cell death, autophagy, and mitophagy. Cell Death Differ. 2009, 16, 939–946. [Google Scholar] [CrossRef]
- Lowery, E.M.; Brubaker, A.L.; Kuhlmann, E.; Kovacs, E.J. The aging lung. Clin. Interv. Aging 2013, 8, 1489–1496. [Google Scholar] [CrossRef]
- Skloot, G.S. The Effects of Aging on Lung Structure and Function. Clin. Geriatr. Med. 2017, 33, 447–457. [Google Scholar] [CrossRef]
- Cho, S.J.; Stout-Delgado, H.W. Aging and Lung Disease. Annu. Rev. Physiol. 2020, 82, 433–459. [Google Scholar] [CrossRef]
- Janssens, J.P.; Pache, J.C.; Nicod, L.P. Physiological changes in respiratory function associated with ageing. Eur. Respir. J. 1999, 13, 197–205. [Google Scholar] [CrossRef]
- Meiners, S.; Eickelberg, O.; Königshoff, M. Hallmarks of the ageing lung. Eur. Respir. J. 2015, 45, 807–827. [Google Scholar] [CrossRef]
- Ma, K.; Chen, G.; Li, W.; Kepp, O.; Zhu, Y.; Chen, Q. Mitophagy, Mitochondrial Homeostasis, and Cell Fate. Front. Cell Dev. Biol. 2020, 8, 467. [Google Scholar] [CrossRef]
- Youle, R.J.; Narendra, D.P. Mechanisms of mitophagy. Nat. Rev. Mol. Cell Biol. 2011, 12, 9–14. [Google Scholar] [CrossRef] [PubMed]
- Cloonan, S.M.; Kim, K.; Esteves, P.; Trian, T.; Barnes, P.J. Mitochondrial dysfunction in lung ageing and disease. Eur. Respir. Rev. 2020, 29, 157. [Google Scholar] [CrossRef]
- Lee, H.C.; Lu, C.Y.; Fahn, H.J.; Wei, Y.H. Aging- and smoking-associated alteration in the relative content of mitochondrial DNA in human lung. FEBS Lett. 1998, 441, 292–296. [Google Scholar] [CrossRef] [PubMed]
- Fahn, H.J.; Wang, L.S.; Hsieh, R.H.; Chang, S.C.; Kao, S.H.; Huang, M.H.; Wei, Y.H. Age-related 4977 bp deletion in human lung mitochondrial DNA. Am. J. Respir. Crit. Care Med. 2012, 154, 1141–1145. [Google Scholar] [CrossRef]
- Kwon, Y.; Kim, J.; Lee, C.Y.; Kim, H. Expression of SIRT1 and SIRT3 varies according to age in mice. Anat. Cell Biol. 2015, 48, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Álvarez, D.; Cárdenes, N.; Sellarés, J.; Bueno, M.; Corey, C.; Hanumanthu, V.S.; Peng, Y.; D’cunha, H.; Sembrat, J.; Nouraie, M.; et al. IPF lung fibroblasts have a senescent phenotype. Am. J. Physiol. Lung Cell Mol. Physiol. 2017, 313, L1164–L1173. [Google Scholar] [CrossRef]
- Xu, Y.; Mizuno, T.; Sridharan, A.; Du, Y.; Guo, M.; Tang, J.; Wikenheiser-Brokamp, K.A.; Perl, A.-K.T.; Funari, V.A.; Gokey, J.J.; et al. Single-cell RNA sequencing identifies diverse roles of epithelial cells in idiopathic pulmonary fibrosis. JCI Insight 2017, 1, 90558. [Google Scholar] [CrossRef]
- Bueno, M.; Calyeca, J.; Rojas, M.; Mora, A.L. Mitochondria dysfunction and metabolic reprogramming as drivers of idiopathic pulmonary fibrosis. Redox Biol. 2020, 33, 101509. [Google Scholar] [CrossRef]
- Bratic, A.; Larsson, N.G. The role of mitochondria in aging. J. Clin. Invest. 2013, 123, 951–957. [Google Scholar] [CrossRef]
