

Phenotypic Characterization of Female Carrier Mice Heterozygous for Tafazzin Deletion
 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Mice
2.2. Comprehensive Laboratory Animal Monitoring System Measures
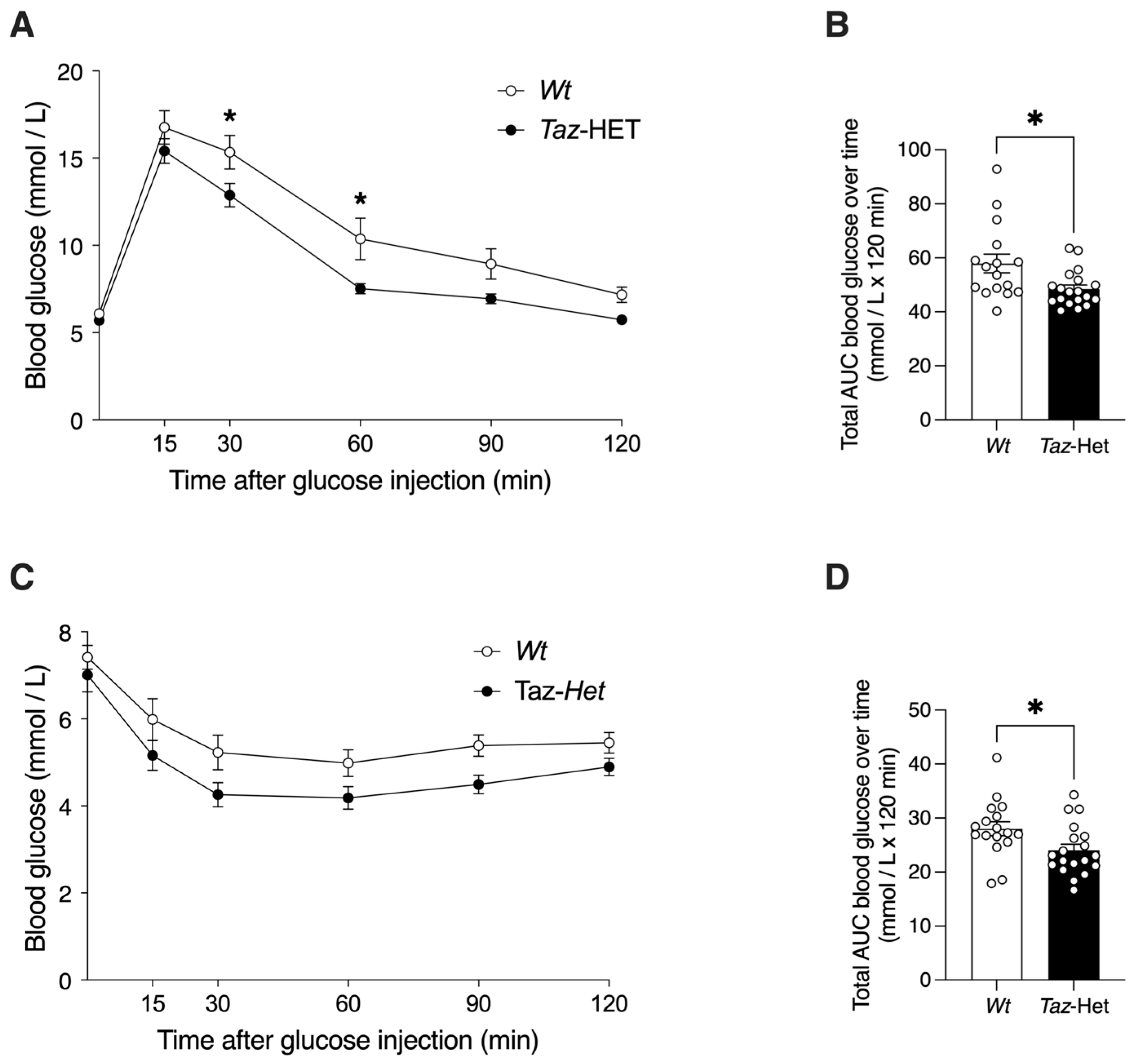
2.3. Glucose Tolerance Testing (GTT)
2.4. Insulin Tolerance Testing (ITT)
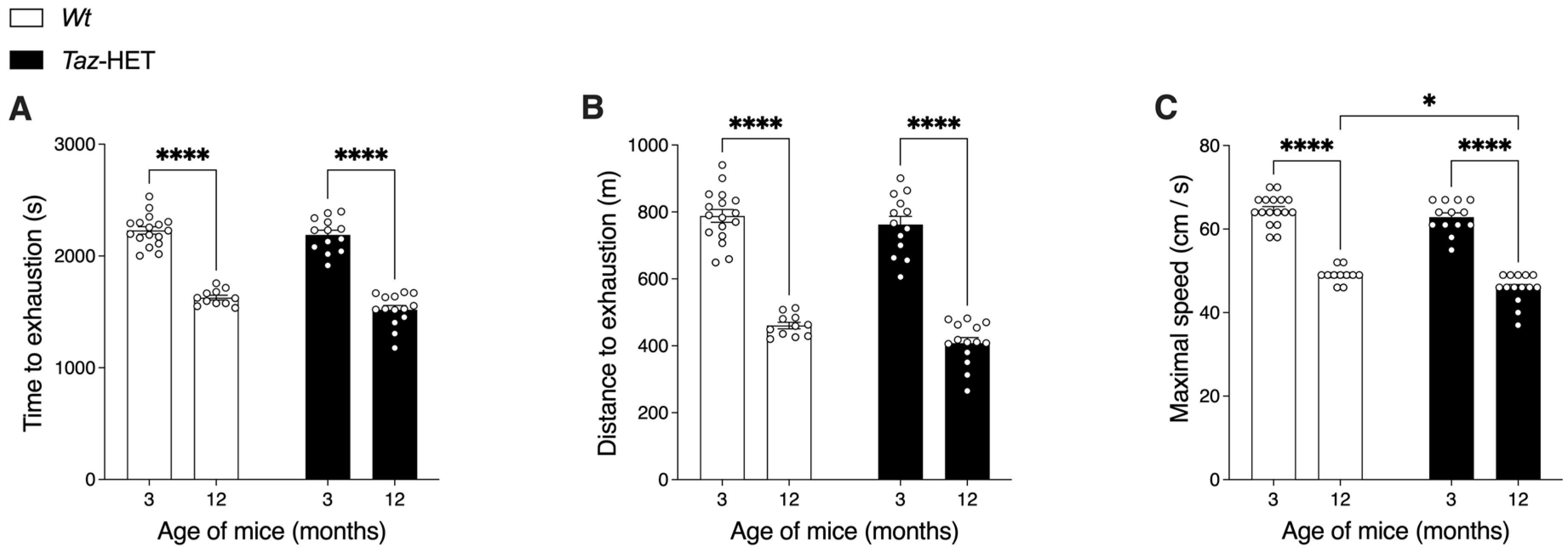
2.5. Treadmill Exercise Capacity Test
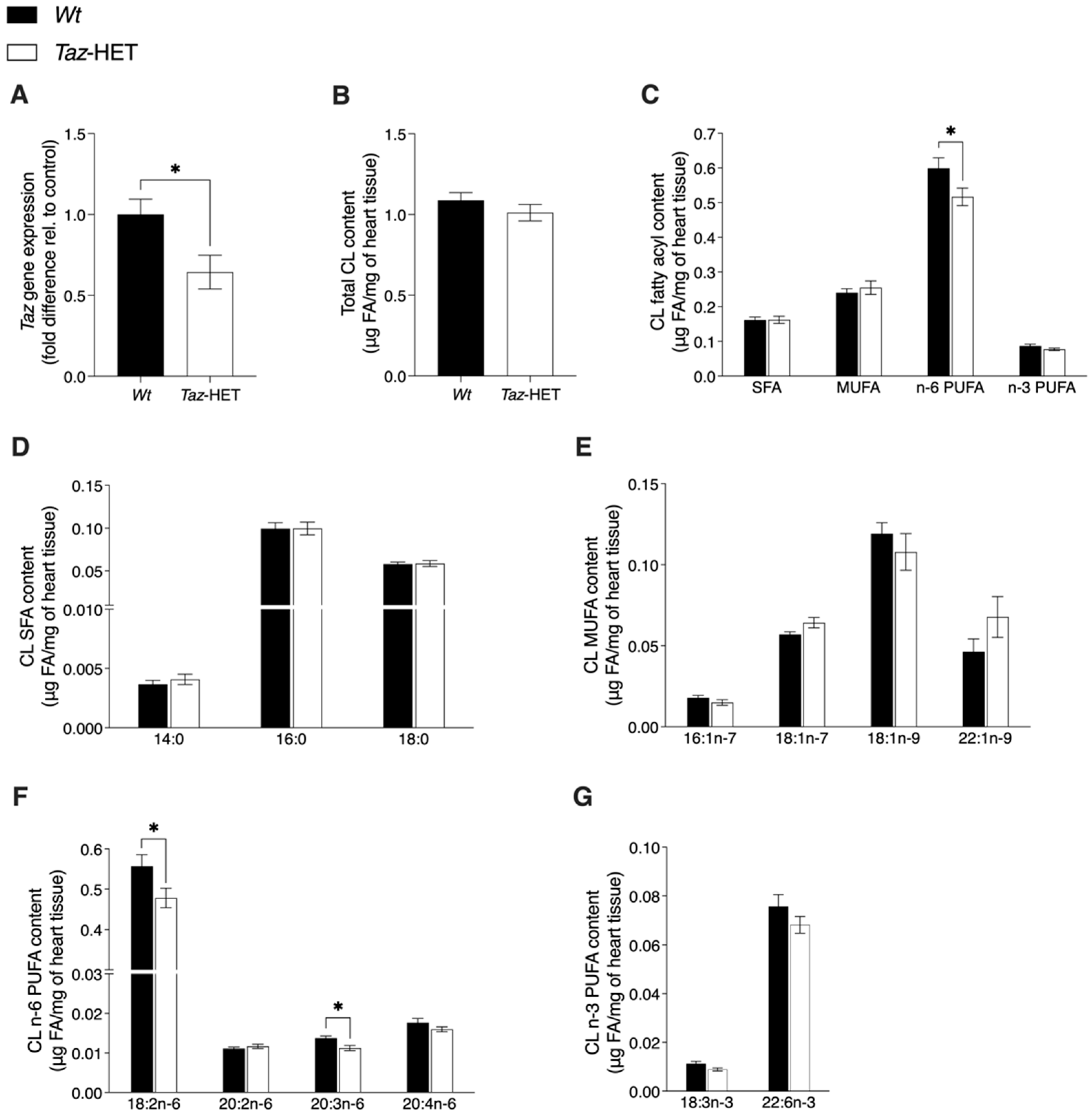
2.6. Tafazzin Gene Expression
2.7. Gas Chromatography Analysis of Cardiolipin
2.8. Statistical Analyses
3. Results
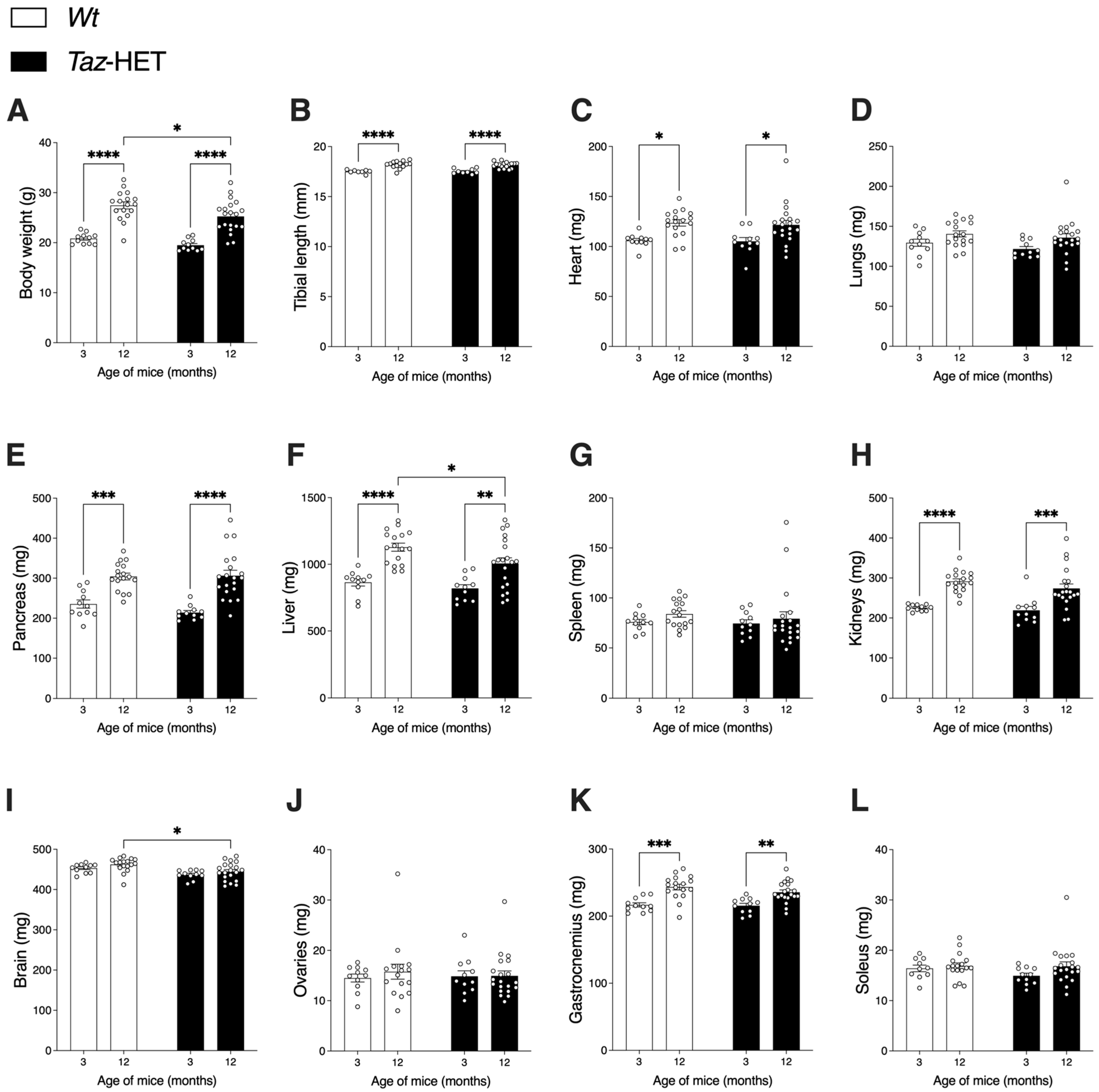
3.1. Body, Organ, and Lean Tissue Weights
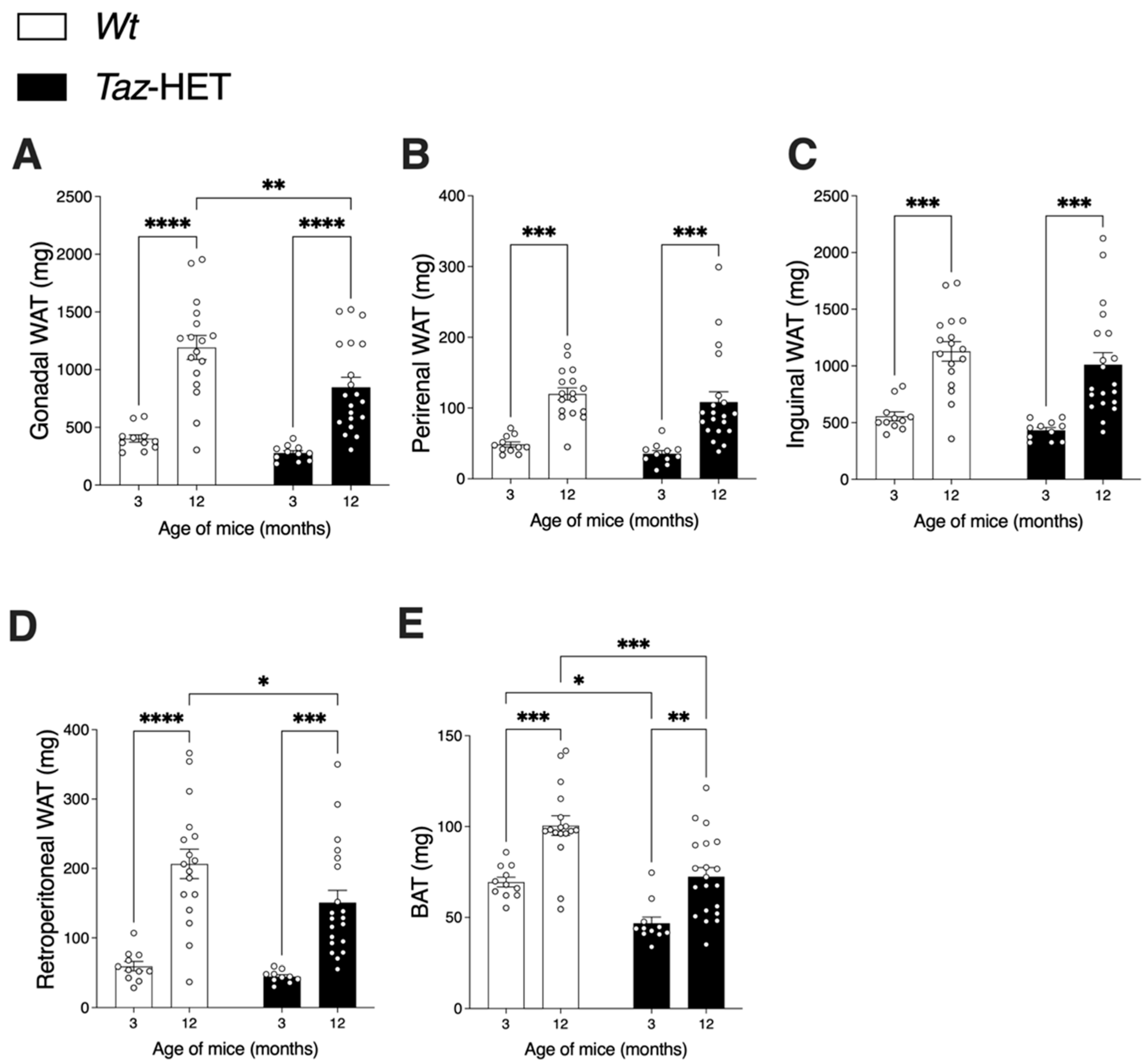
3.2. Adipose Tissue Masses
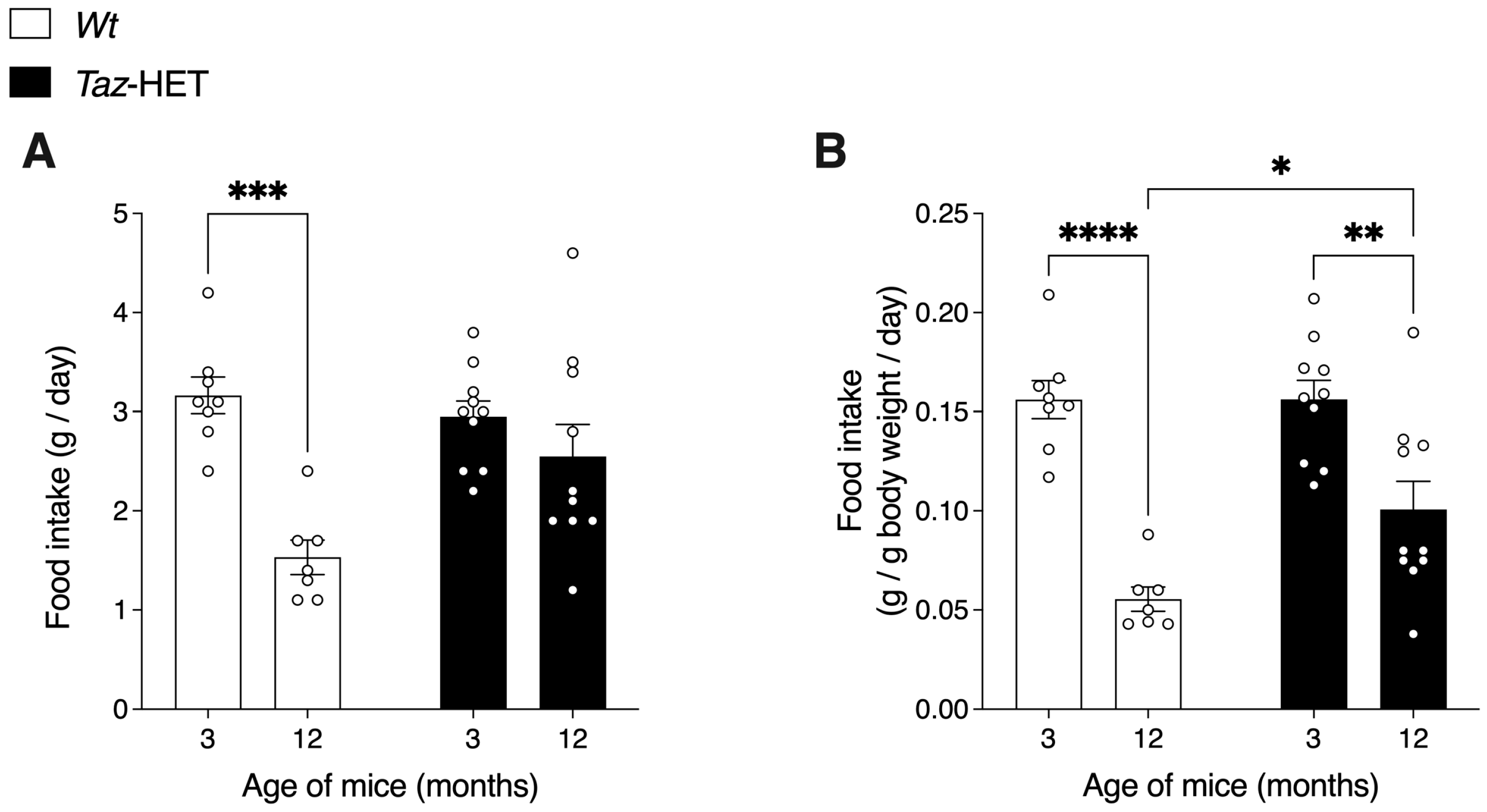
3.3. Food Intake
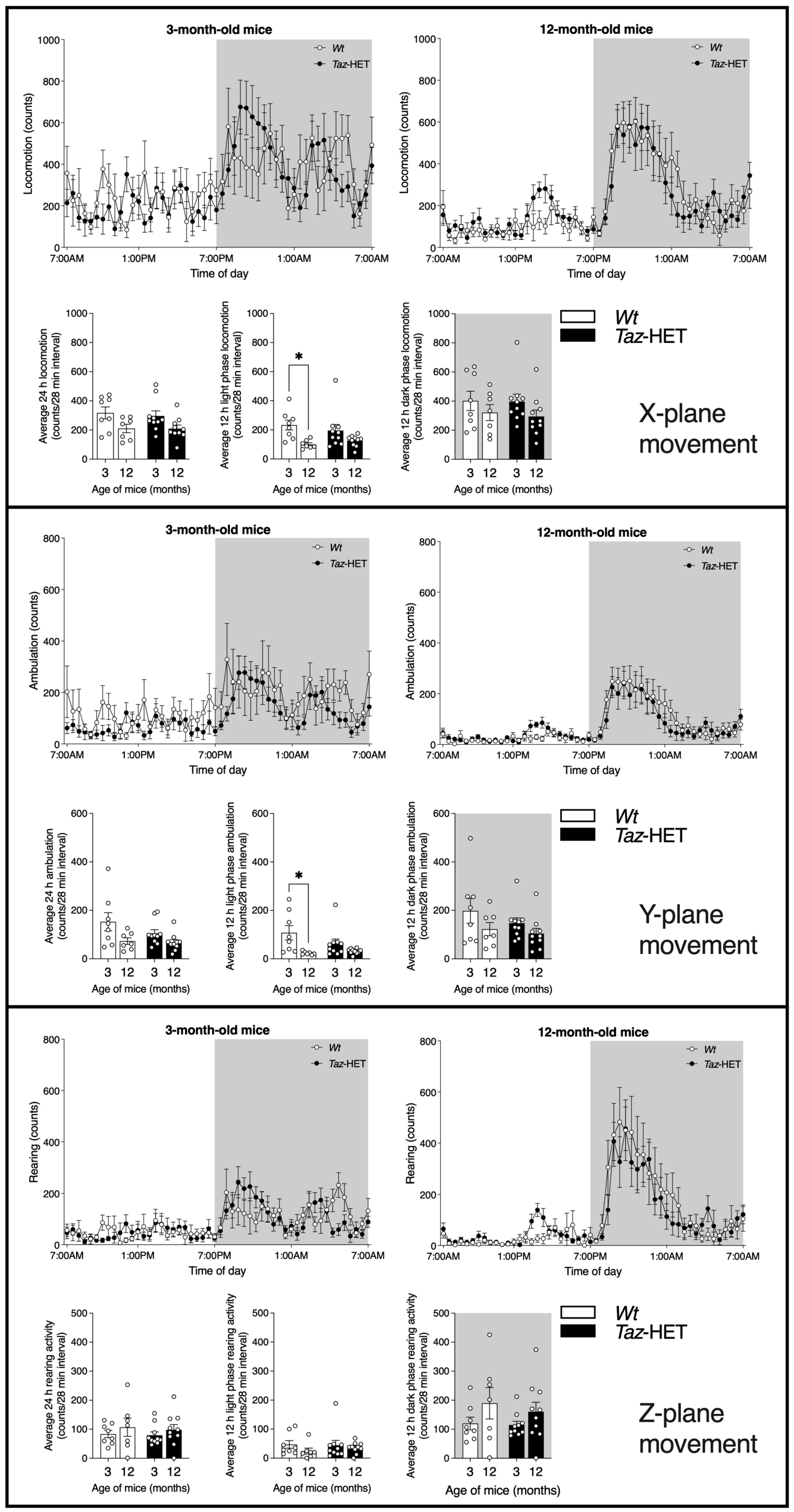
3.4. Movement
3.5. Respiratory Exchange Ratio and Total Energy Expenditure
3.6. VO2 and VCO2
3.7. Glucose Tolerance and Insulin Sensitivity
3.8. Exercise Endurance Capacity
3.9. Tafazzin Gene Expression and Cardiolipin Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pennington, E.R.; Funai, K.; Brown, D.A.; Shaikh, S.R. The role of cardiolipin concentration and acyl chain composition on mitochondrial inner membrane molecular organization and function. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2019, 1864, 1039–1052. [Google Scholar] [CrossRef] [PubMed]
- Bradley, R.M.; Stark, K.D.; Duncan, R.E. Influence of tissue, diet, and enzymatic remodeling on cardiolipin fatty acyl profile. Mol. Nutr. Food Res. 2016, 60, 1804–1818. [Google Scholar] [CrossRef]
- Han, X.; Yang, K.; Yang, J.; Cheng, H.; Gross, R.W. Shotgun lipidomics of cardiolipin molecular species in lipid extracts of biological samples. J. Lipid Res. 2006, 47, 864–879. [Google Scholar] [CrossRef] [PubMed]
- Schlame, M.; Towbin, J.A.; Heerdt, P.M.; Jehle, R.; DiMauro, S.; Blanck, T.J. Deficiency of tetralinoleoyl-cardiolipin in Barth syndrome. Ann. Neurol. 2002, 51, 634–637. [Google Scholar] [CrossRef] [PubMed]
- Acehan, D.; Vaz, F.; Houtkooper, R.H.; James, J.; Moore, V.; Tokunaga, C.; Kulik, W.; Wansapura, J.; Toth, M.J.; Strauss, A.; et al. Cardiac and skeletal muscle defects in a mouse model of human Barth syndrome. J. Biol. Chem. 2011, 286, 899–908. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, E.P.; Weiss, S.B. The function of cytidine coenzymes in the biosynthesis of phospholipides. J. Biol. Chem. 1956, 222, 193–214. [Google Scholar] [CrossRef]
