Simple Summary
This study provides a comprehensive review and evaluation of computational methods for the prediction of N6-methyladenosine (m6A) sites, crucial in regulating cellular functions and gene expression. Advances in high-confidence m6A site mapping have enabled the development of robust computational approaches. We assess 52 computational methods, including machine learning, deep learning, and ensemble-based techniques, using 13 benchmark datasets from nine different species. The evaluation reveals that deep learning methods generally surpass traditional scoring function-based approaches. This systematic analysis aims to guide the design and refinement of computational tools for m6A identification, facilitating rigorous method comparison and supporting future research in RNA modifications. The findings are vital in understanding m6A-dependent mRNA regulation, with implications in addressing diseases like cancer, where m6A plays a regulatory role.
Abstract
N6-methyladenosine (m6A) plays a crucial regulatory role in the control of cellular functions and gene expression. Recent advances in sequencing techniques for transcriptome-wide m6A mapping have accelerated the accumulation of m6A site information at a single-nucleotide level, providing more high-confidence training data to develop computational approaches for m6A site prediction. However, it is still a major challenge to precisely predict m6A sites using in silico approaches. To advance the computational support for m6A site identification, here, we curated 13 up-to-date benchmark datasets from nine different species (i.e., H. sapiens, M. musculus, Rat, S. cerevisiae, Zebrafish, A. thaliana, Pig, Rhesus, and Chimpanzee). This will assist the research community in conducting an unbiased evaluation of alternative approaches and support future research on m6A modification. We revisited 52 computational approaches published since 2015 for m6A site identification, including 30 traditional machine learning-based, 14 deep learning-based, and 8 ensemble learning-based methods. We comprehensively reviewed these computational approaches in terms of their training datasets, calculated features, computational methodologies, performance evaluation strategy, and webserver/software usability. Using these benchmark datasets, we benchmarked nine predictors with available online websites or stand-alone software and assessed their prediction performance. We found that deep learning and traditional machine learning approaches generally outperformed scoring function-based approaches. In summary, the curated benchmark dataset repository and the systematic assessment in this study serve to inform the design and implementation of state-of-the-art computational approaches for m6A identification and facilitate more rigorous comparisons of new methods in the future.
1. Introduction
N6-methyladenosine (m6A), the best-characterized RNA modification, is formed by the methylation of adenosine at position 6. This modification is the most abundant within mRNA in mammalian cells [1] and could be also found in primary miRNA (pri-miRNA) [2], long intergenic ncRNAs (lincRNAs) [3], and rRNA [4]. m6A has recently been shown to alter the nuclear export of RNA, RNA splicing, RNA stability, and translation efficiency [5]; moreover, it has been demonstrated to link to a wide range of cellular functions [6,7,8,9,10,11,12,13]. The aberrant regulation of m6A has also been implicated in a variety of cancers, such as lung cancer, liver cancer, breast cancer, glioblastoma, and acute myeloid leukemia [14,15], highlighting the crucial regulatory roles of m6A modification. Consequently, the precise identification of m6A modification sites at the transcriptome level is of vital importance to understand and explore underlying m6A-dependent mRNA regulation mechanisms and biological functions.
A variety of sequencing techniques have been developed for transcriptome-wide m6A mapping, spanning three important periods from non-single-nucleotide resolution to single-base resolution in bulk cellular populations to single-base resolution within single cells. During its first developmental period, methylated RNA immunoprecipitation and sequencing such as MeRIP-seq or m6A-seq [16] emerged as a classical technique widely used to detect m6A. However, this mapping technique only localizes m6A residues to approximate regions with a ~100 nucleotide (nt) length, and it cannot identify the exact positions of individual m6A sites at a transcriptome-wide level. This disadvantage was addressed during the second development period, when a variety of refined methods were developed to improve the resolution for whole-transcriptome m6A identification and quantification, such as PA-m6A-seq [17], miCLIP [18], UV-CLIP [19], m6A-REF-seq [20], and DART-seq [21]. Recently, a novel method called scDART-seq [22] has been developed to identify single-nucleotide m6A sites at a transcriptome-wide level in single cells, marking the advent of a more sophisticated m6A profiling era. Although such advanced experimental methods are too time-consuming and costly to perform genome-wide analysis, they open the door to research on precise m6A mapping and provide sufficient data for the development of in silico methods of m6A site identification.
During the past few years, a number of computational approaches have been developed for the prediction of m6A sites in the RNA of different species, such as H. sapiens, M. musculus, Chimpanzee, Rhesus, Pig, Rat, Zebrafish, D. melanogaster, S. cerevisiae, and A. thaliana (Supplementary Material Figure S1A). Such computational methods can be generally categorized into three groups according to their computational methodologies, including (i) traditional machine learning-based methods (accounting for 60%), (ii) deep learning-based methods (24%), and (iii) ensemble learning-based methods (16%) (Supplementary Material Figure S1B). Several surveys [23,24,25] of RNA methylation prediction have been published (Supplementary Material Figure S1C); however, there is almost no comprehensive and specific overview of m6A site prediction. Only one focuses on tools published prior to 2019 for the prediction of m6A sites in the RNA of S. cerevisiae [24]. More recent tools for S. cerevisiae, as well as predictors of other species, have not been systematically reviewed; in particular, there is a lack of comprehensive assessments of the prediction performance of the compared approaches via the implementation of extensive and independent benchmarking tests.
To overcome the issues mentioned above, here, we conduct a comparative and systematic analysis by summarizing the most up-to-date research progress in m6A site prediction. For this purpose, we have manually curated 13 up-to-date and large-scale benchmark datasets with high-confidence single-nucleotide m6A site sequences to accompany this comprehensive survey analysis, thereby helping the community to carry out an unbiased evaluation of alternative approaches and support future research on m6A modification. A total number of 52 computational methods, collected from Web of Science and PubMed with the keywords ‘m6A prediction’ or ‘m6A identification’, are carefully assessed, benchmarked, and extensively discussed in terms of feature extraction, model construction, performance evaluation strategies, and webserver/software usability. More importantly, using the 13 independent test datasets, which have never been seen in the training data of the existing approaches, we have systemically evaluated the generalization and robustness of the investigated methods with available online webservers or locally stand-alone software. We expect that the comparative analysis in this study will serve as a critical analysis of state-of-the-art m6A prediction approaches and pave the way for future development to accurately predict m6A sites in single cells.
The structure of this paper consists of four main parts. The first part provides an overview of the research background and significance of m6A site prediction, introducing existing sequencing technologies and computational methods. The second part systematically compares computational methods for m6A site prediction, including the construction of training datasets, feature engineering, and prediction algorithms. The third part presents the experimental results, covering species-specific predictions, cross-species validation, and the performance comparison of single-cell m6A site prediction. The fourth part concludes with a summary of the research findings and considerations for future work.
2. Systematic Comparison of Computational Approaches for m6A Site Prediction
2.1. Existing Methods for m6A Site Prediction
Among the 52 computational approaches for m6A site prediction analyzed in this study, 32 were designed and implemented based on m6A data at a non-single-nucleotide resolution. For the prediction of m6A sites in the S. cerevisiae genome, Chen et al. constructed a balanced dataset termed Smet1307, which consisted of 1307 positive samples and 1307 negative samples with a 51 nt length after removing sequence redundancy. They proposed the first computational predictor, iRNA-Methyl [26], in this field. To further improve the prediction accuracy, scholars have developed several predictors based on machine learning, such as support vector machine (SVM) [27,28,29,30,31,32], random forest (RF) [33], and eXtreme Gradient Boosting (XGBoost) [34], as well as ensemble learning [35,36], using various sequence-based feature extraction methods. Additionally, two SVM-based predictors [37,38] were successively developed based on a high-confidence subset, named Smet1307sub, which was generated after filtering m6A sites with distances to the detected m6A-seq peaks greater than 10 bp from the dataset Smet1307. Recently, a new deep learning-based predictor named iMethyl-Deep [39] was proposed using Smet1307 and another dataset, Smet3270 [24], which contained 3270 experimentally verified m6A sites and 3270 non-m6A sites.
For the prediction of m6A sites in the RNA of H. sapiens, Chen et al. first developed an SVM-based model called iRNA-PseColl [40] using a dataset, Hmet1130, which consisted of 1130 experimentally verified positive samples and 1130 negative samples with a 41 nt length. Moreover, this predictor could implement other prediction functions for m1A and m5C sites after being trained on the corresponding benchmark dataset. Based on Chen’s data, a deep learning-based model called iRNA-Mod-CNN [41] was proposed to further improve the prediction performance for m6A sites in the RNA of H. sapiens. For the identification of m6A sites in the A. thaliana transcriptome, Chen et al. developed an SVM-based predictor, M6ATH [42], based on a dataset, Amet394, that contained 394 experimentally verified m6A sites and 394 non-m6A sites. In the same year, a new model called AthMethPre [43] was developed on another larger dataset that covered 5081 experimentally verified positive samples and 5081 negative samples. Additionally, another 14 computational approaches [44,45,46,47,48,49,50,51,52,53,54,55,56,57] were proposed using m6A data at a non-single-nucleotide resolution from multiple species.
Seventeen computational approaches for m6A site prediction analyzed in this study were proposed based on single-nucleotide m6A data. Such computational models can be categorized into two types, namely species-specific predictors [58,59,60,61,62,63,64,65,66,67,68,69] and tissue-specific predictors [70,71,72,73,74]. Four public databases, namely RMVar [75], RMBase [76,77], Met-DB [78,79], and the Ensembl database “http://www.ensembl.org (accessed on 12 September 2022)”, were used as the mainstream data resources for training dataset construction for species-specific predictors, while the latest data generated by m6A-REF-seq [20] were used for the training of tissue-specific predictors. Significantly, among these predictors, MASS [68] and MultiRM [58] are the two most important and recommended methods in view of their innovation. MASS, a multi-task framework embedding a convolutional neural network (CNN) and bi-directional long short-term memory (BiLSTM), is the first computational method based on a multi-task curriculum learning strategy for the extraction of shared sequence features across multiple species in the prediction of m6A sites simultaneously. MultiRM, an attention-based multi-label deep learning framework, is the first computational model aimed at simultaneously predicting the putative sites of twelve widely occurring transcriptome modifications, including m6A modifications.