- Herbener, C.H. A Morphometric Study of Age-Dependent Changes in Mitochondrial Populations of Mouse Liver and Heart. J. Gerontol. 1976, 31, 8–12. [Google Scholar] [CrossRef] [PubMed]
- Sanz, A. Mitochondrial reactive oxygen species: Do they extend or shorten animal lifespan? Biochim. Biophys. Acta 2016, 1857, 1116–1126. [Google Scholar] [CrossRef]
- Vendelbo, M.H.; Nair, K.S. Mitochondrial longevity pathways. Biochim. Biophys. Acta Mol. Cell Res. 2011, 1813, 634–644. [Google Scholar] [CrossRef] [PubMed]
- Bárcena, C.; Mayoral, P.; Quirós, P.M. Mitohormesis, an Antiaging Paradigm. Int. Rev. Cell Mol. Biol. 2018, 340, 35–77. [Google Scholar] [CrossRef] [PubMed]
- Yun, J.; Finkel, T. Mitohormesis. Cell Metab. 2014, 19, 757–766. [Google Scholar] [CrossRef]
- Mora, A.L.; Bueno, M.; Rojas, M. Mitochondria in the spotlight of aging and idiopathic pulmonary fibrosis. J. Clin. Investig. 2017, 127, 405–414. [Google Scholar] [CrossRef]
- Parimon, T.; Yao, C.; Stripp, B.R.; Noble, P.W.; Chen, P. Alveolar Epithelial Type II Cells as Drivers of Lung Fibrosis in Idiopathic Pulmonary Fibrosis. Int. J. Mol. Sci. 2020, 21, 2269. [Google Scholar] [CrossRef]
- Hawkins, A.; Guttentag, S.H.; Deterding, R.; Funkhouser, W.K.; Goralski, J.L.; Chatterjee, S.; Mulugeta, S.; Beers, M.F.; Solaligue, D.E.S.; Rodríguez-Castillo, J.A.; et al. A non-BRICHOS SFTPC mutant (SP-CI73T) linked to interstitial lung disease promotes a late block in macroautophagy disrupting cellular proteostasis and mitophagy. Am. J. Physiol. Lung Cell Mol. Physiol. 2015, 308, L33–L47. [Google Scholar] [CrossRef]
- Bueno, M.; Lai, Y.-C.; Romero, Y.; Brands, J.; Croix, C.M.S.; Kamga, C.; Corey, C.; Herazo-Maya, J.D.; Sembrat, J.; Lee, J.S.; et al. PINK1 deficiency impairs mitochondrial homeostasis and promotes lung fibrosis. J. Clin. Investig. 2015, 125, 521–538. [Google Scholar] [CrossRef]
- Sosulski, M.L.; Gongora, R.; Danchuk, S.; Dong, C.; Luo, F.; Sanchez, C.G. Deregulation of selective autophagy during aging and pulmonary fibrosis: The role of TGFβ1. Aging Cell 2015, 14, 774–783. [Google Scholar] [CrossRef]
- Patel, A.S.; Song, J.W.; Chu, S.G.; Mizumura, K.; Osorio, J.C.; Shi, Y.; El-Chemaly, S.; Lee, C.G.; Rosas, I.O.; Elias, J.A.; et al. Epithelial Cell Mitochondrial Dysfunction and PINK1 Are Induced by Transforming Growth Factor- Beta1 in Pulmonary Fibrosis. PLoS ONE 2015, 10, e0121246. [Google Scholar] [CrossRef]
- Yang, W.; Guo, X.; Tu, Z.; Chen, X.; Han, R.; Liu, Y.; Yan, S.; Wang, Q.; Wang, Z.; Zhao, X.; et al. PINK1 kinase dysfunction triggers neurodegeneration in the primate brain without impacting mitochondrial homeostasis. Protein Cell 2022, 13, 26–46. [Google Scholar] [CrossRef] [PubMed]