- Bione, S.; D’Adamo, P.; Maestrini, E.; Gedeon, A.K.; Bolhuis, P.A.; Toniolo, D. A novel X-linked gene, G4.5. is responsible for Barth syndrome. Nat. Genet. 1996, 12, 385–389. [Google Scholar] [CrossRef]
- Bradley, R.M.; Hashemi, A.; Aristizabal-Henao, J.J.; Stark, K.D.; Duncan, R.E. PLAAT1 Exhibits Phosphatidylcholine: Monolysocardiolipin Transacylase Activity. Int. J. Mol. Sci. 2022, 23, 6714. [Google Scholar] [CrossRef]
- Ma, B.J.; Taylor, W.A.; Dolinsky, V.W.; Hatch, G.M. Acylation of monolysocardiolipin in rat heart. J. Lipid Res. 1999, 40, 1837–1845. [Google Scholar] [CrossRef]
- Taylor, W.A.; Hatch, G.M. Identification of the human mitochondrial linoleoyl-coenzyme A monolysocardiolipin acyltransferase (MLCL AT-1). J. Biol. Chem. 2009, 284, 30360–30371. [Google Scholar] [CrossRef]
- Taylor, W.A.; Mejia, E.M.; Mitchell, R.W.; Choy, P.C.; Sparagna, G.C.; Hatch, G.M. Human trifunctional protein alpha links cardiolipin remodeling to beta-oxidation. PLoS ONE 2012, 7, e48628. [Google Scholar] [CrossRef]
- Cao, J.; Liu, Y.; Lockwood, J.; Burn, P.; Shi, Y. A novel cardiolipin-remodeling pathway revealed by a gene encoding an endoplasmic reticulum-associated acyl-CoA:lysocardiolipin acyltransferase (ALCAT1) in mouse. J. Biol. Chem. 2004, 279, 31727–31734. [Google Scholar] [CrossRef] [PubMed]
- Zegallai, H.M.; Hatch, G.M. Barth syndrome: Cardiolipin, cellular pathophysiology, management, and novel therapeutic targets. Mol. Cell. Biochem. 2021, 476, 1605–1629. [Google Scholar] [CrossRef]
- Clarke, S.L.; Bowron, A.; Gonzalez, I.L.; Groves, S.J.; Newbury-Ecob, R.; Clayton, N.; Martin, R.P.; Tsai-Goodman, B.; Garratt, V.; Ashworth, M.; et al. Barth syndrome. Orphanet J. Rare Dis. 2013, 8, 23. [Google Scholar] [CrossRef] [PubMed]
- Rigaud, C.; Lebre, A.-S.; Touraine, R.; Beaupain, B.; Ottolenghi, C.; Chabli, A.; Ansquer, H.; Ozsahin, H.; Di Filippo, S.; De Lonlay, P.; et al. Natural history of Barth syndrome: A national cohort study of 22 patients. Orphanet J. Rare Dis. 2013, 8, 70. [Google Scholar] [CrossRef] [PubMed]
- Huhta, J.C.; Pomerance, H.H.; Barness, E.G. Clinicopathologic conference: Barth Syndrome. Fetal Pediatr. Pathol. 2005, 24, 239–254. [Google Scholar] [CrossRef] [PubMed]
- Engelen, M.; Barbier, M.; Dijkstra, I.M.; Schür, R.; de Bie, R.M.; Verhamme, C.; Dijkgraaf, M.G.; Aubourg, P.A.; Wanders, R.J.; van Geel, B.M.; et al. X-linked adrenoleukodystrophy in women: A cross-sectional cohort study. Brain 2014, 137, 693–706. [Google Scholar] [CrossRef]