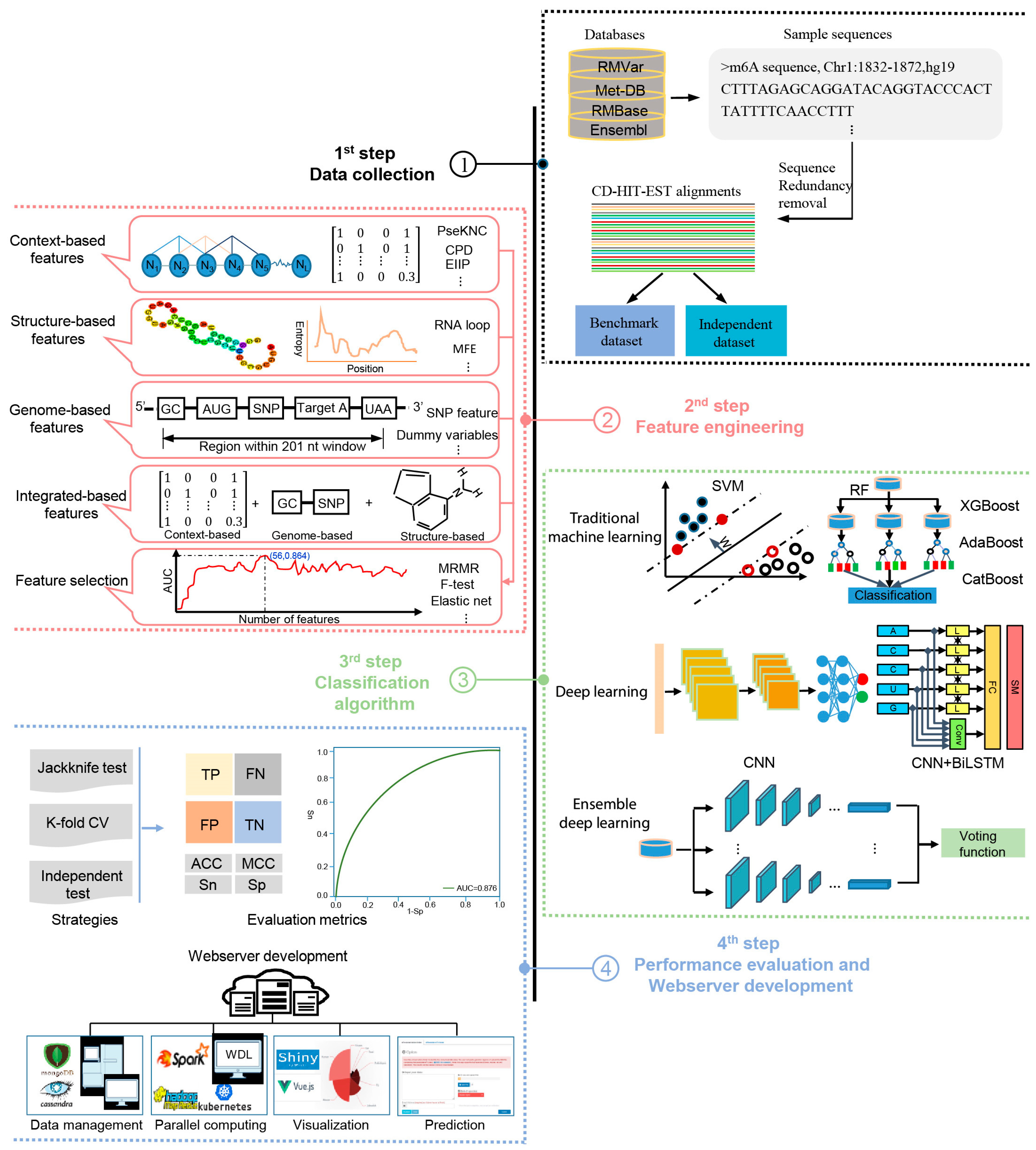
The methods analyzed in this study are systematically described and summarized in Table 1 in terms of the targeted species, sequence length, algorithm, features, evaluation strategy, webserver availability, and data size. The four generic steps used by these computational approaches to identify m6A sites are illustrated in Figure 1; these will be discussed in detail in the following sections.

Table 1.
Detailed information of existing models.
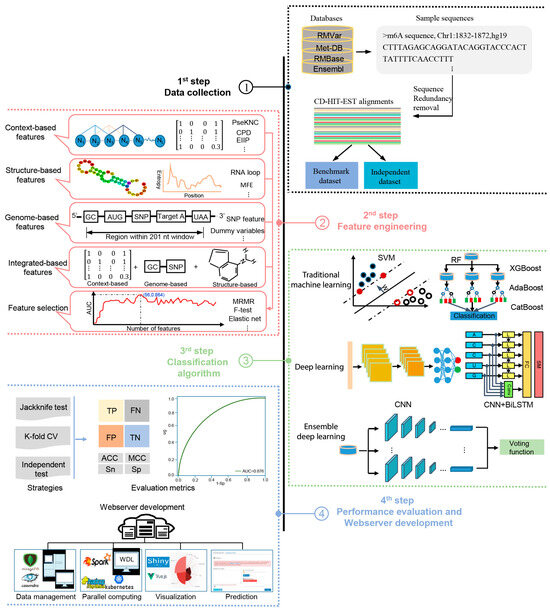
Figure 1.
A graphical illustration of four common steps in the construction and evaluation of computational approaches for the prediction of m6A sites.
2.2. Construction of Training Dataset
As mentioned above, the data used for training and benchmarking in m6A site prediction are composed of non-single-nucleotide data collected from the literature and single-nucleotide data derived from public databases (Table 2). The generic steps to construct the training dataset for m6A site prediction are given as follows. (i) m6A site information including the chromosome type, m6A site location, strand sequence, species, and reference genome version was collected from the literature or public databases. (ii) Based on such m6A site information, a (2ξ+1)-nt-long sliding window was used to extract sample sequences with adenosine at the center along each of the RNA segments. Only m6A sites containing the motif DRACH (where D = A, G or U, R = A or G, H =A, C, or U) were retained as positive samples, while those unmethylated adenosines with the motif DRACH along the whole transcriptome were used as negative samples. (iii) Subsequently, the CD-HIT-EST tool [83] was employed to remove those sequences with sequence similarity greater than a certain threshold (typically 80%) to reduce the redundancy in the sample sequences. (iv) Finally, negative samples were randomly sampled as a certain positive-to-negative ratio to construct a balanced or unbalanced dataset. (v) Generally, the obtained dataset could be divided into two subsets, including a training dataset and testing dataset. This construction process for training datasets could be also applicable to other post-transcriptional modifications and protein post-translational modifications.
Table 2.
Detailed information of public databases.
Table 2.
Detailed information of public databases.
| Database | Species | Latest Version | Feature | Website (URL) |
|---|---|---|---|---|
| Met-DB [78,79] | H. sapiens, M. musculus | 1.0 (November 2014) | MeT-DB is the first comprehensive resource for m6A in the mammalian transcriptome and provides ∼300 k m6A methylation sites in 74 MeRIP-Seq samples from 22 different experimental conditions. | http://compgenomics.utsa.edu/methylation/ (accessed on 12 September 2022) |
| RMBase [76,77] | H. sapiens, M. musculus, Rhesus, Chimpanzee, Rat, Pig, Zebrafish, S. cerevisiae, Fly, A. thaliana, S. pombe, E. coli, P. aetuginosa | 2.0 (October 2017) | RMBase v2.0 was expanded with ∼600 datasets and ∼1,397,000 modification sites from 47 studies among 13 species, including ∼1,373,000 m6A sites at a single nucleotide or very high resolution. | http://rna.sysu.edu.cn/rmbase/ (accessed on 12 September 2022) |
| RMVar [75] | H. sapiens, M. musculus | 2.0 (October 2020) | RMVar is an updated version of m6Avar and contains 179,270 high-confidence m6A sites from H. sapiens and 10,760 from M. musculus in total. | http://rmvar.renlab.org (accessed on 12 September 2022) |
| m6A-Atlas [84] | H. sapiens, M. musculus, A. thaliana, Fly, Rat, Yeast, Zebrafish, virus | 1.0 (August 2020) | m6A-Atlas is a comprehensive knowledge base for the unraveling of the m6A epitranscriptome and provides 442,162 high-confidence m6A sites identified from seven base-resolution technologies. | www.xjtlu.edu.cn/biologicalsciences/atlas (accessed on 12 September 2022) |
| ConsRM [65] | H. sapiens | 1.0 (February 2021) | ConsRM is a database on the collection and large-scale prediction of evolutionarily conserved RNA methylation sites and includes 177,998 base-resolution human m6A RNA methylation sites with ConsRM scores. | https://www.xjtlu.edu.cn/biologicalsciences/con (accessed on 12 September 2022) |
| Ensembl | H. sapiens, M. musculus | 106 (April 2022) | Ensembl annotates genes, collects disease data, and provides m6A site information from mammalian species. | https://asia.ensembl.org/index.html (accessed on 12 September 2022) |
2.3. Construction of Independent Test Dataset
In order to objectively evaluate the generalization performance of the current and existing methods, we collected single-base-resolution m6A sites recently validated by experiments and then constructed a total of 13 balanced independent test datasets after removing the overlapping sequences with the training datasets of the compared methods. The resulting independent test datasets contained sequences for eukaryotes, including H. sapiens, M. musculus, Rat, S. cerevisiae, Zebrafish, A. thaliana, Pig, Rhesus, and Chimpanzee. Herein, we describe the detailed procedures for the curation of the independent test datasets (Table 3).
Specifically, we collected m6A site sequences containing the motif DRACH with a consistent length with the above-mentioned computational approaches from the recent literature and according to m6A site information produced by m6A-seq2 [85], m6A-RFE-seq [86], m6A-SAC-seq [87], miCLIP [88], and scDART-seq [22], respectively. Moreover, the m6A site sequences with a single-base resolution deposited in the recently published database m6A-Atlas [84] were also included. After removing those sequences with sequence similarity greater than 80% using CD-HIT-EST and further removing the overlapping sequences with positive samples of the training datasets of the compared methods, the remaining sequences were retained as the positive samples of the independent test datasets.
Non-m6A sequences (i.e., negative samples) refer to RNA sequences of which the central positions are confirmed not to be m6A sites. In accordance with the approaches for the selection of negative samples in the majority of studies about m6A site prediction, all possible non-m6A sequences containing unmethylated adenosines in the motif DRACH at sequence centers could be extracted from the reference genomes of species corresponding to positive samples. Next, the overlapping sequences with the training datasets of the compared methods, as well as the positive samples of the independent test datasets, were removed, thereby enabling the remaining sequences to not be seen in these two types of datasets. As the distribution of m6A sites and non-m6A sites in the transcriptome is unbalanced, after removing those sequences with greater than 80% similarity, the non-m6A sequences were randomly selected from the remaining sequences in terms of a certain positive-to-negative ratio to construct balanced and unbalanced independent test datasets. In the current study, the positive-to-negative ratio was set to 1:1. The numbers of non-m6A sequences in the independent test datasets for seven species are shown in Table 3.
Table 3.
Detailed information of independent test datasets.
Table 3.
Detailed information of independent test datasets.
| Species | Dataset Name | Source | Positive-to-Negative Ratio |
|---|---|---|---|
| 1:1 | |||
| H. sapiens | Hg38_Human | scDART-seq data containing single-nucleotide m6A sites in single cells [22] | 22,248 |
| hg19_Human1 | Single-nucleotide m6A data [86] | 2064 | |
| hg19_Human2 | Single-nucleotide m6A data [85] | 37,372 | |
| hg19_Human3 | Single-nucleotide m6A data [88] | 930 | |
| hg19_Human4 | Data intersection between ConsRM, RMBase, and m6A-Atlas | 12,588 | |
| M. musculus | mm10_Mouse | Data intersection between RMVar, RMBase, and m6A-Atlas | 3330 |
| Rhesus | rheMac8_Rhesus | RMBase | 12,098 |
| Chimpanzee | panTro4_Chimpanzee | RMBase | 15,424 |
| Rat | rn5_Rat | RMBase | 24,380 |
| Pig | susScr3_Pig | RMBase | 42,838 |
| Zebrafish | danRer10_Zebrafish | RMBase | 8946 |
| S. cerevisiae | sacCer3_S_cerevisiae | Data intersection between m6A-Atlas and RMBase | 14,876 |
| A. thaliana | TAIR10_A_thaliana | Data intersection between m6A-Atlas and RMBase | 4516 |
2.4. Feature Engineering and Representation
To develop robust and reliable computational approaches for m6A prediction, it is vitally important to carefully design and extract features for the representation and conversion of the RNA sequences into numeric vectors. The features applied in the 52 computational approaches that we revisited in this study can be categorized into four groups, i.e., context-, structure-, genome-based, and integrated features.