- Quinn, P.M.J.; Moreira, P.I.; Ambrósio, A.F.; Alves, C.H. PINK1/PARKIN signalling in neurodegeneration and neuroinflammation. Acta Neuropathol. Commun. 2020, 8, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Sosulski, M.L.; Gongora, R.; Feghali-Bostwick, C.; Lasky, J.A.; Sanchez, C.G. Sirtuin 3 Deregulation Promotes Pulmonary Fibrosis. J. Gerontol. Ser. A 2017, 72, 595–602. [Google Scholar] [CrossRef]
- Rehan, M.; Kurundkar, D.; Kurundkar, A.R.; Logsdon, N.J.; Smith, S.R.; Chanda, D.; Bernard, K.; Sanders, Y.Y.; Deshane, J.S.; Dsouza, K.G.; et al. Restoration of SIRT3 gene expression by airway delivery resolves age-associated persistent lung fibrosis in mice. Nat. Aging 2021, 1, 205–217. [Google Scholar] [CrossRef]
- Tashiro, J.; Rubio, G.A.; Limper, A.H.; Williams, K.; Elliot, S.J.; Ninou, I.; Aidinis, V.; Tzouvelekis, A.; Glassberg, M.K. Exploring animal models that resemble idiopathic pulmonary fibrosis. Front. Med. 2017, 4, 118. [Google Scholar] [CrossRef]
- Liu, T.; De Los Santos, F.G.; Phan, S.H. The Bleomycin Model of Pulmonary Fibrosis. Methods Mol. Biol. 2017, 1627, 27–42. [Google Scholar] [CrossRef]
- Cheresh, P.; Kim, S.-J.; Jablonski, R.; Watanabe, S.; Lu, Z.; Chi, M.; Helmin, K.A.; Gius, D.; Budinger, G.R.S.; Kamp, D.W. SIRT3 overexpression ameliorates asbestos-induced pulmonary fibrosis, mt-DNA damage and lung fibrogenic monocyte recruitment. Int. J. Mol. Sci. 2021, 22, 6856. [Google Scholar] [CrossRef] [PubMed]
- Hayakawa, T.; Iwai, M.; Aoki, S.; Takimoto, K.; Maruyama, M.; Maruyama, W.; Motoyama, N. SIRT1 Suppresses the Senescence-Associated Secretory Phenotype through Epigenetic Gene Regulation. PLoS ONE 2015, 10, e0116480. [Google Scholar] [CrossRef]
- Rangarajan, S.; Bernard, K.; Thannickal, V.J. Mitochondrial dysfunction in pulmonary fibrosis. Ann. Am. Thorac. Soc. 2017, 14, S383–S388. [Google Scholar] [CrossRef]
- Trifunovic, A.; Wredenberg, A.; Falkenberg, M.; Spelbrink, J.N.; Rovio, A.T.; Bruder, C.E.; Bohlooly-Y, M.; Gidlöf, S.; Oldfors, A.; Wibom, R.; et al. Premature ageing in mice expressing defective mitochondrial DNA polymerase. Nature 2004, 429, 417–423. [Google Scholar] [CrossRef] [PubMed]
- Vohwinkel, C.U.; Lecuona, E.; Sun, H.; Sommer, N.; Vadász, I.; Chandel, N.S.; Sznajder, J.I. Elevated CO2 levels cause mitochondrial dysfunction and impair cell proliferation. J. Biol. Chem. 2011, 286, 37067–37076. [Google Scholar] [CrossRef]
- Zhou, G.; Dada, L.A.; Wu, M.; Kelly, A.; Trejo, H.; Zhou, Q.; Varga, J.; Sznajder, J.I.; Bernard, O.; Jeny, F.; et al. Hypoxia-induced alveolar epithelial-mesenchymal transition requires mitochondrial ROS and hypoxia-inducible factor 1. Am. J. Physiol. Lung Cell Mol. Physiol. 2009, 297, L1120–L1130. [Google Scholar] [CrossRef]