- Wang, S.; Li, Y.; Xu, Y.; Ma, Q.; Lin, Z.; Schlame, M.; Bezzerides, V.J.; Strathdee, D.; Pu, W.T. AAV Gene Therapy Prevents and Reverses Heart Failure in a Murine Knockout Model of Barth Syndrome. Circ. Res. 2020, 126, 1024–1039. [Google Scholar] [CrossRef]
- Ren, M.; Xu, Y.; Erdjument-Bromage, H.; Donelian, A.; Phoon, C.K.L.; Terada, N.; Strathdee, D.; Neubert, T.A.; Schlame, M. Extramitochondrial cardiolipin suggests a novel function of mitochondria in spermatogenesis. J. Cell Biol. 2019, 218, 1491–1502. [Google Scholar] [CrossRef]
- Tomczewski, M.V.; Chan, J.Z.; Campbell, Z.E.; Strathdee, D.; Duncan, R.E. Phenotypic Characterization of Male Tafazzin-Knockout Mice at 3, 6, and 12 Months of Age. Biomedicines 2023, 11, 638. [Google Scholar] [CrossRef]
- Even, P.C.; Nadkarni, N.A. Indirect calorimetry in laboratory mice and rats: Principles, practical considerations, interpretation and perspectives. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2012, 303, R459–R476. [Google Scholar] [CrossRef] [PubMed]
- Lusk, G. ANIMAL CALORIMETRY Twenty-Fourth Paper. Analysis of the oxidation of mixtures of carbohydrate and fat. J. Biol. Chem. 1924, 59, 41–42. [Google Scholar] [CrossRef]
- Virtue, S.; Vidal-Puig, A. GTTs and ITTs in mice: Simple tests, complex answers. Nat. Metab. 2021, 3, 883–886. [Google Scholar] [CrossRef] [PubMed]
- Handschin, C.; Chin, S.; Li, P.; Liu, F.; Maratos-Flier, E.; LeBrasseur, N.K.; Yan, Z.; Spiegelman, B.M. Skeletal Muscle Fiber-type Switching, Exercise Intolerance, and Myopathy in PGC-1α Muscle-specific Knock-out Animals. J. Biol. Chem. 2007, 282, 30014–30021. [Google Scholar] [CrossRef] [PubMed]
- Claghorn, G.C.; Fonseca, I.A.T.; Thompson, Z.; Barber, C.; Garland, T. Serotonin-mediated central fatigue underlies increased endurance capacity in mice from lines selectively bred for high voluntary wheel running. Physiol. Behav. 2016, 161, 145–154. [Google Scholar] [CrossRef]
- Chan, J.Z.; Fernandes, M.F.; Steckel, K.E.; Bradley, R.M.; Hashemi, A.; Groh, M.R.; Sciaini, G.; Stark, K.D.; Duncan, R.E. N-oleoylethanolamide treatment of lymphoblasts deficient in Tafazzin improves cell growth and mitochondrial morphology and dynamics. Sci. Rep. 2022, 12, 9466. [Google Scholar] [CrossRef]
- Schlame, M.; Kelley, R.I.; Feigenbaum, A.; Towbin, J.A.; Heerdt, P.M.; Schieble, T.; Wanders, R.J.; DiMauro, S.; Blanck, T.J. Phospholipid abnormalities in children with Barth syndrome. J. Am. Coll. Cardiol. 2003, 42, 1994–1999. [Google Scholar] [CrossRef]