2.4.1. Context-Based Features
Context-based features are designed and extracted to describe the genomic contexts of the m6A/non-m6A sequences. These context-based features of the 52 approaches systematically described and summarized in Table 1 can be classified into three groups: (i) nucleotide physicochemical properties, such as the pseudo-dinucleotide composition (PseDNC) [26,30,35,55], general PseDNC [44], pseudo-trinucleotide composition (PseTNC) [45], dinucleotide-based auto-/cross-covariance (DAC/DCC) [27,29,32,70], chemical property with density (CPD) [33,37,40,42,46,47,54,59,70], and electron–ion interaction pseudopotential (EIIP) [53,72]; (ii) RNA primary sequence-derived features, including one-hot or binary encoding [31,39,54,56,58,66,67,68,71,74], RNA word embedding [49,50,67,69], dinucleotide binary encoding [57], the k-mer composition [33,34,41,43,45,52,62], the KNN score [28,45], features related to entropy information [60], and the local position-specific dinucleotide frequency (LPSDF) [57]; and (iii) position-specific scoring matrices, such as the nucleotide pair position specificity (NPPS) [48,53], position-specific k-mer nucleotide propensity (PS(k-mer)NP) [34,36,38,64,73], probability matrix [51], and bi-profile Bayes (BPB) [53].
Context-based features are the most widely applied feature type for m6A site prediction. Most of these features can be easily generated by useful bioinformatics pipelines without following complex mathematical formulas, such as Pse-in-One [89], iFeature, PseKNC [90], PseKNC-General [91], iLearn [92], and iLearnPlus [93]. Among the above-mentioned features, the chemical property with density (CPD), which describes three distinct structural chemical properties of nucleotides, including functional groups, ring structures, and hydrogen bonds, and the density of the i-th nucleotide along the sample sequence, have been extensively used in computational biology [94,95,96,97]. Compared to earlier published models, the majority of the computational methods for the identification of m6A sites since 2019 have favored binary encoding features.
2.4.2. Structure-Based Features
m6A/non-m6A sequences can also be characterized by structure-based features, which are calculated based on 3D RNA structures. Several structure-based features have been applied to four predictors, namely WHISTLE [59], RNAMethPre [62], SRAMP [63], and ConsRM [65], and have been confirmed to be effective in improving their prediction performance. These structure-based features, predicted by the RNAfold [98] tool, can be categorized into two major types: (i) the predicted RNA loop region [59,63,65], such as bulged loop (B), hairpin loop (H), interior loop (I), multiple loop (M), and paired (P), which can be encoded into the binary vectors (0,0,0,0,1), (1,0,0,0,0), (0,0,1,0,0), (0,1,0,0,0), (0,0,0,1,0), respectively; (ii) the minimum free energy (MFE) [62], which is calculated to measure the secondary structure strength of the region around the site; and (iii) the predicted RNA hybridized region [59,65], which is computed by minimizing the free energy.
2.4.3. Genome-Based Features
Genome-based features [59,60,62,65] can contribute to m6A site prediction because such features may capture the adequate attributes of the m6A modification topology. In total, there are eight major types of genome-based features, namely (i) dummy variables indicating whether the site is overlapped with the topological region on the major RNA transcript, such as the 5′ UTR, 3′ UTR, coding sequence, stop/start codons flanked by 100 bp, downstream 100 bp of TSS on A, and so on; (ii) the relative position in the region, including the relative position in the 5′ UTR/3′ UTR, the relative position in the coding sequence, and the relative position in an exon; (iii) the nucleotide distances to the splicing junctions, such as the distance to the 5′/3′ splicing junction; (iv) the region length, including the 5′ UTR/3′ UTR length, coding sequence length, gene length in exons, full gene length, mature transcript length, and full transcript length; (v) clustering information, such as the count of neighboring input sites at 1001/101 nt, the count of neighboring As within a 2001/201 nt window, the distance to the closest neighboring input site at 2001/201 nt, and the distance to the closest neighboring A at 2001/201 nt; (vi) scores related to evolutionary conservation, such as the phastCons/fitCons scores of the nucleotide, and the average phastCons/fitCons scores within the flanking 101 nt; (vii) the attributes of the genes or transcripts, such as the miRNA targeted genes, miRNA targeted sites verified by experiments, overlapped binding regions of METTL3, and so on; (viii) genomic properties, including sncRNA, lncRNA, the number of isoforms/exons, housekeeping genes, the GC composition of genes, and the GC composition of 101 nt; (ix) SNP features [60], i.e., the distribution of the single-nucleotide polymorphism sites of the mRNA sequence around m6A sites. It is noteworthy that most of these features can be generated by the GenomicFeatures R package.
2.4.4. Integrated Features
To further boost the prediction performance, many m6A site predictors [59,60,62,65] integrate multiple types of features, rather than only employing a single type of feature mentioned above. For example, by integrating context-, structure-, and genome-based features using SVM, WHISTLE [59] achieved promising prediction performance with an average AUROC of 0.948 and 0.880 on the independent test datasets from full transcripts and mature mRNA, respectively. SRAMP [63] used context- and structure-based features, including the predicted binary encoding, KNN encoding, spectrum encoding, and RNA loop region, to describe sample sequences and achieved high specificity.
2.4.5. Feature Selection
Feature selection is an effective way to eliminate redundant features and reduce the large calculation burden and overfitting caused by high-dimensional vectors. Several filter-based feature selection methods have been adopted and proven to be effective in boosting the computational efficiency and prediction performance. There are three major types of filter-based feature selection methods: (i) the minimum redundancy maximum relevance (mRMR) [32,70], which is applied to calculate and rank the contribution values of each feature to achieve the optimal feature subset; (ii) elastic net (EN) [73], which is an approach that can implement feature selection based on regularized terms; (iii) statistical algorithms, such as the F-test [38], and F-score [57,72], which are employed to measure and rank the significance of all of the extracted feature components.
2.5. Predictive Algorithms Employed
There are three types of major prediction algorithms for m6A prediction (Supplementary Material Figure S1 and Table 1), namely (i) traditional machine learning-based methods, such as support vector machine (SVM) [26,27,28,29,30,31,32,37,38,40,42,43,45,46,47,48,59,62,64,65,70], random forest (RF) [33,63], XGBoost [34,57,60], AdaBoost [61], CatBoost [53], and second-order Markov models [51]; (ii) deep learning-based methods, such as deep neural networks (DNNs) [73], convolutional neural networks (CNNs) [39,41,49,52,54,71,74], and CNNs in combination with bidirectional long short-term memory (BiLSTM) [58,66,68]; and (iii) ensemble learning-based methods, such as machine learning-based ensemble methods [35,36,44,55] and ensemble deep learning [50,56,67,69,72].
2.5.1. Traditional Machine Learning-Based Methods
Among the predictors analyzed in this study, SVM is the most widely used machine learning algorithm (Supplementary Material Figure S1B). When constructing these SVM-based m6A site predictors, the radial basis function (RBF) was generally considered as the kernel function, while a grid search strategy and K-fold cross-validation were employed to optimize the two parameters of SVM, i.e., the regularization parameter C and the kernel width parameter . However, it is time-consuming to perform this parameter optimization process, especially if the sample size is large. For large m6A datasets, the boosting method is an excellent choice, which generates strong learners by integrating weak learners based on the idea of ensemble learning. Five predictors were built using the boosting method: three for XGBoost [34,57,60], one for AdaBoost [61], and one for CatBoost [53]. All of them illustrated excellent prediction on their own datasets in comparison to other machine learning methods, such as SVM, RF, or KNN. At the same time, random forest [33], by integrating multiple decision trees, can reduce the risk of overfitting associated with a single model and improve the predictive accuracy, making it a good choice for datasets with high-dimensional features. Given the robust performance of SVM, RF, boost, and Markov models, we categorized these four classical models separately (Supplementary Material Figure S1B).
2.5.2. Deep Learning-Based Methods
Deep learning techniques, advancing from traditional artificial neural network frameworks, have been widely applied in bioinformatics, such as in protein modification sites [99,100,101], gene ontology function prediction [102,103], medical image analysis [104], drug design [105,106,107], HLA epitope prediction [108,109,110], and so on. Among them, a typical feed-forward artificial neural network, the CNN, has attracted widespread attention and has been widely used in m6A site prediction. Many predictors [40,41,49,52,54,58,66,68,71,74] have been successively proposed using CNNs or CNNs in combination with other strategies (BiLSTM) based on single-nucleotide m6A data.
2.5.3. Ensemble Learning-Based Methods
At the forefront of machine learning, ensemble learning, which integrates the output of multiple models, has exerted a significant impact on various field of bioinformatics as this technology can generally obtain better generalizability in comparison to a single model. Among the m6A predictors analyzed, in addition to homogeneous ensemble models—such as RF and XGBoost, which use decision trees as base learners—there are also heterogeneous ensemble methods based on different machine learning models [35,36,44,55], as well as heterogeneous ensemble methods combining various deep learning models [50,56,67,69,72]. For example, Zhang et al. constructed a machine learning-based ensemble model named M6A-GSMS [44], which integrated the output of six base classifiers, namely RF, extra trees (ET), bagging, SVM, Adaboost, and LightGBM, as the input of a meta-classifier, i.e., Gaussian Naïve Bayes (GaussianNB), to achieve the final prediction result. Chen et al. developed an ensemble deep learning framework, DeepM6Aseq-EL [69], consisting of five sub-networks, each of which was stacked by LSTM and a CNN.
2.6. Strategies and Measures for Performance Assessment
Three performance evaluation strategies, namely the jackknife validation test, K-fold cross-validation (CV), and independent test, are usually adopted to evaluate the prediction performance of the proposed predictors. The jackknife validation test, also termed leave-one-out CV, refers to a strategy where each sample is in turn used as test data to evaluate the predictor trained by the remaining data. K-fold CV is another similar strategy in which the dataset is randomly divided into k equal or almost equal subsets, and then each subset is in turn used as test data to evaluate the prediction performance of the predictor trained on the remaining subsets. Although the jackknife test is stricter and more objective, K-fold CV has more advantages in terms of the computing time, especially for large sample sizes. For this reason, most predictors surveyed in this study employ K-fold CV (k = 5 or 10) to construct performance matrices. Moreover, in order to compare them with existing methods fairly and objectively, an independent test is usually implemented using newly constructed data that have not been seen in the training data of the compared predictors.
In the field of bioinformatics [95,111,112,113,114,115,116,117], several widely used performance metrics have been employed to quantitatively measure the prediction performance, including the accuracy (Acc), sensitivity (Sn), specificity (Sp), Matthew’s correlation coefficient (MCC), F1-score, precision, area under the precision–recall curve (AUPR), and area under the receiver operating characteristic curve (AUROC) [110,118,119]. These metrics are formulated as follows:
where TP, FP, TN, and FN stand for the numbers of true positives, false positives, true negatives, and false negatives, respectively.
2.7. Webserver/Software Availability and Usability
A user-friendly webserver and/or local software can significantly facilitate scholars’ implementation of m6A prediction using the proposed computational methods. In total, 67.3% (35 out of 52) of the m6A site predictors surveyed in this study have available online websites or stand-alone software, and 60.0% (21 out of 35) of them are still active (Table 1).
The query sequences in the FASTA format are allowed to be submitted to the above-mentioned webservers for the implementation of m6A site prediction online. However, some servers limit the length and number of the submitted sequences. For example, TS-m6A-DL only allows a fixed length of 41 nt for each sequence, while m6A-NeuralTool was designed to analyze fixed 101-nt-long sequences from A. thaliana, 51 nt from S. cerevisiae, and 41 nt from M. musculus/H. sapiens. iRNA(m6A)-PseDNC allows query sequences with a minimum length of 51 nt to be submitted, while they must be no shorter than 41 nt for iRNA-PseColl, iRNA-3typeA, iRNA-m6A, and MethyRNA; M6ATH requires no more than 100 sequences for each submission.