- Kobayashi, K.; Araya, J.; Minagawa, S.; Hara, H.; Saito, N.; Kadota, T.; Sato, N.; Yoshida, M.; Tsubouchi, K.; Kurita, Y.; et al. Involvement of PARK2-Mediated Mitophagy in Idiopathic Pulmonary Fibrosis Pathogenesis. J. Immunol. 2016, 197, 504–516. [Google Scholar] [CrossRef] [PubMed]
- Larson-Casey, J.L.; Murthy, S.; Ryan, A.J.; Carter, A.B. Modulation of the Mevalonate Pathway by Akt Regulates Macrophage Survival and Development of Pulmonary Fibrosis. J. Biol. Chem. 2014, 289, 36204. [Google Scholar] [CrossRef] [PubMed]
- Larson-Casey, J.L.; Deshane, J.S.; Ryan, A.J.; Thannickal, V.J.; Carter, A.B. Macrophage Akt1 Kinase-Mediated Mitophagy Modulates Apoptosis Resistance and Pulmonary Fibrosis. Immunity 2016, 44, 582–596. [Google Scholar] [CrossRef] [PubMed]
- Tsitoura, E.; Vasarmidi, E.; Bibaki, E.; Trachalaki, A.; Koutoulaki, C.; Papastratigakis, G.; Papadogiorgaki, S.; Chalepakis, G.; Tzanakis, N.; Antoniou, K.M. Accumulation of damaged mitochondria in alveolar macrophages with reduced OXPHOS related gene expression in IPF. Respir Res. 2019, 20, 264. [Google Scholar] [CrossRef]
- He, C.; Larson-Casey, J.L.; Davis, D.; Hanumanthu, V.S.; Longhini, A.L.F.; Thannickal, V.J.; Gu, L.; Carter, A.B. NOX4 modulates macrophage phenotype and mitochondrial biogenesis in asbestosis. JCI Insight 2019, 4, e126551. [Google Scholar] [CrossRef]
- Bernard, K.; Logsdon, N.J.; Miguel, V.; Benavides, G.A.; Zhang, J.; Carter, A.B.; Darley-Usmar, V.M.; Thannickal, V.J. NADPH Oxidase 4 (Nox4) Suppresses Mitochondrial Biogenesis and Bioenergetics in Lung Fibroblasts via a Nuclear Factor Erythroid-derived 2-like 2 (Nrf2)-dependent Pathway. J. Biol. Chem. 2017, 292, 3029–3038. [Google Scholar] [CrossRef]
- Amara, N.; Goven, D.; Prost, F.; Muloway, R.; Crestani, B.; Boczkowski, J. NOX4/NADPH oxidase expression is increased in pulmonary fibroblasts from patients with idiopathic pulmonary fibrosis and mediates TGFbeta1-induced fibroblast differentiation into myofibroblasts. Thorax 2010, 65, 733–738. [Google Scholar] [CrossRef]
- Aversa, Z.; Atkinson, E.J.; Carmona, E.M.; White, T.A.; Heeren, A.A.; Jachim, S.K.; Zhang, X.; Cummings, S.R.; Chiarella, S.E.; Limper, A.H.; et al. Biomarkers of cellular senescence in idiopathic pulmonary fibrosis. Respir. Res. 2023, 24, 101. [Google Scholar] [CrossRef]
- Haque, A.; Engel, J.; Teichmann, S.A.; Lönnberg, T. A practical guide to single-cell RNA-sequencing for biomedical research and clinical applications. Genome Med. 2017, 9, 75. [Google Scholar] [CrossRef]
- Yao, C.; Guan, X.; Carraro, G.; Parimon, T.; Liu, X.; Huang, G.; Mulay, A.; Soukiasian, H.J.; David, G.; Weigt, S.S.; et al. Senescence of Alveolar Type 2 Cells Drives Progressive Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2021, 203, 707–717. [Google Scholar] [CrossRef] [PubMed]