- Houtkooper, R.H.; Rodenburg, R.J.; Thiels, C.; van Lenthe, H.; Stet, F.; Poll-The, B.T.; Stone, J.E.; Steward, C.G.; Wanders, R.J.; Smeitink, J.; et al. Cardiolipin and monolysocardiolipin analysis in fibroblasts, lymphocytes, and tissues using high-performance liquid chromatography-mass spectrometry as a diagnostic test for Barth syndrome. Anal. Biochem. 2009, 387, 230–237. [Google Scholar] [CrossRef]
- Vernon, H.J.; Sandlers, Y.; McClellan, R.; Kelley, R.I. Clinical laboratory studies in Barth Syndrome. Mol. Genet. Metab. 2014, 112, 143–147. [Google Scholar] [CrossRef]
- Płatek, T.; Sordyl, M.; Polus, A.; Olszanecka, A.; Kroczka, S.; Solnica, B. Analysis of tafazzin and deoxyribonuclease 1 like 1 transcripts and X chromosome sequencing in the evaluation of the effect of mosaicism in the TAZ gene on phenotypes in a family affected by Barth syndrome. Mutat. Res. 2023, 826, 111812. [Google Scholar] [CrossRef]
- Johnston, J.; Kelley, R.I.; Feigenbaum, A.; Cox, G.F.; Iyer, G.S.; Funanage, V.L.; Proujansky, R. Mutation characterization and genotype-phenotype correlation in Barth syndrome. Am. J. Hum. Genet. 1997, 61, 1053–1058. [Google Scholar] [CrossRef]
- Orstavik, K.H.; Orstavik, R.E.; Naumova, A.K.; D’Adamo, P.; Gedeon, A.; Bolhuis, P.A.; Barth, P.G.; Toniolo, D. X chromosome inactivation in carriers of Barth syndrome. Am. J. Hum. Genet. 1998, 63, 1457–1463. [Google Scholar] [CrossRef] [PubMed]
- Bakšienė, M.; Benušienė, E.; Morkūnienė, A.; Ambrozaitytė, L.; Utkus, A.; Kučinskas, V. A novel intronic splice site tafazzin gene mutation detected prenatally in a family with Barth syndrome. Balkan. J. Med. Genet. 2016, 19, 95–100. [Google Scholar] [CrossRef] [PubMed]
- Barth, P.G.; Scholte, H.R.; Berden, J.A.; Van der Klei-Van Moorsel, J.M.; Luyt-Houwen, I.E.; Van’t Veer-Korthof, E.T.; Van der Harten, J.J.; Sobotka-Plojhar, M.A. An X-linked mitochondrial disease affecting cardiac muscle, skeletal muscle and neutrophil leucocytes. J. Neurol. Sci. 1983, 62, 327–355. [Google Scholar] [CrossRef] [PubMed]
- Cosson, L.; Toutain, A.; Simard, G.; Kulik, W.; Matyas, G.; Guichet, A.; Blasco, H.; Maakaroun-Vermesse, Z.; Vaillant, M.C.; Le Caignec, C.; et al. Barth syndrome in a female patient. Mol. Genet. Metab. 2012, 106, 115–120. [Google Scholar] [CrossRef]