In addition, it is critical for users that the output format of webservers is well designed to facilitate the understanding and interpretation of the prediction results. All of the available servers surveyed in this study can offer immediate prediction results on their webpages after submitting the query sequences. iRNA-3typeA, m6A-NeuralTool, and TS-m6A-DL can provide the output containing the predicted labels (e.g., m6A or non-m6A sequences). Most methods, such as iRNA-Methyl, m6Apred, iRNA(m6A)-PseDNC, iRNA-PseColl, MethyRNA, and SRAMP, offer the probability scores and predicted positions of m6A or non-m6A as well.
3. Experimental Results
3.1. Performance Comparison of Species-Specific Predictors
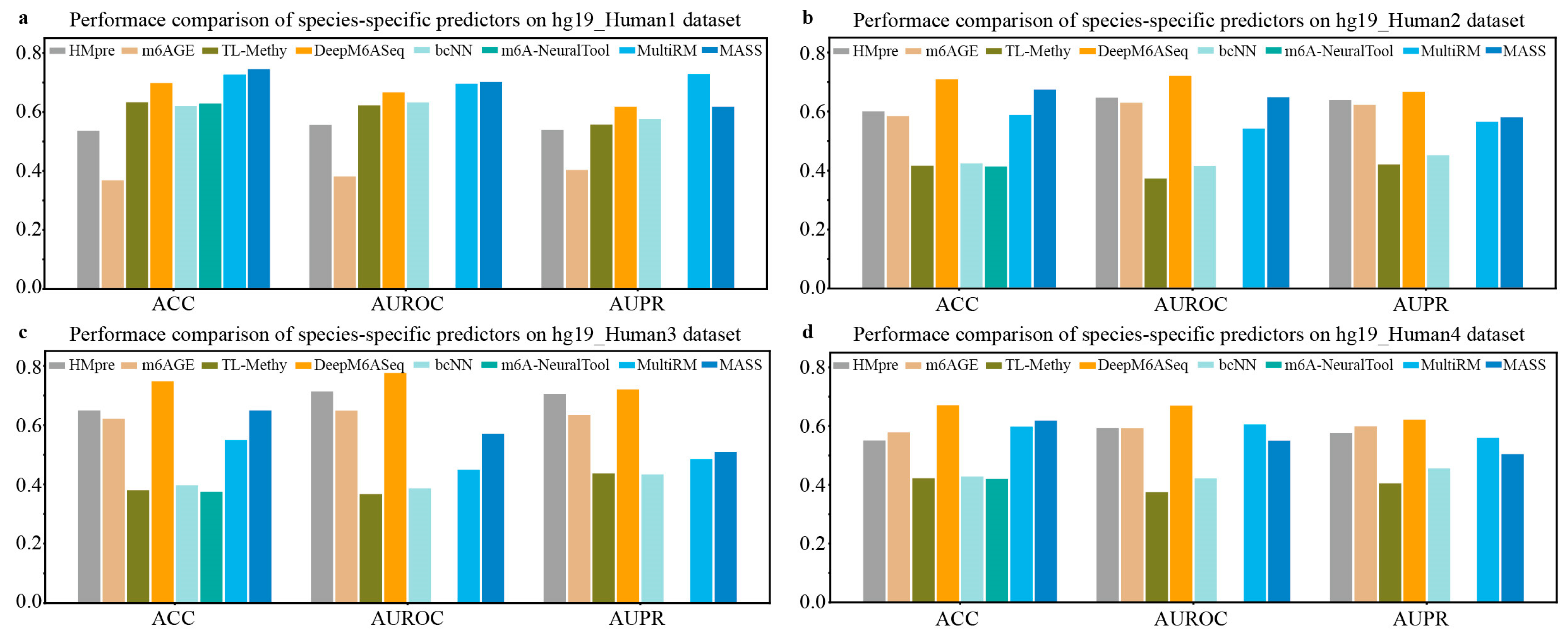
We collected m6A site sequences for nine species, as depicted in Table 3, and conducted a series of experiments to evaluate the performance of species-specific predictors. First, we utilized independent m6A data from four different sources of H. sapiens to evaluate the generalization performance of the existing species-specific predictors available, including DeepM6ASeq, HMpre, bCNN, m6AGE, m6A-NeuralTool, MultiRM, TL-Methy, and MASS. The results, shown in Figure 2, indicate that the predictor DeepM6ASeq demonstrates outstanding predictive performance on independent datasets from different sources. On the hg19_Human2 dataset, DeepM6ASeq achieves an ACC of 70.92% and an AUROC of 0.7211, surpassing the second-best predictor, MASS, by 3.5% and 7.36%, respectively. Additionally, its AUPR is 0.6663, exceeding the second-best predictor, m6AGE, by 4.41%. On the hg19_Human3 and hg9_Human4 datasets, the ACC, AUROC, and AUPR of DeepM6ASeq are all higher than those of the other predictors, while its performance on the hg19_Human1 dataset is also comparable to that of the other predictors. Furthermore, as seen from Figure 2, MultiRM and MASS also exhibit stable predictive performance in predicting the m6A sites of H. sapiens.
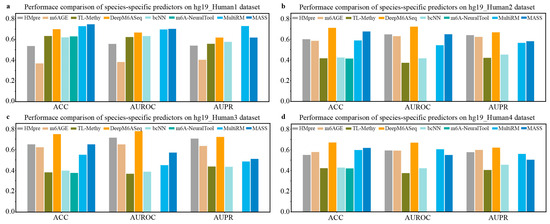
Figure 2.
Performance comparison of species-specific predictors on independent datasets of H. sapiens.
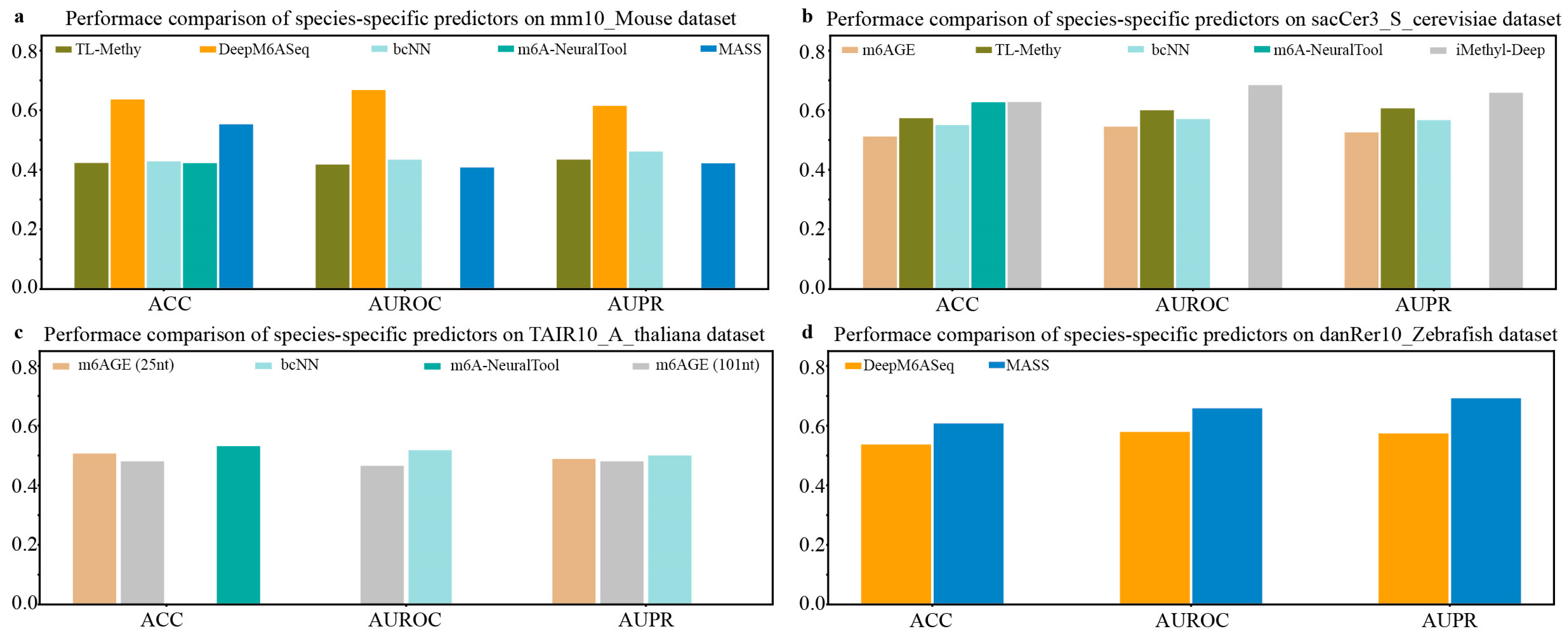
Next, we compared the prediction performance of species-specific predictors on independent datasets of M. musculus, S. cerevisiae, A. thaliana, and Zebrafish. As shown in Figure 3a, among the available species-specific predictors for the prediction of the m6A sites of M. musculus, DeepM6ASeq achieves the best predictive performance, followed by MASS. It can be seen from Figure 3b that iMethy-Deep demonstrates the most superior performance for m6A site prediction on the independent dataset sacCer3_S_cerevisiae of S. cerevisiae. The results depicted in Figure 3c suggest that the available species-specific predictors, such as m6AGE (25nt), m6AGE (101nt), bCNN, and m6A-NeuralTool, exhibit comparable prediction performance in predicting the m6A sites of A. thaliana. Additionally, on the independent dataset danRer10_Zebrafish, the species-specific predictor MASS exhibits slightly higher values for the ACC, AUROC, and AUPR compared to another predictor, DeepM6ASeq (Figure 3d).
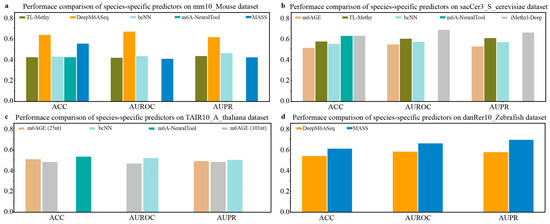
Figure 3.
Performance comparison of species-specific predictors on independent datasets of M. musculus, S. cerevisiae, A. thaliana, and Zebrafish.
Overall, species-specific deep learning-based methods, such as DeepM6ASeq, MultiRM, and MASS, demonstrate superior predictive performance compared to traditional machine learning methods, such as HMpre, m6AGE, and TL-Methy. In particular, among them, DeepM6ASeq and MASS can predict m6A sites across multiple species.
3.2. Cross-Species Validation of State-of-the-Art Predictors
Considering that the performance of DeepM6ASeq and MASS is superior to that of the current methods, we next evaluate the cross-species prediction performance of these predictors on the independent datasets of nine species. DeepM6ASeq consists of three sub-models, DeepM6ASeq-HSA, DeepM6ASeq-MMU, and DeepM6ASeq-ZF, trained on m6A site data from H. sapiens, M. musculus, and Zebrafish, respectively. Thus, we first merge the four H. sapiens datasets, namely hg19_Human1, hg19_Human2, hg19_Human3, and hg19_Human4, into a larger dataset and then test the cross-species prediction performance of these sub-models of DeepM6ASeq on nine independent datasets. The results depicted in Figure 4 indicate that when species-specific predictors are used to predict cross-species m6A sites, their predictive performance shows a significant decrease. For example, DeepM6ASeq-HSA achieves an ACC of 0.7 on the H. sapiens datasets, while its ACC drops to 0.626, 0.529, 0.544, 0.507, 0.472, 0.55, 0.663, and 0.547 on the other cross-species datasets, respectively.

Figure 4.
Cross-species performance comparison of DeepM6ASeq predictor on independent datasets of 9 species. (a–c) respectively show the values for ACC, AUROC, and AUPR.