- Adams, T.S.; Schupp, J.C.; Poli, S.; Ayaub, E.A.; Neumark, N.; Ahangari, F.; Chu, S.G.; Raby, B.A.; DeIuliis, G.; Januszyk, M.; et al. Single-cell RNA-seq reveals ectopic and aberrant lung-resident cell populations in idiopathic pulmonary fibrosis. Sci. Adv. 2020, 6, eaba1983. [Google Scholar] [CrossRef]
- Moss, B.J.; Ryter, S.W.; Rosas, I.O. Pathogenic Mechanisms Underlying Idiopathic Pulmonary Fibrosis. Annu. Rev. Pathol. 2020, 17, 515–546. [Google Scholar] [CrossRef] [PubMed]
- Sakamuri, S.S.V.P.; Sperling, J.A.; Sure, V.N.; Dholakia, M.H.; Peterson, N.R.; Rutkai, I.; Mahalingam, P.S.; Satou, R.; Katakam, P.V.G. Measurement of respiratory function in isolated cardiac mitochondria using Seahorse XFe24 Analyzer: Applications for aging research. Geroscience 2018, 40, 347. [Google Scholar] [CrossRef]
- Sure, V.N.; Sakamuri, S.S.V.P.; Sperling, J.A.; Evans, W.R.; Merdzo, I.; Mostany, R.; Murfee, W.L.; Busija, D.W.; Katakam, P.V.G. A novel high-throughput assay for respiration in isolated brain microvessels reveals impaired mitochondrial function in the aged mice. Geroscience 2018, 40, 365–375. [Google Scholar] [CrossRef] [PubMed]
- Bueno, M.; Zank, D.; Buendia-Roldán, I.; Fiedler, K.; Mays, B.G.; Alvarez, D.; Sembrat, J.; Kimball, B.; Bullock, J.K.; Martin, J.L.; et al. PINK1 attenuates mtDNA release in alveolar epithelial cells and TLR9 mediated profibrotic responses. PLoS ONE 2019, 14, e0218003. [Google Scholar] [CrossRef] [PubMed]
- Bueno, M.; Rojas, M. Lost in Translation: Endoplasmic Reticulum-Mitochondria Crosstalk in Idiopathic Pulmonary Fibrosis. Am. J. Respir. Cell Mol. Biol. 2020, 63, 408–409. [Google Scholar] [CrossRef]
- Quirós, P.M.; Mottis, A.; Auwerx, J. Mitonuclear communication in homeostasis and stress. Nat. Rev. Mol. Cell Biol. 2016, 17, 213–226. [Google Scholar] [CrossRef]
- Bueno, M.; Brands, J.; Voltz, L.; Fiedler, K.; Mays, B.; Croix, C.S.; Sembrat, J.; Mallampalli, R.K.; Rojas, M.; Mora, A.L. ATF3 represses PINK1 gene transcription in lung epithelial cells to control mitochondrial homeostasis. Aging Cell 2018, 17, e12720. [Google Scholar] [CrossRef] [PubMed]
- Jiang, D.; Cui, H.; Xie, N.; Banerjee, S.; Liu, R.-M.; Dai, H.; Thannickal, V.J.; Liu, G. ATF4 Mediates Mitochondrial Unfolded Protein Response in Alveolar Epithelial Cells. Am. J. Respir. Cell Mol. Biol. 2020, 63, 478–489. [Google Scholar] [CrossRef] [PubMed]
- Caporarello, N.; A Meridew, J.; Jones, D.L.; Tan, Q.; Haak, A.J.; Choi, K.M.; Manlove, L.J.; Prakash, Y.S.; Tschumperlin, D.J.; Ligresti, G. PGC1α repression in IPF fibroblasts drives a pathologic metabolic, secretory and fibrogenic state. Thorax 2019, 74, 749–760. [Google Scholar] [CrossRef]
- Pope, J.E.; Denton, C.P.; Johnson, S.R.; Fernandez-Codina, A.; Hudson, M.; Nevskaya, T. State-of-the-art evidence in the treatment of systemic sclerosis. Nat. Rev. Rheumatol. 2023, 19, 212–226. [Google Scholar] [CrossRef] [PubMed]