- Ichida, F.; Tsubata, S.; Bowles, K.R.; Haneda, N.; Uese, K.; Miyawaki, T.; Dreyer, W.J.; Messina, J.; Li, H.; Bowles, N.E.; et al. Novel gene mutations in patients with left ventricular noncompaction or Barth syndrome. Circulation 2001, 103, 1256–1263. [Google Scholar] [CrossRef]
- Alquier, T.; Poitout, V. Considerations and guidelines for mouse metabolic phenotyping in diabetes research. Diabetologia 2018, 61, 526–538. [Google Scholar] [CrossRef]
- Brewer, R.A.; Gibbs, V.K.; Smith, D.L., Jr. Targeting glucose metabolism for healthy aging. Nutr. Healthy Aging 2016, 4, 31–46. [Google Scholar] [CrossRef]
- Orstavik, K.H. X chromosome inactivation in clinical practice. Hum. Genet. 2009, 126, 363–373. [Google Scholar] [CrossRef]
- Schlame, M.; Ren, M.; Xu, Y.; Greenberg, M.L.; Haller, I. Molecular symmetry in mitochondrial cardiolipins. Chem. Phys. Lipids 2005, 138, 38–49. [Google Scholar] [CrossRef]
- Soustek, M.S.; Falk, D.J.; Mah, C.S.; Toth, M.J.; Schlame, M.; Lewin, A.S.; Byrne, B.J. Characterization of a transgenic short hairpin RNA-induced murine model of Tafazzin deficiency. Hum. Gene Ther. 2011, 22, 865–871. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.; Chen, Z.e.; Zhu, M.; Shen, Y.; Leon, L.J.; Chi, L.; Spinozzi, S.; Tan, C.; Gu, Y.; Nguyen, A.; et al. Cardiolipin Remodeling Defects Impair Mitochondrial Architecture and Function in a Murine Model of Barth Syndrome Cardiomyopathy. Circ. Heart Fail. 2021, 14, e008289. [Google Scholar] [CrossRef]
- Powers, C.; Huang, Y.; Strauss, A.; Khuchua, Z. Diminished Exercise Capacity and Mitochondrial bc1 Complex Deficiency in Tafazzin-Knockdown Mice. Front. Physiol. 2013, 4, 74. [Google Scholar] [CrossRef] [PubMed]
- Bashir, A.; Bohnert, K.L.; Reeds, D.N.; Peterson, L.R.; Bittel, A.J.; de Las Fuentes, L.; Pacak, C.A.; Byrne, B.J.; Cade, W.T. Impaired cardiac and skeletal muscle bioenergetics in children, adolescents, and young adults with Barth syndrome. Physiol. Rep. 2017, 5, e13130. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Phoon, C.K.L.; Ren, M.; Schlame, M. A simple mechanistic explanation for Barth syndrome and cardiolipin remodeling. J. Inherit. Metab. Dis. 2022, 45, 51–59. [Google Scholar] [CrossRef]
- Ozcelik, T. X chromosome inactivation and female predisposition to autoimmunity. Clin. Rev. Allergy Immunol. 2008, 34, 348–351. [Google Scholar] [CrossRef]
- Ostan, R.; Monti, D.; Gueresi, P.; Bussolotto, M.; Franceschi, C.; Baggio, G. Gender, aging and longevity in humans: An update of an intriguing/neglected scenario paving the way to a gender-specific medicine. Clin. Sci. 2016, 130, 1711–1725. [Google Scholar] [CrossRef]