Although DeepM6ASeq-HSA performs well on the H. sapiens dataset, its performance declines on the cross-species datasets. Despite some genes being conserved across multiple species, significant differences in gene expression regulation and biological functions may exist between species, leading to the model’s inability to effectively generalize from H. sapiens datasets to other species. In contrast, the DeepM6ASeq-MMU model achieved the top ACC values of 0.671 and 0.688 on the H. sapiens and S. cerevisiae datasets, respectively, and the DeepM6ASeq-ZF model achieved the top ACC values of 0.604 and 0.688 on the M. musculus and S. cerevisiae datasets, demonstrating their good generalization abilities. This indicates that both models can effectively capture and utilize shared sequence information among different species, thereby improving the prediction accuracy.
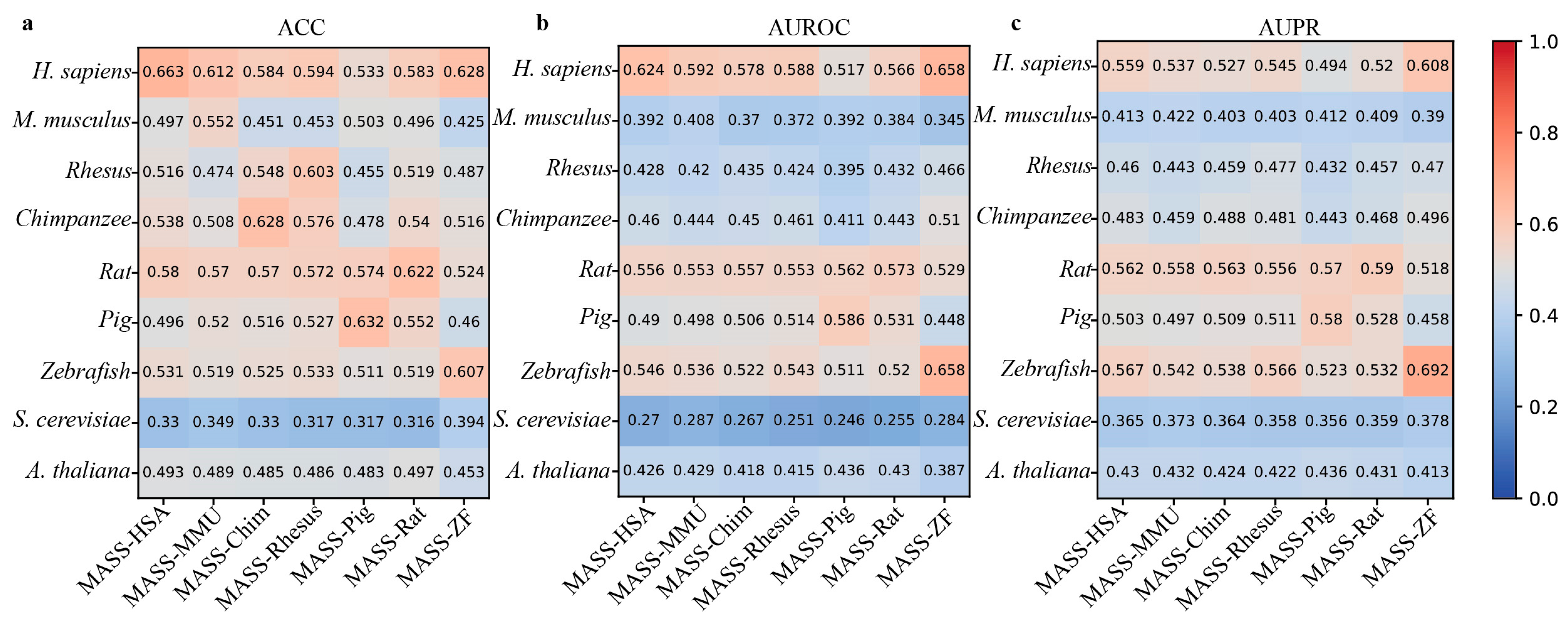
In addition, MASS contains seven species-specific sub-models. Therefore, we next use the independent datasets derived from nine species to evaluate the cross-species prediction performance of these sub-models. From Figure 5, it can be observed that these species-specific sub-models exhibit varying degrees of performance decline when predicting m6A sites in different species. For example, the MASS-HSA predictor trained on human m6A data achieves an ACC of 0.663 when predicting human m6A sites, while the accuracy on the independent datasets of the other eight species is 0.497, 0.516, 0.538, 0.58, 0.486, 0.531, 0.33, and 0.493, respectively.
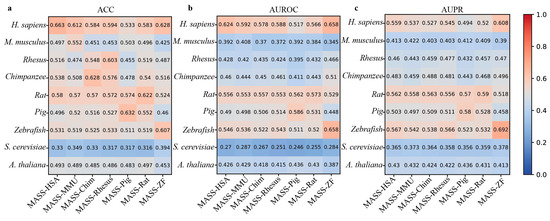
Figure 5.
Cross-species performance comparison of MASS predictor on independent datasets of 9 species. (a–c) respectively show the values for ACC, AUROC, and AUPR.
Furthermore, to make our evaluation more comprehensive, we also assessed the cross-species prediction performance of seven other predictors—MultiRM, TL-Methy, m6AGE, iMethyl-Deep, HMpre, bCNN, and m6A-NeuralTool—and present the results in Supplementary Material S2. Similar to the findings for DeepM6ASeq and MASS, the performance metrics of these predictors across different species, such as the ACC, AUROC, and AUPR, also indicate that species-specific predictors perform better when predicting m6A sites within their own species, while their performance often declines in cross-species predictions. For example, MultiRM and iMethyl-Deep exhibit high prediction performance on their training species but show significantly reduced performance on other species. This further suggests that constructing species-specific prediction models is necessary for the accurate identification of m6A sites.
3.3. Performance Comparison for Prediction of m6A Sites in Single Cells
The current computational methods for m6A site prediction are all based on bulk-cell sequencing data, while the emerging scDART-seq technology can achieve the identification of single-nucleotide m6A sites at a transcriptome-wide level in single cells. Therefore, we need to evaluate whether the current methods can accurately predict m6A sites at the single-cell level.
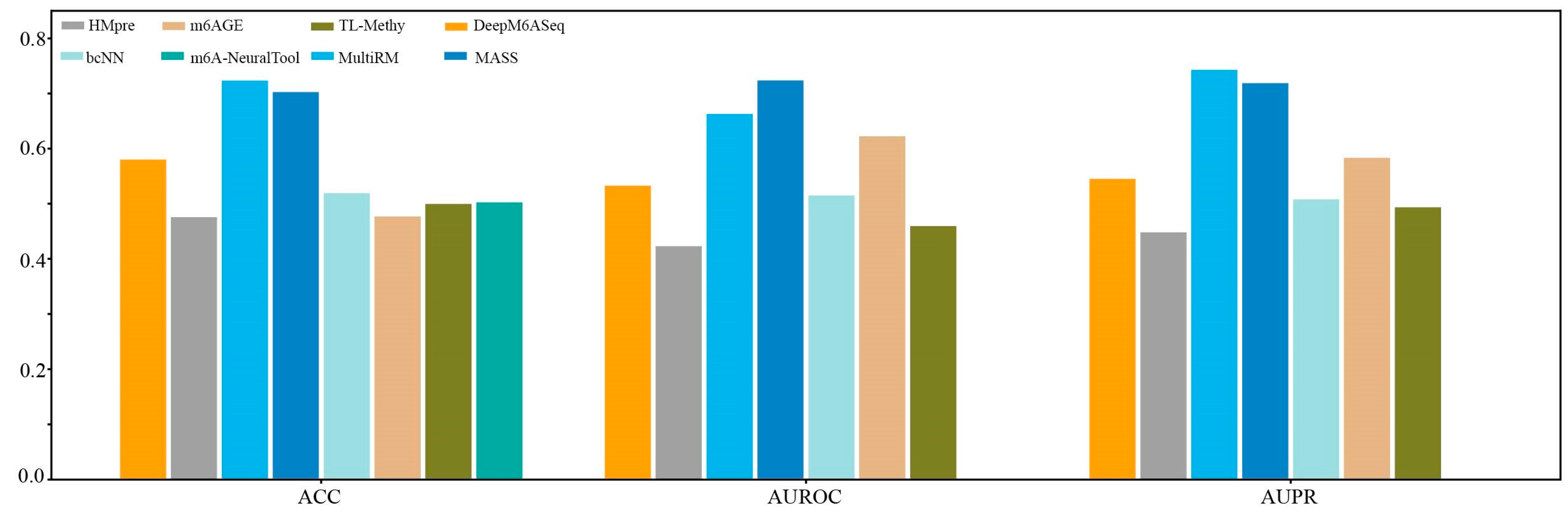
We first constructed a dataset, named Hg38_Human, which contained 11124 single-cell m6A site sequences from scDART-seq data and 11124 non-m6A site sequences. Subsequently, we compared the prediction performance of the currently available species-specific predictors in identifying m6A sites in H. sapiens at a single-cell level on the independent dataset Hg38_Human. As shown in Figure 6, MultiRM achieves an ACC of 0.7233, an AUROC of 0.6628, and an AUPR of 0.7428, while MASS achieves an ACC of 0.7025, an AUROC of 0.7236, and an AUPR of 0.7118. Compared with other prediction methods, MultiRM has the best prediction performance in predicting m6A sites in single cells, followed by MASS. Furthermore, among these predictors, the predictive performance of deep learning-based methods, including MultiRM, MASS, DeepM6ASeq, bCNN, and m6A-NeuralTool, is significantly better than that of traditional machine learning methods, such as HMpre, m6AGE, and TL-Methy.
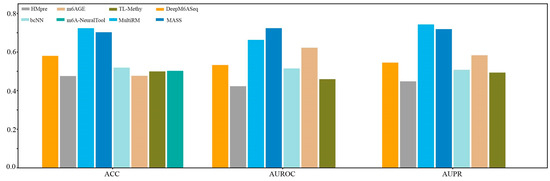
Figure 6.
Comparison of 8 predictors on the H. sapiens m6A single-cell dataset.
4. Conclusions
In this study, we have surveyed 52 state-of-the-art computational approaches for the prediction of m6A sequences and benchmarked nine species-specific predictors with available and functioning webservers/local tools. To the best of our knowledge, this study represents the most comprehensive and large-scale benchmarking test of predictors for the identification of m6A sites. A wide range of aspects were summarized in detail, including the employed algorithms, calculated features, performance evaluation strategies, and software usability. By curating 13 independent test datasets for various species, we extensively benchmarked nine available predictors and demonstrated their prediction performance, including species-specific prediction, cross-species prediction, and the identification of m6A sites in single cells. The prediction results show that deep learning-based methods generally outperform traditional machine learning-based methods. Although the performance of the surveyed predictors is subject to change in cases where different test datasets and new data are used to test the models, DeepM6ASeq, MultiRM, and MASS have the best generalization performance. We expect this analysis to be a stepping stone toward the design and implementation of more accurate predictors for m6A sites in the future.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/biology13100777/s1. All 13 independent test datasets used for the benchmarking of various species-specific predictors in this study are available at “https://github.com/ztLuo-bioinfo/m6A/ (accessed on 20 September 2024)”.