- Akamata, K.; Wei, J.; Bhattacharyya, M.; Cheresh, P.; Bonner, M.Y.; Arbiser, J.L.; Raparia, K.; Gupta, M.P.; Kamp, D.W.; Varga, J. SIRT3 is attenuated in systemic sclerosis skin and lungs, and its pharmacologic activation mitigates organ fibrosis. Oncotarget 2016, 7, 69321–69336. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, C.N.; Baughman, J.M.; Phu, L.; Tea, J.S.; Yu, C.; Coons, M.; Kirkpatrick, D.S.; Bingol, B.; Corn, J.E. USP30 and parkin homeostatically regulate atypical ubiquitin chains on mitochondria. Nat. Cell Biol. 2015, 17, 160–169. [Google Scholar] [CrossRef] [PubMed]
- Roque, W.; Cuevas-Mora, K.; Romero, F. Mitochondrial Quality Control in Age-Related Pulmonary Fibrosis. Int. J. Mol. Sci. 2020, 21, 643. [Google Scholar] [CrossRef]
- Jain, M.; Rivera, S.; Monclus, E.A.; Synenki, L.; Zirk, A.; Eisenbart, J.; Feghali-Bostwick, C.; Mutlu, G.M.; Budinger, G.R.S.; Chandel, N.S. Mitochondrial reactive oxygen species regulate transforming growth factor-β signaling. J. Biol. Chem. 2013, 288, 770–777. [Google Scholar] [CrossRef]
- Rangarajan, S.; Bone, N.B.; Zmijewska, A.A.; Jiang, S.; Park, D.W.; Bernard, K.; Locy, M.L.; Ravi, S.; Deshane, J.; Mannon, R.B.; et al. Metformin reverses established lung fibrosis in a bleomycin model. Nat. Med. 2018, 24, 1121–1127. [Google Scholar] [CrossRef]

{kind=link}
| Cellular Type | Mitochondrial Dysfunction Features |
|---|---|
| ATII [29,36] | Increased:
|
| Fibroblasts [59] | Increased:
|
| Myofibroblasts [54,60] | Increased:
|
| Macrophages [57,58] | Increased:
|
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cala-Garcia, J.D.; Medina-Rincon, G.J.; Sierra-Salas, P.A.; Rojano, J.; Romero, F. The Role of Mitochondrial Dysfunction in Idiopathic Pulmonary Fibrosis: New Perspectives for a Challenging Disease. Biology 2023, 12, 1237. https://doi.org/10.3390/biology12091237
Cala-Garcia JD, Medina-Rincon GJ, Sierra-Salas PA, Rojano J, Romero F. The Role of Mitochondrial Dysfunction in Idiopathic Pulmonary Fibrosis: New Perspectives for a Challenging Disease. Biology. 2023; 12(9):1237. https://doi.org/10.3390/biology12091237
Chicago/Turabian StyleCala-Garcia, Juan David, German Jose Medina-Rincon, Paula Andrea Sierra-Salas, Julio Rojano, and Freddy Romero. 2023. "The Role of Mitochondrial Dysfunction in Idiopathic Pulmonary Fibrosis: New Perspectives for a Challenging Disease" Biology 12, no. 9: 1237. https://doi.org/10.3390/biology12091237
APA StyleCala-Garcia, J. D., Medina-Rincon, G. J., Sierra-Salas, P. A., Rojano, J., & Romero, F. (2023). The Role of Mitochondrial Dysfunction in Idiopathic Pulmonary Fibrosis: New Perspectives for a Challenging Disease. Biology, 12(9), 1237. https://doi.org/10.3390/biology12091237