- Avdjieva-Tzavella, D.M.; Todorova, A.P.; Kathom, H.M.; Ivanova, M.B.; Yordanova, I.T.; Todorov, T.P.; Litvinenko, I.O.; Dasheva-Dimitrova, A.T.; Tincheva, R.S. Barth Syndrome in Male and Female Siblings Caused by a Novel Mutation in the Taz Gene. Genet. Couns. 2016, 27, 495–501. [Google Scholar]
- Bachou, T.; Giannakopoulos, A.; Trapali, C.; Vazeou, A.; Kattamis, A. A novel mutation in the G4.5 (TAZ) gene in a Greek patient with Barth syndrome. Blood Cells Mol. Dis. 2009, 42, 262–264. [Google Scholar] [CrossRef]
- Brión, M.; de Castro López, M.J.; Santori, M.; Pérez Muñuzuri, A.; López Abel, B.; Baña Souto, A.M.; Martínez Soto, M.I.; Couce, M.L. Prospective and Retrospective Diagnosis of Barth Syndrome Aided by Next-Generation Sequencing. Am. J. Clin. Pathol. 2016, 145, 507–513. [Google Scholar] [CrossRef]
- Kuijpers, T.W.; Maianski, N.A.; Tool, A.T.; Becker, K.; Plecko, B.; Valianpour, F.; Wanders, R.J.; Pereira, R.; Van Hove, J.; Verhoeven, A.J.; et al. Neutrophils in Barth syndrome (BTHS) avidly bind annexin-V in the absence of apoptosis. Blood 2004, 103, 3915–3923. [Google Scholar] [CrossRef] [PubMed]
- Dutta, S.; Sengupta, P. Men and mice: Relating their ages. Life Sci. 2016, 152, 244–248. [Google Scholar] [CrossRef] [PubMed]
- Ishizaki, M.; Kobayashi, M.; Adachi, K.; Matsumura, T.; Kimura, E. Female dystrophinopathy: Review of current literature. Neuromuscul. Disord. 2018, 28, 572–581. [Google Scholar] [CrossRef] [PubMed]
- Politano, L.; Nigro, V.; Nigro, G.; Petretta, V.R.; Passamano, L.; Papparella, S.; Di Somma, S.; Comi, L.I. Development of cardiomyopathy in female carriers of Duchenne and Becker muscular dystrophies. Jama 1996, 275, 1335–1338. [Google Scholar] [CrossRef]
- Adachi, K.; Kawai, H.; Saito, M.; Naruo, T.; Kimura, C.; Mine, H.; Inui, T.; Kashiwagi, S.; Akaike, M. Plasma levels of brain natriuretic peptide as an index for evaluation of cardiac function in female gene carriers of Duchenne muscular dystrophy. Intern. Med. 1997, 36, 497–500. [Google Scholar] [CrossRef]
- Au, W.Y.; Ma, E.S.; Lam, V.M.; Chan, J.L.; Pang, A.; Kwong, Y.L. Glucose 6-phosphate dehydrogenase (G6PD) deficiency in elderly Chinese women heterozygous for G6PD variants. Am. J. Med. Genet. A 2004, 129a, 208–211. [Google Scholar] [CrossRef]