Author Contributions
Z.X., L.G. and K.L. conceived and designed the project. Z.X., Z.L., L.Y. and K.L. conducted the data analysis and independent tests and drafted the manuscript. L.G. provided critical comments and useful insights to improve the scientific quality of the work. Z.X., L.G. and K.L. revised the manuscript, which was approved by all other authors. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the National Natural Science Foundation of China (Nos. 62062043, 32270789, and 31771679), the University Synergy Innovation Program of Anhui Province under Grants GXXT-2022-046 and GXXT-2022-055, and the Natural Science Foundation of Anhui Province under Grant 2308085MF217.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding authors.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Li, X.; Xiong, X.; Yi, C. Epitranscriptome sequencing technologies: Decoding RNA modifications. Nat. Methods 2016, 14, 23–31. [Google Scholar] [CrossRef] [PubMed]
- Alarcón, C.R.; Lee, H.; Goodarzi, H.; Halberg, N.; Tavazoie, S.F. N6-methyladenosine marks primary microRNAs for processing. Nature 2015, 519, 482–485. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Q.; Ren, J.; Li, L.; Li, S.; Xiang, K.; Shang, D. Development and validation of a novel N6-methyladenosine (m6A)-related multi- long non-coding RNA (lncRNA) prognostic signature in pancreatic adenocarcinoma. Bioengineered 2021, 12, 2432–2448. [Google Scholar] [CrossRef] [PubMed]
- Maden, B.E. Locations of methyl groups in 28 S rRNA of Xenopus laevis and man. Clustering in the conserved core of molecule. J. Mol. Biol. 1988, 201, 289–314. [Google Scholar] [CrossRef] [PubMed]
- Wilson, C.; Chen, P.J.; Miao, Z.; Liu, D.R. Programmable m(6)A modification of cellular RNAs with a Cas13-directed methyltransferase. Nat. Biotechnol. 2020, 38, 1431–1440. [Google Scholar] [CrossRef] [PubMed]
- Roundtree, I.A.; Evans, M.E.; Pan, T.; He, C. Dynamic RNA Modifications in Gene Expression Regulation. Cell 2017, 169, 1187–1200. [Google Scholar] [CrossRef]
- Zhong, X.; Yu, J.; Frazier, K.; Weng, X.; Li, Y.; Cham, C.M.; Dolan, K.; Zhu, X.; Hubert, N.; Tao, Y.; et al. Circadian Clock Regulation of Hepatic Lipid Metabolism by Modulation of m(6)A mRNA Methylation. Cell Rep. 2018, 25, 1816–1828.e1814. [Google Scholar] [CrossRef]
- Zhang, Z.; Wang, M.; Xie, D.; Huang, Z.; Zhang, L.; Yang, Y.; Ma, D.; Li, W.; Zhou, Q.; Yang, Y.G.; et al. METTL3-mediated N(6)-methyladenosine mRNA modification enhances long-term memory consolidation. Cell Res. 2018, 28, 1050–1061. [Google Scholar] [CrossRef]
- Patil, D.P.; Chen, C.K.; Pickering, B.F.; Chow, A.; Jackson, C.; Guttman, M.; Jaffrey, S.R. m(6)A RNA methylation promotes XIST-mediated transcriptional repression. Nature 2016, 537, 369–373. [Google Scholar] [CrossRef]
- Zhou, J.; Wan, J.; Gao, X.; Zhang, X.; Jaffrey, S.R.; Qian, S.B. Dynamic m(6)A mRNA methylation directs translational control of heat shock response. Nature 2015, 526, 591–594. [Google Scholar] [CrossRef]
- Xu, K.; Yang, Y.; Feng, G.H.; Sun, B.F.; Chen, J.Q.; Li, Y.F.; Chen, Y.S.; Zhang, X.X.; Wang, C.X.; Jiang, L.Y.; et al. Mettl3-mediated m(6)A regulates spermatogonial differentiation and meiosis initiation. Cell Res. 2017, 27, 1100–1114. [Google Scholar] [CrossRef] [PubMed]
- Han, D.; Liu, J.; Chen, C.; Dong, L.; Liu, Y.; Chang, R.; Huang, X.; Liu, Y.; Wang, J.; Dougherty, U.; et al. Anti-tumour immunity controlled through mRNA m(6)A methylation and YTHDF1 in dendritic cells. Nature 2019, 566, 270–274. [Google Scholar] [CrossRef] [PubMed]
- Xiang, Y.; Laurent, B.; Hsu, C.H.; Nachtergaele, S.; Lu, Z.; Sheng, W.; Xu, C.; Chen, H.; Ouyang, J.; Wang, S.; et al. RNA m(6)A methylation regulates the ultraviolet-induced DNA damage response. Nature 2017, 543, 573–576. [Google Scholar] [CrossRef]
- Hong, K. Emerging function of N6-methyladenosine in cancer. Oncol. Lett. 2018, 16, 5519–5524. [Google Scholar] [CrossRef]
- Liu, J.; Li, K.; Cai, J.; Zhang, M.; Zhang, X.; Xiong, X.; Meng, H.; Xu, X.; Huang, Z.; Peng, J.; et al. Landscape and Regulation of m(6)A and m(6)Am Methylome across Human and Mouse Tissues. Mol. Cell 2020, 77, 426–440.e426. [Google Scholar] [CrossRef]
- Dominissini, D.; Moshitch-Moshkovitz, S.; Schwartz, S.; Salmon-Divon, M.; Ungar, L.; Osenberg, S.; Cesarkas, K.; Jacob-Hirsch, J.; Amariglio, N.; Kupiec, M.; et al. Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature 2012, 485, 201–206. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Lu, Z.; Wang, X.; Fu, Y.; Luo, G.Z.; Liu, N.; Han, D.; Dominissini, D.; Dai, Q.; Pan, T.; et al. High-resolution N(6)-methyladenosine (m(6) A) map using photo-crosslinking-assisted m(6) A sequencing. Angew. Chem. Int. Ed. Engl. 2015, 54, 1587–1590. [Google Scholar] [CrossRef]
- Linder, B.; Grozhik, A.V.; Olarerin-George, A.O.; Meydan, C.; Mason, C.E.; Jaffrey, S.R. Single-nucleotide-resolution mapping of m6A and m6Am throughout the transcriptome. Nat. Methods 2015, 12, 767–772. [Google Scholar] [CrossRef]
- Ule, J.; Jensen, K.B.; Ruggiu, M.; Mele, A.; Ule, A.; Darnell, R.B. CLIP identifies Nova-regulated RNA networks in the brain. Science 2003, 302, 1212–1215. [Google Scholar] [CrossRef]
- Zhang, Z.; Chen, L.Q.; Zhao, Y.L.; Yang, C.G.; Roundtree, I.A.; Zhang, Z.; Ren, J.; Xie, W.; He, C.; Luo, G.Z. Single-base mapping of m(6)A by an antibody-independent method. Sci. Adv. 2019, 5, eaax0250. [Google Scholar] [CrossRef]
- Meyer, K.D. DART-seq: An antibody-free method for global m(6)A detection. Nat. Methods 2019, 16, 1275–1280. [Google Scholar] [CrossRef] [PubMed]
- Tegowski, M.; Flamand, M.N.; Meyer, K.D. scDART-seq reveals distinct m(6)A signatures and mRNA methylation heterogeneity in single cells. Mol. Cell 2022, 82, 868–878.e810. [Google Scholar] [CrossRef]
- Chen, X.; Sun, Y.Z.; Liu, H.; Zhang, L.; Li, J.Q.; Meng, J. RNA methylation and diseases: Experimental results, databases, Web servers and computational models. Brief Bioinform. 2019, 20, 896–917. [Google Scholar] [CrossRef]
- Zhu, X.; He, J.; Zhao, S.; Tao, W.; Xiong, Y.; Bi, S. A comprehensive comparison and analysis of computational predictors for RNA N6-methyladenosine sites of Saccharomyces cerevisiae. Brief Funct. Genom. 2019, 18, 367–376. [Google Scholar] [CrossRef]
- Zhang, S.Y.; Zhang, S.W.; Zhang, T.; Fan, X.N.; Meng, J. Recent advances in functional annotation and prediction of the epitranscriptome. Comput. Struct. Biotechnol. J. 2021, 19, 3015–3026. [Google Scholar] [CrossRef]
- Chen, W.; Feng, P.; Ding, H.; Lin, H.; Chou, K.C. iRNA-Methyl: Identifying N(6)-methyladenosine sites using pseudo nucleotide composition. Anal. Biochem. 2015, 490, 26–33. [Google Scholar] [CrossRef]
- Liu, Z.; Xiao, X.; Yu, D.J.; Jia, J.; Qiu, W.R.; Chou, K.C. pRNAm-PC: Predicting N(6)-methyladenosine sites in RNA sequences via physical-chemical properties. Anal. Biochem. 2016, 497, 60–67. [Google Scholar] [CrossRef] [PubMed]
- Jia, C.Z.; Zhang, J.J.; Gu, W.Z. RNA-MethylPred: A high-accuracy predictor to identify N6-methyladenosine in RNA. Anal. Biochem. 2016, 510, 72–75. [Google Scholar] [CrossRef]
- Zhang, M.; Sun, J.W.; Liu, Z.; Ren, M.W.; Shen, H.B.; Yu, D.J. Improving N(6)-methyladenosine site prediction with heuristic selection of nucleotide physical-chemical properties. Anal. Biochem. 2016, 508, 104–113. [Google Scholar] [CrossRef]
- Chen, W.; Ding, H.; Zhou, X.; Lin, H.; Chou, K.C. iRNA(m6A)-PseDNC: Identifying N(6)-methyladenosine sites using pseudo dinucleotide composition. Anal. Biochem. 2018, 561–562, 59–65. [Google Scholar] [CrossRef] [PubMed]
- Wei, L.; Su, R.; Wang, B.; Li, X.; Zou, Q.; Gao, X. Integration of deep feature representations and handcrafted features to improve the prediction of N6-methyladenosine sites. Neurocomputing 2019, 324, 3–9. [Google Scholar] [CrossRef]
- Khan, A.; Rehman, H.U.; Habib, U.; Ijaz, U. m6A-Finder: Detecting m6A methylation sites from RNA transcriptomes using physical and statistical properties based features. Comput. Biol. Chem. 2022, 97, 107640. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.; Rehman, H.U.; Habib, U.; Ijaz, U. Detecting N6-methyladenosine sites from RNA transcriptomes using random forest. J. Comput. Sci. 2020, 47, 101238. [Google Scholar] [CrossRef]
- Zhao, X.; Zhang, Y.; Ning, Q.; Zhang, H.; Ji, J.; Yin, M. Identifying N(6)-methyladenosine sites using extreme gradient boosting system optimized by particle swarm optimizer. J. Theor. Biol. 2019, 467, 39–47. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Xing, P.; Zou, Q. Detecting N(6)-methyladenosine sites from RNA transcriptomes using ensemble Support Vector Machines. Sci. Rep. 2017, 7, 40242. [Google Scholar] [CrossRef]
- Wei, L.; Chen, H.; Su, R. M6APred-EL: A Sequence-Based Predictor for Identifying N6-methyladenosine Sites Using Ensemble Learning. Mol. Ther. Nucleic Acids 2018, 12, 635–644. [Google Scholar] [CrossRef]
- Chen, W.; Tran, H.; Liang, Z.; Lin, H.; Zhang, L. Identification and analysis of the N(6)-methyladenosine in the Saccharomyces cerevisiae transcriptome. Sci. Rep. 2015, 5, 13859. [Google Scholar] [CrossRef]
- Li, G.Q.; Liu, Z.; Shen, H.B.; Yu, D.J. TargetM6A: Identifying N(6)-Methyladenosine Sites From RNA Sequences via Position-Specific Nucleotide Propensities and a Support Vector Machine. IEEE Trans. Nanobioscience 2016, 15, 674–682. [Google Scholar] [CrossRef] [PubMed]