- Au, W.Y.; Lam, V.; Pang, A.; Lee, W.M.; Chan, J.L.; Song, Y.Q.; Ma, E.S.; Kwong, Y.L. Glucose-6-phosphate dehydrogenase deficiency in female octogenarians, nanogenarians, and centenarians. J. Gerontol. A Biol. Sci. Med. Sci. 2006, 61, 1086–1089. [Google Scholar] [CrossRef]
- Schmidt, S.; Träber, F.; Block, W.; Keller, E.; Pohl, C.; von Oertzen, J.; Schild, H.; Schlegel, U.; Klockgether, T. Phenotype assignment in symptomatic female carriers of X-linked adrenoleukodystrophy. J. Neurol. 2001, 248, 36–44. [Google Scholar] [CrossRef]
- Davis, L.E.; Snyder, R.D.; Orth, D.N.; Nicholson, W.E.; Kornfeld, M.; Seelinger, D.F. Adrenoleukodystrophy and adrenomyeloneuropathy associated with partial adrenal insufficiency in three generations of a kindred. Am. J. Med. 1979, 66, 342–347. [Google Scholar] [CrossRef]
- Heffungs, W.; Hameister, H.; Ropers, H.H. Addison disease and cerebral sclerosis in an apparently heterozygous girl: Evidence for inactivation of the adrenoleukodystrophy locus. Clin. Genet. 1980, 18, 184–188. [Google Scholar] [CrossRef]
- Jangouk, P.; Zackowski, K.M.; Naidu, S.; Raymond, G.V. Adrenoleukodystrophy in female heterozygotes: Underrecognized and undertreated. Mol. Genet. Metab. 2012, 105, 180–185. [Google Scholar] [CrossRef]
- Moser, H.W.; Moser, A.B.; Naidu, S.; Bergin, A. Clinical aspects of adrenoleukodystrophy and adrenomyeloneuropathy. Dev. Neurosci. 1991, 13, 254–261. [Google Scholar] [CrossRef]
- Costello, D.J.; Eichler, A.F.; Eichler, F.S. Leukodystrophies: Classification, diagnosis, and treatment. Neurologist 2009, 15, 319–328. [Google Scholar] [CrossRef] [PubMed]
- Dooley, J.M.; Wright, B.A. Adrenoleukodystrophy mimicking multiple sclerosis. Can. J. Neurol. Sci. 1985, 12, 73–74. [Google Scholar] [CrossRef] [PubMed]
- Stöckler, S.; Millner, M.; Molzer, B.; Ebner, F.; Körner, E.; Moser, H.W. Multiple sclerosis-like syndrome in a woman heterozygous for adrenoleukodystrophy. Eur. Neurol. 1993, 33, 390–392. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, B.P.; Moser, H.W.; Saxena, K.M.; Marmion, L.C. Adrenoleukodystrophy: Clinical and biochemical manifestations in carriers. Neurology 1984, 34, 798–801. [Google Scholar] [CrossRef]
- van Geel, B.M.; Koelman, J.H.; Barth, P.G.; Ongerboer de Visser, B.W. Peripheral nerve abnormalities in adrenomyeloneuropathy: A clinical and electrodiagnostic study. Neurology 1996, 46, 112–118. [Google Scholar] [CrossRef]
- Horn, M.A.; Retterstøl, L.; Abdelnoor, M.; Skjeldal, O.H.; Tallaksen, C.M. Adrenoleukodystrophy in Norway: High rate of de novo mutations and age-dependent penetrance. Pediatr. Neurol. 2013, 48, 212–219. [Google Scholar] [CrossRef]
- Jung, H.H.; Wimplinger, I.; Jung, S.; Landau, K.; Gal, A.; Heppner, F.L. Phenotypes of female adrenoleukodystrophy. Neurology 2007, 68, 960–961. [Google Scholar] [CrossRef]
- Maier, E.M.; Kammerer, S.; Muntau, A.C.; Wichers, M.; Braun, A.; Roscher, A.A. Symptoms in carriers of adrenoleukodystrophy relate to skewed X inactivation. Ann. Neurol. 2002, 52, 683–688. [Google Scholar] [CrossRef]
- Migeon, B.R.; Moser, H.W.; Moser, A.B.; Axelman, J.; Sillence, D.; Norum, R.A. Adrenoleukodystrophy: Evidence for X linkage, inactivation, and selection favoring the mutant allele in heterozygous cells. Proc. Natl. Acad. Sci. USA 1981, 78, 5066–5070. [Google Scholar] [CrossRef] [PubMed]
- Watkiss, E.; Webb, T.; Bundey, S. Is skewed X inactivation responsible for symptoms in female carriers for adrenoleucodystrophy? J. Med. Genet. 1993, 30, 651–654. [Google Scholar] [CrossRef] [PubMed]
- Flurkey, K.; Currer, J.M.; Harrison, D.E. Chapter 20—Mouse Models in Aging Research. In the Mouse in Biomedical Research, 2nd ed.; Fox, J.G., Davisson, M.T., Quimby, F.W., Barthold, S.W., Newcomer, C.E., Smith, A.L., Eds.; Academic Press: Burlington, NJ, USA, 2007; pp. 637–672. [Google Scholar]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tomczewski, M.V.; Chan, J.Z.; Al-Majmaie, D.M.; Liu, M.R.; Cocco, A.D.; Stark, K.D.; Strathdee, D.; Duncan, R.E. Phenotypic Characterization of Female Carrier Mice Heterozygous for Tafazzin Deletion. Biology 2023, 12, 1238. https://doi.org/10.3390/biology12091238
Tomczewski MV, Chan JZ, Al-Majmaie DM, Liu MR, Cocco AD, Stark KD, Strathdee D, Duncan RE. Phenotypic Characterization of Female Carrier Mice Heterozygous for Tafazzin Deletion. Biology. 2023; 12(9):1238. https://doi.org/10.3390/biology12091238
Chicago/Turabian StyleTomczewski, Michelle V., John Z. Chan, Duaa M. Al-Majmaie, Ming Rong Liu, Alex D. Cocco, Ken D. Stark, Douglas Strathdee, and Robin E. Duncan. 2023. "Phenotypic Characterization of Female Carrier Mice Heterozygous for Tafazzin Deletion" Biology 12, no. 9: 1238. https://doi.org/10.3390/biology12091238
APA StyleTomczewski, M. V., Chan, J. Z., Al-Majmaie, D. M., Liu, M. R., Cocco, A. D., Stark, K. D., Strathdee, D., & Duncan, R. E. (2023). Phenotypic Characterization of Female Carrier Mice Heterozygous for Tafazzin Deletion. Biology, 12(9), 1238. https://doi.org/10.3390/biology12091238