- Mahmoudi, O.; Wahab, A.; Chong, K.T. iMethyl-Deep: N6 Methyladenosine Identification of Yeast Genome with Automatic Feature Extraction Technique by Using Deep Learning Algorithm. Genes 2020, 11, 529. [Google Scholar] [CrossRef]
- Feng, P.; Ding, H.; Yang, H.; Chen, W.; Lin, H.; Chou, K.C. iRNA-PseColl: Identifying the Occurrence Sites of Different RNA Modifications by Incorporating Collective Effects of Nucleotides into PseKNC. Mol. Ther. Nucleic Acids 2017, 7, 155–163. [Google Scholar] [CrossRef]
- Tahir, M.; Hayat, M.; Chong, K.T. A convolution neural network-based computational model to identify the occurrence sites of various RNA modifications by fusing varied features. Chemom. Intell. Lab. Syst. 2021, 211, 104233. [Google Scholar] [CrossRef]
- Chen, W.; Feng, P.; Ding, H.; Lin, H. Identifying N (6)-methyladenosine sites in the Arabidopsis thaliana transcriptome. Mol. Genet. Genom. 2016, 291, 2225–2229. [Google Scholar] [CrossRef] [PubMed]
- Xiang, S.; Yan, Z.; Liu, K.; Zhang, Y.; Sun, Z. AthMethPre: A web server for the prediction and query of mRNA m(6)A sites in Arabidopsis thaliana. Mol. Biosyst. 2016, 12, 3333–3337. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Wang, J.; Li, X.; Liang, Y. M6A-GSMS: Computational identification of N(6)-methyladenosine sites with GBDT and stacking learning in multiple species. J. Biomol. Struct. Dyn. 2022, 40, 12380–12391. [Google Scholar] [CrossRef] [PubMed]
- Akbar, S.; Hayat, M. iMethyl-STTNC: Identification of N(6)-methyladenosine sites by extending the idea of SAAC into Chou’s PseAAC to formulate RNA sequences. J. Theor. Biol. 2018, 455, 205–211. [Google Scholar] [CrossRef]
- Chen, W.; Tang, H.; Lin, H. MethyRNA: A web server for identification of N(6)-methyladenosine sites. J. Biomol. Struct. Dyn. 2017, 35, 683–687. [Google Scholar] [CrossRef]
- Chen, W.; Feng, P.; Yang, H.; Ding, H.; Lin, H.; Chou, K.C. iRNA-3typeA: Identifying Three Types of Modification at RNA’s Adenosine Sites. Mol. Ther. Nucleic Acids 2018, 11, 468–474. [Google Scholar] [CrossRef]
- Xing, P.; Su, R.; Guo, F.; Wei, L. Identifying N(6)-methyladenosine sites using multi-interval nucleotide pair position specificity and support vector machine. Sci. Rep. 2017, 7, 46757. [Google Scholar] [CrossRef]
- Tahir, M.; Hayat, M.; Chong, K.T. Prediction of N6-methyladenosine sites using convolution neural network model based on distributed feature representations. Neural. Netw. 2020, 129, 385–391. [Google Scholar] [CrossRef]
- Nazari, I.; Tahir, M.; Tayara, H.; Chong, K.T. iN6-Methyl (5-step): Identifying RNA N6-methyladenosine sites using deep learning mode via Chou’s 5-step rules and Chou’s general PseKNC. Chemom. Intell. Lab. Syst. 2019, 193, 103811. [Google Scholar] [CrossRef]
- Pian, C.; Yang, Z.; Yang, Y.; Zhang, L.; Chen, Y. Identifying RNA N6-Methyladenine Sites in Three Species Based on a Markov Model. Front. Genet. 2021, 12, 650803. [Google Scholar] [CrossRef] [PubMed]
- Rehman, M.U.; Chong, K.T. A Neural Network Based Computational Model for Post-transcriptional Modification Site Identification. In Proceedings of the 2021 21st International Conference on Control, Automation and Systems (ICCAS), Jeju, Republic of Korea, 12–15 October 2021; pp. 1853–1857. [Google Scholar]
- Wang, Y.; Guo, R.; Huang, L.; Yang, S.; Hu, X.; He, K. m6AGE: A Predictor for N6-Methyladenosine Sites Identification Utilizing Sequence Characteristics and Graph Embedding-Based Geometrical Information. Front. Genet. 2021, 12, 670852. [Google Scholar] [CrossRef] [PubMed]
- Islam, N.; Park, J. bCNN-Methylpred: Feature-Based Prediction of RNA Sequence Modification Using Branch Convolutional Neural Network. Genes 2021, 12, 1155. [Google Scholar] [CrossRef]
- Zhang, Z.; Wang, L. Using Chou’s 5-steps rule to identify N(6)-methyladenine sites by ensemble learning combined with multiple feature extraction methods. J. Biomol. Struct. Dyn. 2022, 40, 796–806. [Google Scholar] [CrossRef]
- Rehman, M.U.; Hong, K.J.; Tayara, H.; to Chong, K. m6A-NeuralTool: Convolution neural tool for RNA N6-methyladenosine site identification in different species. IEEE Access 2021, 9, 17779–17786. [Google Scholar] [CrossRef]
- Qiang, X.; Chen, H.; Ye, X.; Su, R.; Wei, L. M6AMRFS: Robust Prediction of N6-Methyladenosine Sites With Sequence-Based Features in Multiple Species. Front. Genet. 2018, 9, 495. [Google Scholar] [CrossRef] [PubMed]
- Song, Z.; Huang, D.; Song, B.; Chen, K.; Song, Y.; Liu, G.; Su, J.; Magalhães, J.P.; Rigden, D.J.; Meng, J. Attention-based multi-label neural networks for integrated prediction and interpretation of twelve widely occurring RNA modifications. Nat. Commun. 2021, 12, 4011. [Google Scholar] [CrossRef]
- Chen, K.; Wei, Z.; Zhang, Q.; Wu, X.; Rong, R.; Lu, Z.; Su, J.; de Magalhães, J.P.; Rigden, D.J.; Meng, J. WHISTLE: A high-accuracy map of the human N6-methyladenosine (m6A) epitranscriptome predicted using a machine learning approach. Nucleic Acids Res. 2019, 47, e41. [Google Scholar] [CrossRef]
- Zhao, Z.; Peng, H.; Lan, C.; Zheng, Y.; Fang, L.; Li, J. Imbalance learning for the prediction of N(6)-Methylation sites in mRNAs. BMC Genom. 2018, 19, 574. [Google Scholar] [CrossRef]
- Körtel, N.; Rücklé, C.; Zhou, Y.; Busch, A.; Hoch-Kraft, P.; Sutandy, F.X.R.; Haase, J.; Pradhan, M.; Musheev, M.; Ostareck, D.; et al. Deep and accurate detection of m6A RNA modifications using miCLIP2 and m6Aboost machine learning. Nucleic Acids Res. 2021, 49, e92. [Google Scholar] [CrossRef]
- Xiang, S.; Liu, K.; Yan, Z.; Zhang, Y.; Sun, Z. RNAMethPre: A Web Server for the Prediction and Query of mRNA m6A Sites. PLoS ONE 2016, 11, e0162707. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Zeng, P.; Li, Y.H.; Zhang, Z.; Cui, Q. SRAMP: Prediction of mammalian N6-methyladenosine (m6A) sites based on sequence-derived features. Nucleic Acids Res. 2016, 44, e91. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Xie, J.; Xu, S. M6A-BiNP: Predicting N(6)-methyladenosine sites based on bidirectional position-specific propensities of polynucleotides and pointwise joint mutual information. RNA Biol. 2021, 18, 2498–2512. [Google Scholar] [CrossRef]
- Song, B.; Chen, K.; Tang, Y.; Wei, Z.; Su, J.; de Magalhães, J.P.; Rigden, D.J.; Meng, J. ConsRM: Collection and large-scale prediction of the evolutionarily conserved RNA methylation sites, with implications for the functional epitranscriptome. Brief Bioinform. 2021, 22, bbab088. [Google Scholar] [CrossRef]
- Zhang, Y.; Hamada, M. DeepM6ASeq: Prediction and characterization of m6A-containing sequences using deep learning. BMC Bioinform. 2018, 19, 524. [Google Scholar] [CrossRef]
- Zou, Q.; Xing, P.; Wei, L.; Liu, B. Gene2vec: Gene subsequence embedding for prediction of mammalian N(6)-methyladenosine sites from mRNA. Rna 2019, 25, 205–218. [Google Scholar] [CrossRef]
- Xiong, Y.; He, X.; Zhao, D.; Tian, T.; Hong, L.; Jiang, T.; Zeng, J. Modeling multi-species RNA modification through multi-task curriculum learning. Nucleic Acids Res. 2021, 49, 3719–3734. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Zou, Q.; Li, J. DeepM6ASeq-EL: Prediction of human N6-methyladenosine (m 6A) sites with LSTM and ensemble learning. Front. Comput. Sci. 2022, 16, 162302. [Google Scholar] [CrossRef]
- Dao, F.Y.; Lv, H.; Yang, Y.H.; Zulfiqar, H.; Gao, H.; Lin, H. Computational identification of N6-methyladenosine sites in multiple tissues of mammals. Comput. Struct. Biotechnol. J. 2020, 18, 1084–1091. [Google Scholar] [CrossRef]
- Liu, K.; Cao, L.; Du, P.; Chen, W. im6A-TS-CNN: Identifying the N(6)-Methyladenine Site in Multiple Tissues by Using the Convolutional Neural Network. Mol. Ther. Nucleic Acids 2020, 21, 1044–1049. [Google Scholar] [CrossRef]
- Jia, C.; Dong, J.; Wang, X.; Zhao, Q. Tissue specific prediction of N-methyladenine sites based on an ensemble of multi-input hybrid neural network. Biocell 2022, 46, 1105. [Google Scholar] [CrossRef]
- Zhang, L.; Qin, X.; Liu, M.; Xu, Z.; Liu, G. DNN-m6A: A Cross-Species Method for Identifying RNA N6-Methyladenosine Sites Based on Deep Neural Network with Multi-Information Fusion. Genes 2021, 12, 354. [Google Scholar] [CrossRef]
- Abbas, Z.; Tayara, H.; Zou, Q.; Chong, K.T. TS-m6A-DL: Tissue-specific identification of N6-methyladenosine sites using a universal deep learning model. Comput. Struct. Biotechnol. J. 2021, 19, 4619–4625. [Google Scholar] [CrossRef]
- Luo, X.; Li, H.; Liang, J.; Zhao, Q.; Xie, Y.; Ren, J.; Zuo, Z. RMVar: An updated database of functional variants involved in RNA modifications. Nucleic Acids Res. 2021, 49, D1405–D1412. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.J.; Li, J.H.; Liu, S.; Wu, J.; Zhou, H.; Qu, L.H.; Yang, J.H. RMBase: A resource for decoding the landscape of RNA modifications from high-throughput sequencing data. Nucleic Acids Res. 2016, 44, D259–D265. [Google Scholar] [CrossRef]
- Xuan, J.J.; Sun, W.J.; Lin, P.H.; Zhou, K.R.; Liu, S.; Zheng, L.L.; Qu, L.H.; Yang, J.H. RMBase v2.0: Deciphering the map of RNA modifications from epitranscriptome sequencing data. Nucleic Acids Res. 2018, 46, D327–D334. [Google Scholar] [CrossRef]
- Liu, H.; Flores, M.A.; Meng, J.; Zhang, L.; Zhao, X.; Rao, M.K.; Chen, Y.; Huang, Y. MeT-DB: A database of transcriptome methylation in mammalian cells. Nucleic Acids Res. 2015, 43, D197–D203. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Wang, H.; Wei, Z.; Zhang, S.; Hua, G.; Zhang, S.-W.; Zhang, L.; Gao, S.-J.; Meng, J.; Chen, X. MeT-DB V2. 0: Elucidating context-specific functions of N 6-methyl-adenosine methyltranscriptome. Nucleic Acids Res. 2018, 46, D281–D287. [Google Scholar] [CrossRef] [PubMed]
- Huang, G.; Huang, X.; Jiang, J. Deepm6A-MT: A deep learning-based method for identifying RNA N6-methyladenosine sites in multiple tissues. Methods 2024, 226, 1–8. [Google Scholar] [CrossRef]
- Wang, H.; Zeng, W.; Huang, X.; Liu, Z.; Sun, Y.; Zhang, L. MTTLm(6)A: A multi-task transfer learning approach for base-resolution mRNA m(6)A site prediction based on an improved transformer. Math. Biosci. Eng. 2024, 21, 272–299. [Google Scholar] [CrossRef]
- Tu, G.; Wang, X.; Xia, R.; Song, B. m6A-TCPred: A web server to predict tissue-conserved human m(6)A sites using machine learning approach. BMC Bioinform. 2024, 25, 127. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Godzik, A. Cd-hit: A fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 2006, 22, 1658–1659. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Chen, K.; Song, B.; Ma, J.; Wu, X.; Xu, Q.; Wei, Z.; Su, J.; Liu, G.; Rong, R.; et al. m6A-Atlas: A comprehensive knowledgebase for unraveling the N6-methyladenosine (m6A) epitranscriptome. Nucleic Acids Res. 2021, 49, D134–D143. [Google Scholar] [CrossRef]
- Dierks, D.; Garcia-Campos, M.A.; Uzonyi, A.; Safra, M.; Edelheit, S.; Rossi, A.; Sideri, T.; Varier, R.A.; Brandis, A.; Stelzer, Y.; et al. Multiplexed profiling facilitates robust m6A quantification at site, gene and sample resolution. Nat. Methods 2021, 18, 1060–1067. [Google Scholar] [CrossRef]
- Zhang, Z.; Chen, T.; Chen, H.X.; Xie, Y.Y.; Chen, L.Q.; Zhao, Y.L.; Liu, B.D.; Jin, L.; Zhang, W.; Liu, C.; et al. Systematic calibration of epitranscriptomic maps using a synthetic modification-free RNA library. Nat. Methods 2021, 18, 1213–1222. [Google Scholar] [CrossRef]
- Hu, L.; Liu, S.; Peng, Y.; Ge, R.; Su, R.; Senevirathne, C.; Harada, B.T.; Dai, Q.; Wei, J.; Zhang, L.; et al. m(6)A RNA modifications are measured at single-base resolution across the mammalian transcriptome. Nat. Biotechnol 2022, 40, 1210–1219. [Google Scholar] [CrossRef]
- Cheng, W.; Liu, F.; Ren, Z.; Chen, W.; Chen, Y.; Liu, T.; Ma, Y.; Cao, N.; Wang, J. Parallel functional assessment of m(6)A sites in human endodermal differentiation with base editor screens. Nat. Commun. 2022, 13, 478. [Google Scholar] [CrossRef]
- Liu, B.; Liu, F.; Wang, X.; Chen, J.; Fang, L.; Chou, K.C. Pse-in-One: A web server for generating various modes of pseudo components of DNA, RNA, and protein sequences. Nucleic Acids Res. 2015, 43, W65–W71. [Google Scholar] [CrossRef]
- Chen, W.; Lei, T.Y.; Jin, D.C.; Lin, H.; Chou, K.C. PseKNC: A flexible web server for generating pseudo K-tuple nucleotide composition. Anal. Biochem. 2014, 456, 53–60. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Zhang, X.; Brooker, J.; Lin, H.; Zhang, L.; Chou, K.C. PseKNC-General: A cross-platform package for generating various modes of pseudo nucleotide compositions. Bioinformatics 2015, 31, 119–120. [Google Scholar] [CrossRef]
- Chen, Z.; Zhao, P.; Li, F.; Marquez-Lago, T.T.; Leier, A.; Revote, J.; Zhu, Y.; Powell, D.R.; Akutsu, T.; Webb, G.I.; et al. iLearn: An integrated platform and meta-learner for feature engineering, machine-learning analysis and modeling of DNA, RNA and protein sequence data. Brief Bioinform. 2020, 21, 1047–1057. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Zhao, P.; Li, C.; Li, F.; Xiang, D.; Chen, Y.Z.; Akutsu, T.; Daly, R.J.; Webb, G.I.; Zhao, Q.; et al. iLearnPlus: A comprehensive and automated machine-learning platform for nucleic acid and protein sequence analysis, prediction and visualization. Nucleic Acids Res. 2021, 49, e60. [Google Scholar] [CrossRef]
- Chen, W.; Yang, H.; Feng, P.; Ding, H.; Lin, H. iDNA4mC: Identifying DNA N4-methylcytosine sites based on nucleotide chemical properties. Bioinformatics 2017, 33, 3518–3523. [Google Scholar] [CrossRef]
- Xu, Z.C.; Feng, P.M.; Yang, H.; Qiu, W.R.; Chen, W.; Lin, H. iRNAD: A computational tool for identifying D modification sites in RNA sequence. Bioinformatics 2019, 35, 4922–4929. [Google Scholar] [CrossRef]
- Chen, W.; Song, X.; Lv, H.; Lin, H. iRNA-m2G: Identifying N(2)-methylguanosine Sites Based on Sequence-Derived Information. Mol. Ther. Nucleic Acids 2019, 18, 253–258. [Google Scholar] [CrossRef]
- Yang, H.; Lv, H.; Ding, H.; Chen, W.; Lin, H. iRNA-2OM: A Sequence-Based Predictor for Identifying 2′-O-Methylation Sites in Homo sapiens. J. Comput. Biol. 2018, 25, 1266–1277. [Google Scholar] [CrossRef] [PubMed]
- Lorenz, R.; Bernhart, S.H.; Höner Zu Siederdissen, C.; Tafer, H.; Flamm, C.; Stadler, P.F.; Hofacker, I.L. ViennaRNA Package 2.0. Algorithms Mol. Biol 2011, 6, 26. [Google Scholar] [CrossRef]
- Long, H.; Liao, B.; Xu, X.; Yang, J. A Hybrid Deep Learning Model for Predicting Protein Hydroxylation Sites. Int. J. Mol. Sci. 2018, 19, 2817. [Google Scholar] [CrossRef]
- Wang, D.; Liang, Y.; Xu, D. Capsule network for protein post-translational modification site prediction. Bioinformatics 2019, 35, 2386–2394. [Google Scholar] [CrossRef]
- Rao, R.S.P.; Zhang, N.; Xu, D.; Møller, I.M. CarbonylDB: A curated data-resource of protein carbonylation sites. Bioinformatics 2018, 34, 2518–2520. [Google Scholar] [CrossRef] [PubMed]
- Cai, Y.; Wang, J.; Deng, L. SDN2GO: An Integrated Deep Learning Model for Protein Function Prediction. Front. Bioeng. Biotechnol. 2020, 8, 391. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zhang, J.; Cai, Y.; Deng, L. DeepMiR2GO: Inferring Functions of Human MicroRNAs Using a Deep Multi-Label Classification Model. Int. J. Mol. Sci. 2019, 20, 6046. [Google Scholar] [CrossRef] [PubMed]
- Krizhevsky, A.; Sutskever, I.; Hinton, G.E. Imagenet classification with deep convolutional neural networks. Adv. Neural Inf. Process. Syst. 2012, 25. [Google Scholar] [CrossRef]
- Popova, M.; Isayev, O.; Tropsha, A. Deep reinforcement learning for de novo drug design. Sci. Adv. 2018, 4, eaap7885. [Google Scholar] [CrossRef]
- Ståhl, N.; Falkman, G.; Karlsson, A.; Mathiason, G.; Boström, J. Deep Reinforcement Learning for Multiparameter Optimization in de novo Drug Design. J. Chem. Inf. Model. 2019, 59, 3166–3176. [Google Scholar] [CrossRef]
- Deng, L.; Cai, Y.; Zhang, W.; Yang, W.; Gao, B.; Liu, H. Pathway-Guided Deep Neural Network toward Interpretable and Predictive Modeling of Drug Sensitivity. J. Chem. Inf. Model. 2020, 60, 4497–4505. [Google Scholar] [CrossRef]
- Chen, B.; Khodadoust, M.S.; Olsson, N.; Wagar, L.E.; Fast, E.; Liu, C.L.; Muftuoglu, Y.; Sworder, B.J.; Diehn, M.; Levy, R.; et al. Predicting HLA class II antigen presentation through integrated deep learning. Nat. Biotechnol. 2019, 37, 1332–1343. [Google Scholar] [CrossRef]
- Racle, J.; Michaux, J.; Rockinger, G.A.; Arnaud, M.; Bobisse, S.; Chong, C.; Guillaume, P.; Coukos, G.; Harari, A.; Jandus, C.; et al. Robust prediction of HLA class II epitopes by deep motif deconvolution of immunopeptidomes. Nat. Biotechnol. 2019, 37, 1283–1286. [Google Scholar] [CrossRef]
- Xu, Z.; Luo, M.; Lin, W.; Xue, G.; Wang, P.; Jin, X.; Xu, C.; Zhou, W.; Cai, Y.; Yang, W.; et al. DLpTCR: An ensemble deep learning framework for predicting immunogenic peptide recognized by T cell receptor. Brief Bioinform. 2021, 22, bbab335. [Google Scholar] [CrossRef]
- Jiang, Q.; Wang, G.; Jin, S.; Li, Y.; Wang, Y. Predicting human microRNA-disease associations based on support vector machine. Int. J. Data Min. Bioinform. 2013, 8, 282–293. [Google Scholar] [CrossRef]
- Zhang, D.; Xu, Z.C.; Su, W.; Yang, Y.H.; Lv, H.; Yang, H.; Lin, H. iCarPS: A computational tool for identifying protein carbonylation sites by novel encoded features. Bioinformatics 2021, 37, 171–177. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Xiao, X.; Xu, Z.C. iPromoter-5mC: A Novel Fusion Decision Predictor for the Identification of 5-Methylcytosine Sites in Genome-Wide DNA Promoters. Front. Cell Dev. Biol. 2020, 8, 614. [Google Scholar] [CrossRef] [PubMed]
- Xiao, X.; Xu, Z.C.; Qiu, W.R.; Wang, P.; Ge, H.T.; Chou, K.C. iPSW(2L)-PseKNC: A two-layer predictor for identifying promoters and their strength by hybrid features via pseudo K-tuple nucleotide composition. Genomics 2019, 111, 1785–1793. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.C.; Wang, P.; Qiu, W.R.; Xiao, X. iSS-PC: Identifying Splicing Sites via Physical-Chemical Properties Using Deep Sparse Auto-Encoder. Sci. Rep. 2017, 7, 8222. [Google Scholar] [CrossRef]
- Xu, Z.-C.; Qiu, W.-R.; Xiao, X. iRSpotH-TNCPseAAC: Identifying recombination spots in human by using pseudo trinucleotide composition with an ensemble of support vector machine classifiers. Lett. Org. Chem. 2017, 14, 703–713. [Google Scholar] [CrossRef]
- Xu, Z.-C.; Jiang, S.-Y.; Qiu, W.-R.; Liu, Y.-C.; Xiao, X. iDHSs-PseTNC: Identifying DNase I hypersensitive sites with pseuo trinucleotide component by deep sparse auto-encoder. Lett. Org. Chem. 2017, 14, 655–664. [Google Scholar] [CrossRef]
- Huang, Y.; Zhou, D.; Wang, Y.; Zhang, X.; Su, M.; Wang, C.; Sun, Z.; Jiang, Q.; Sun, B.; Zhang, Y. Prediction of transcription factors binding events based on epigenetic modifications in different human cells. Epigenomics 2020, 12, 1443–1456. [Google Scholar] [CrossRef]
- Su, Z.D.; Huang, Y.; Zhang, Z.Y.; Zhao, Y.W.; Wang, D.; Chen, W.; Chou, K.C.; Lin, H. iLoc-lncRNA: Predict the subcellular location of lncRNAs by incorporating octamer composition into general PseKNC. Bioinformatics 2018, 34, 4196–4204. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).